Probing the Occurrence of Soluble Oligomers through Amyloid Aggregation Scaling Laws

 , , ,
, , ,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Transmission Electron Microscopy
2.3. Dynamic Light Scattering
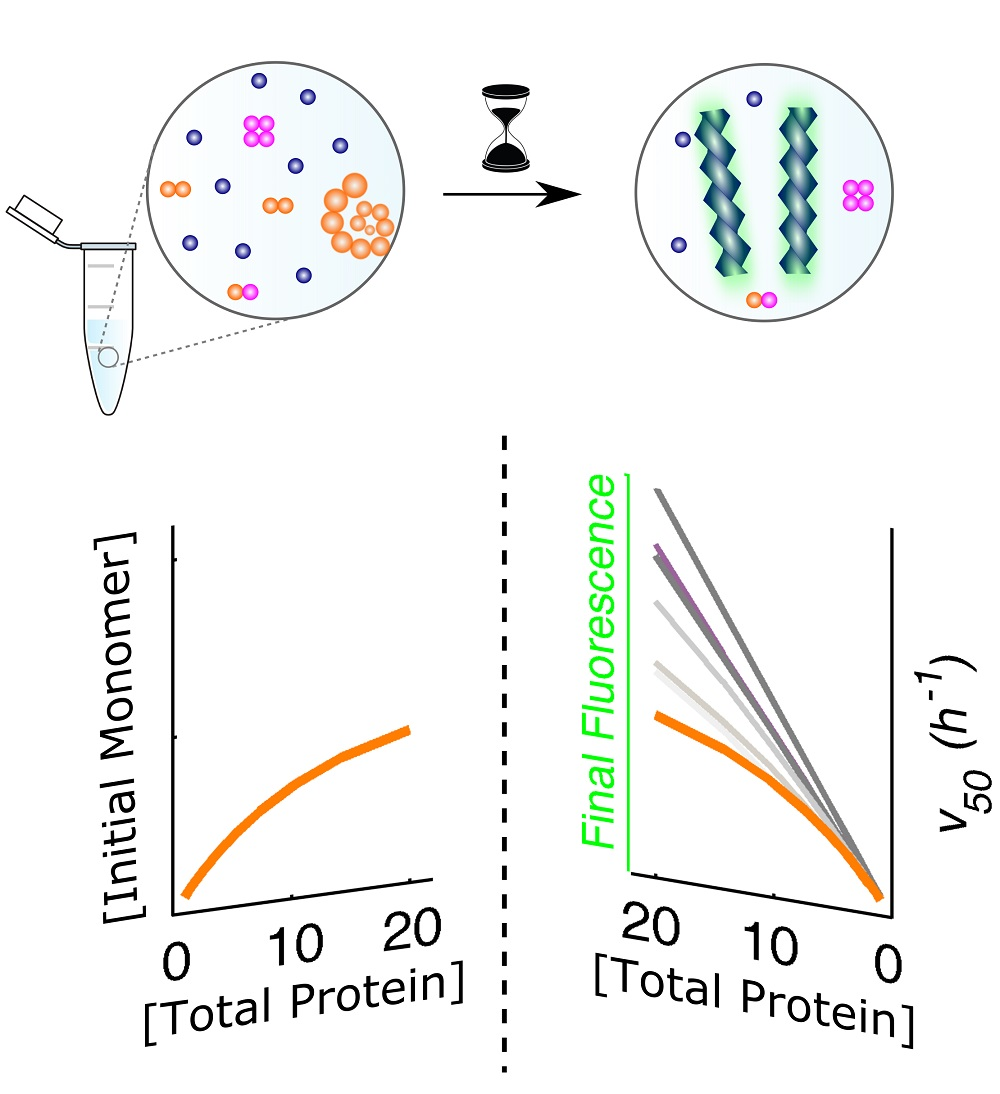
3. Results and Discussion
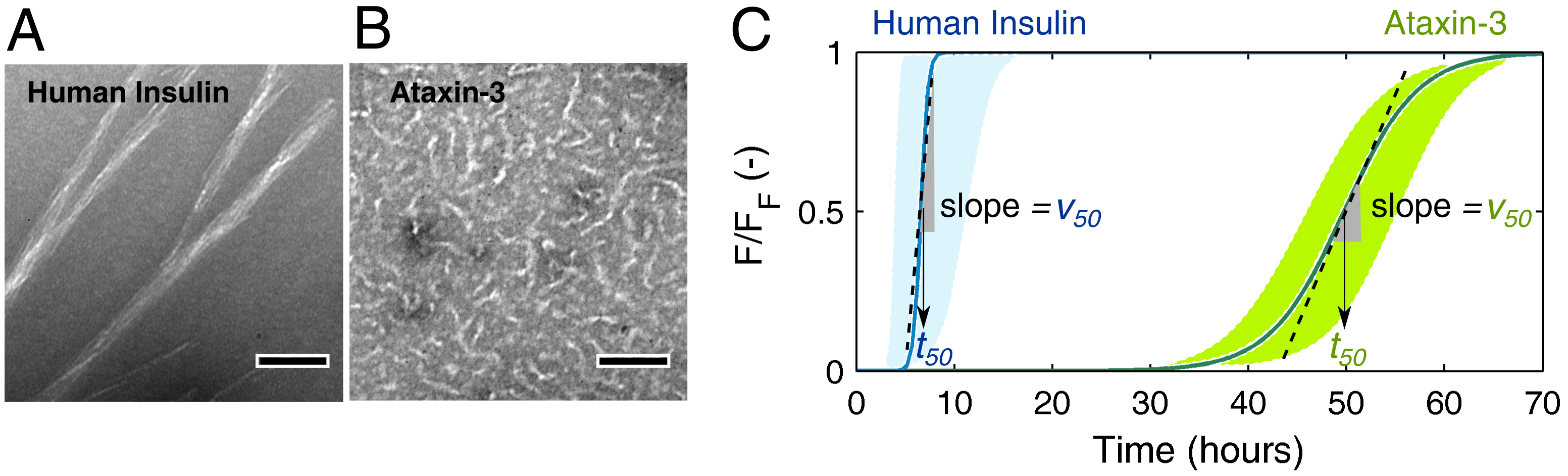
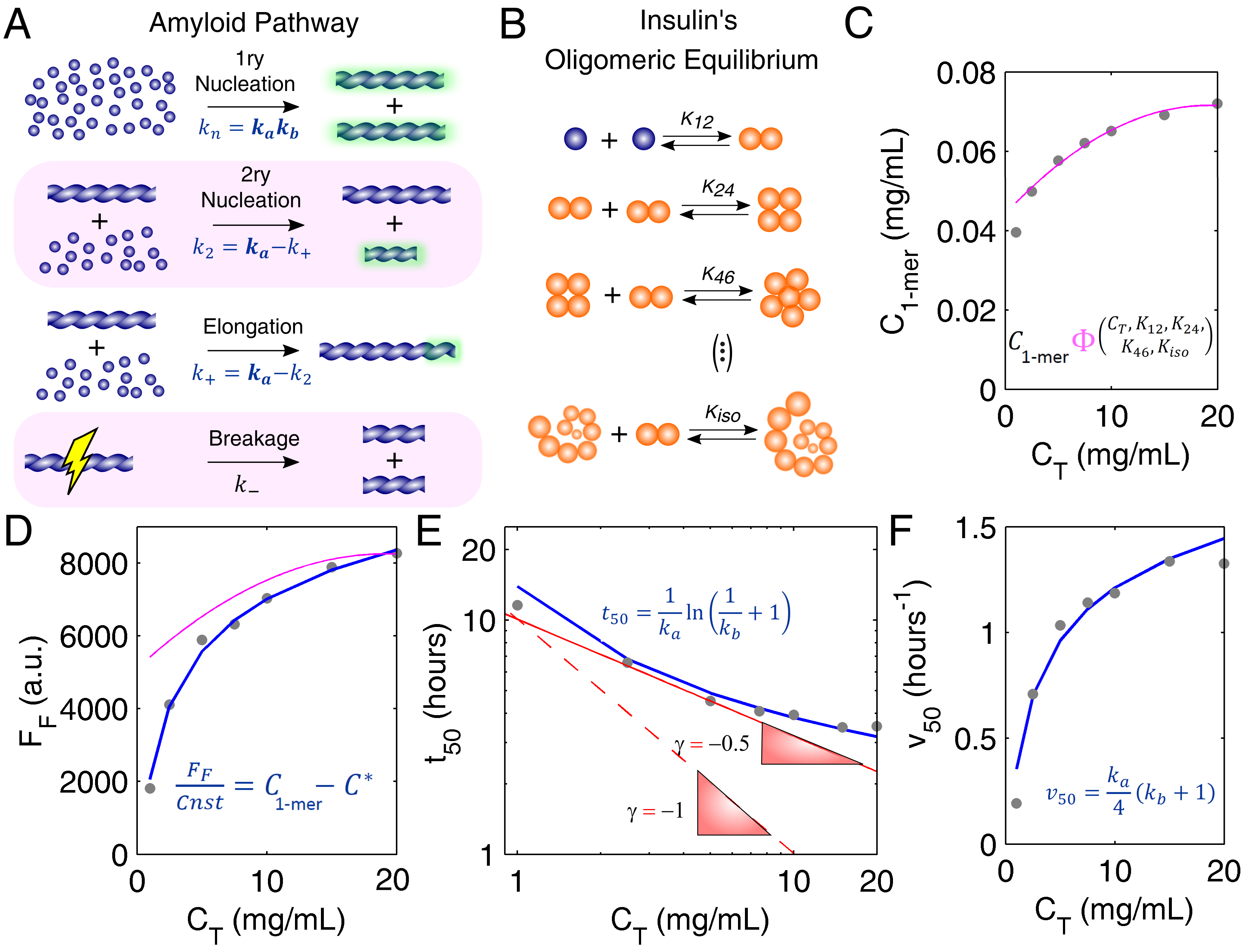
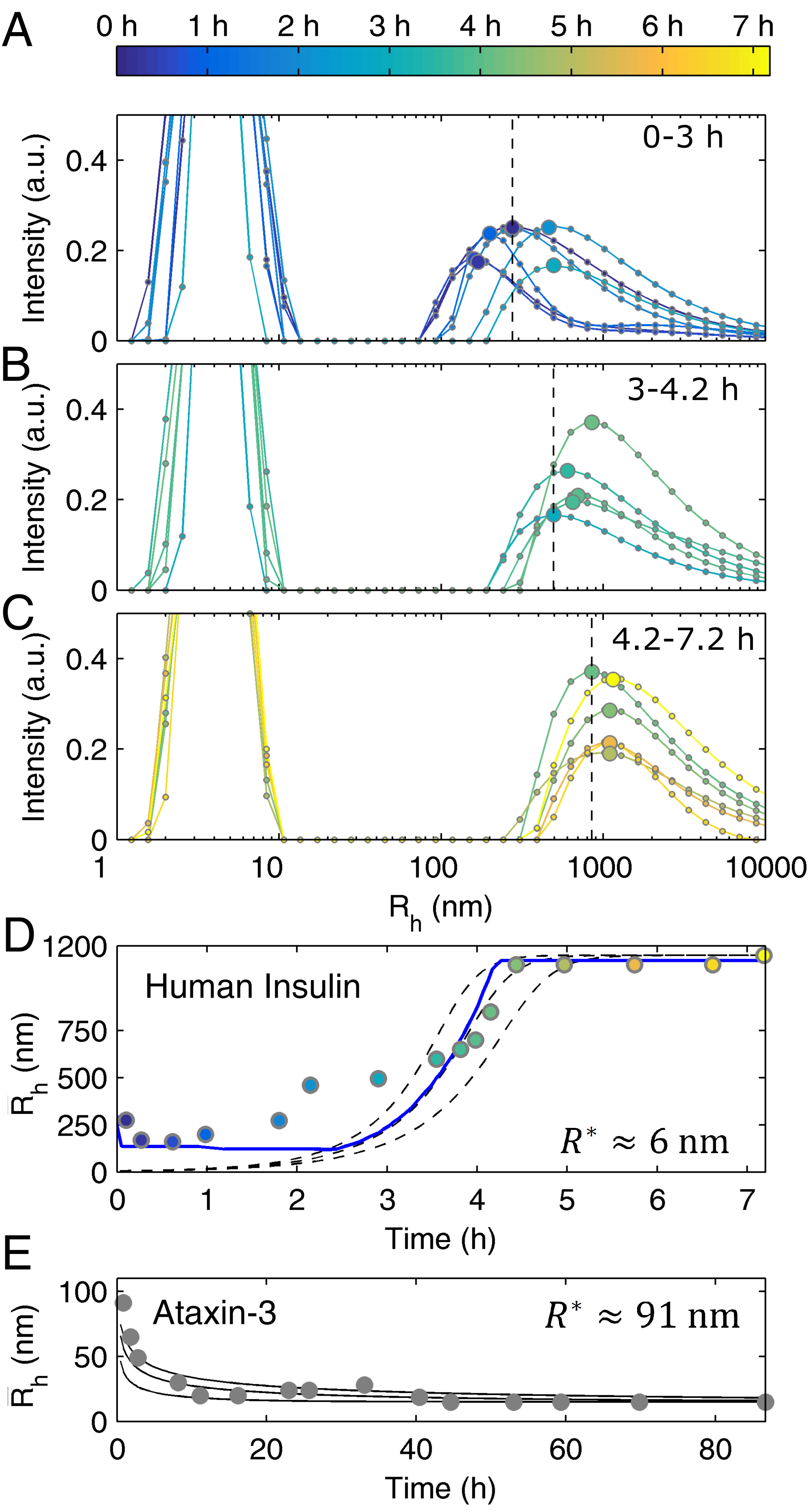
3.1. Mechanistic Analysis of Insulin Aggregation
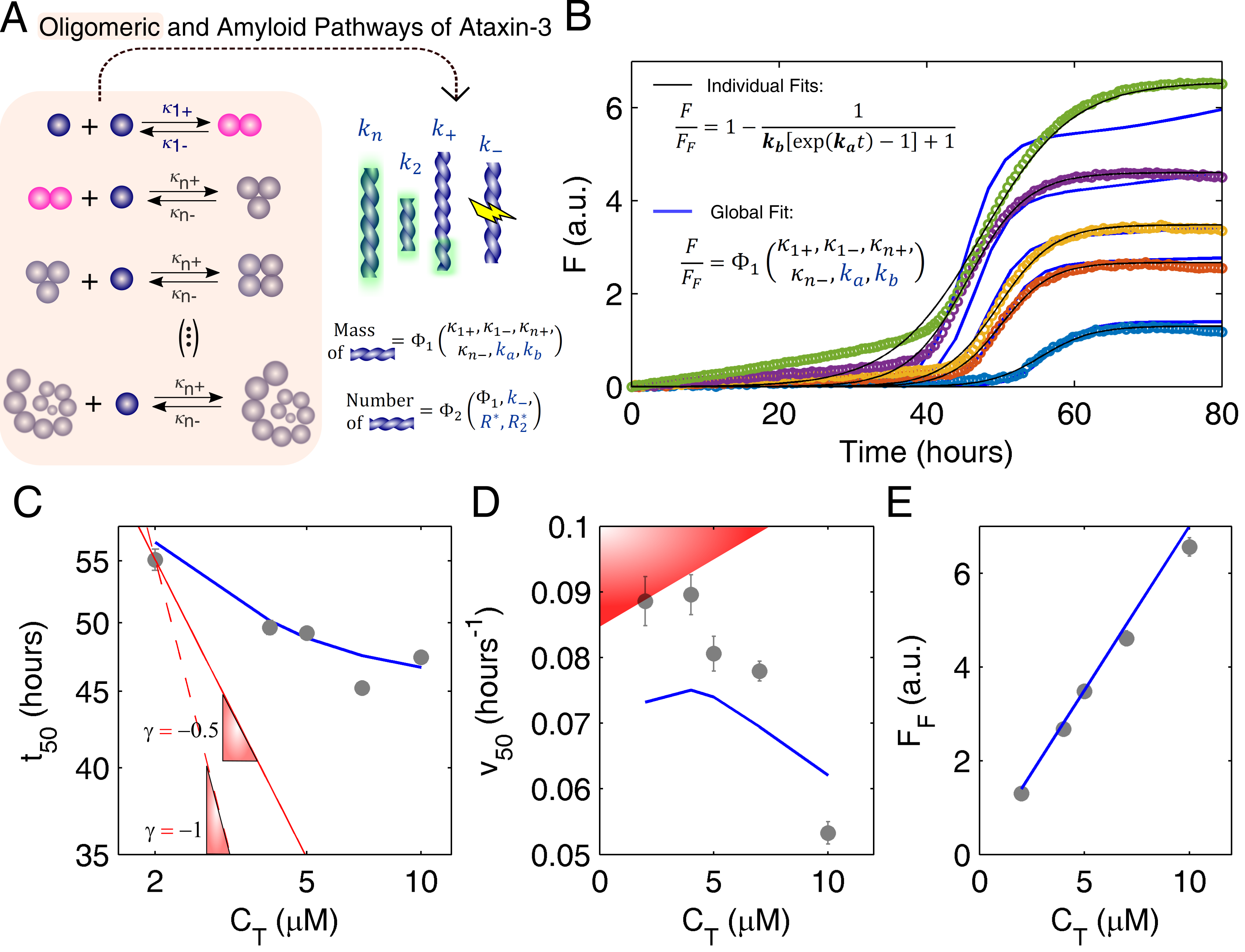
3.2. Mechanistic Analysis of Ataxin-3 Aggregation
3.3. Model Predictions Are Further Confirmed by Size Distribution Analysis of Insulin Aggregation
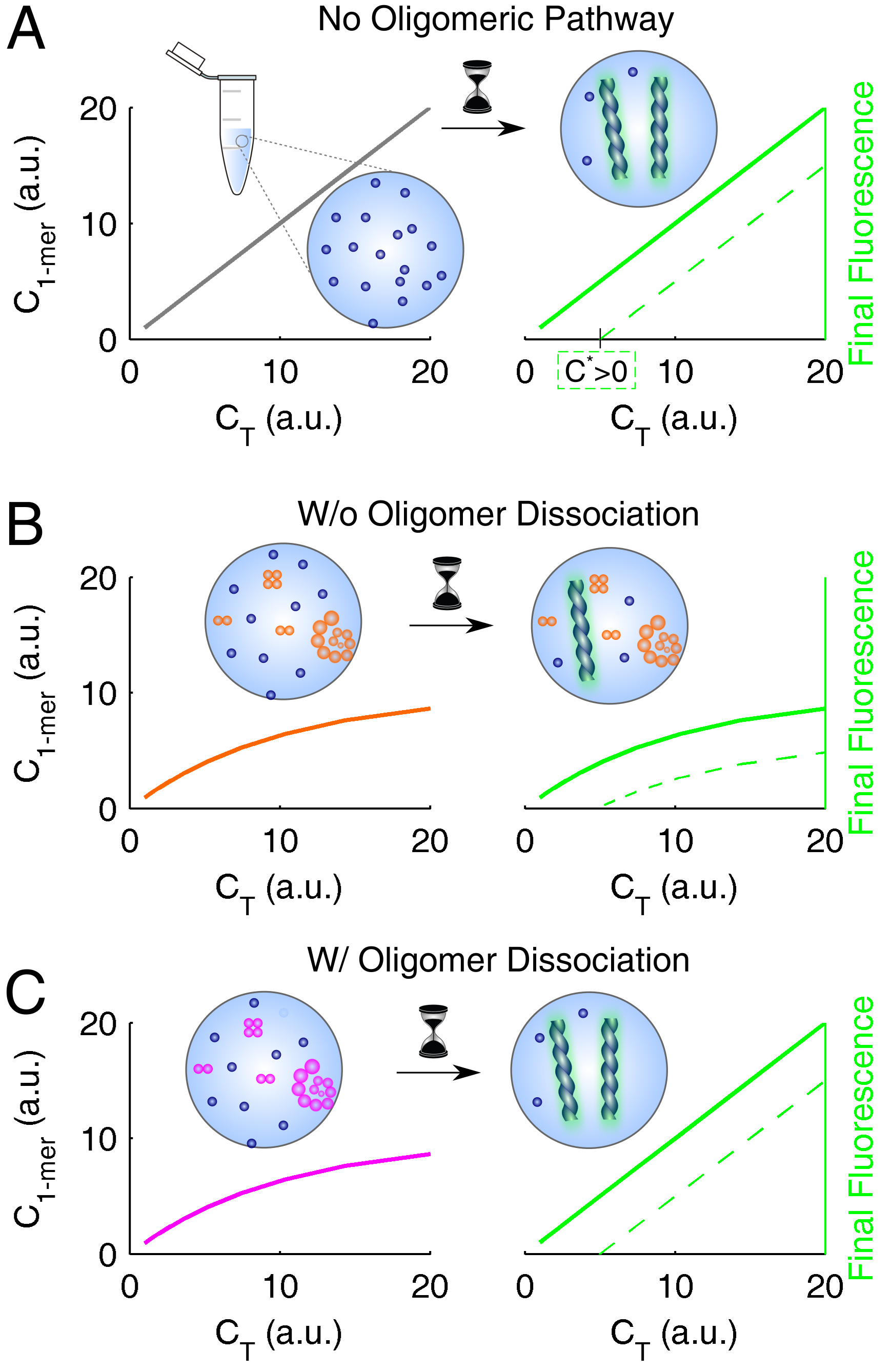
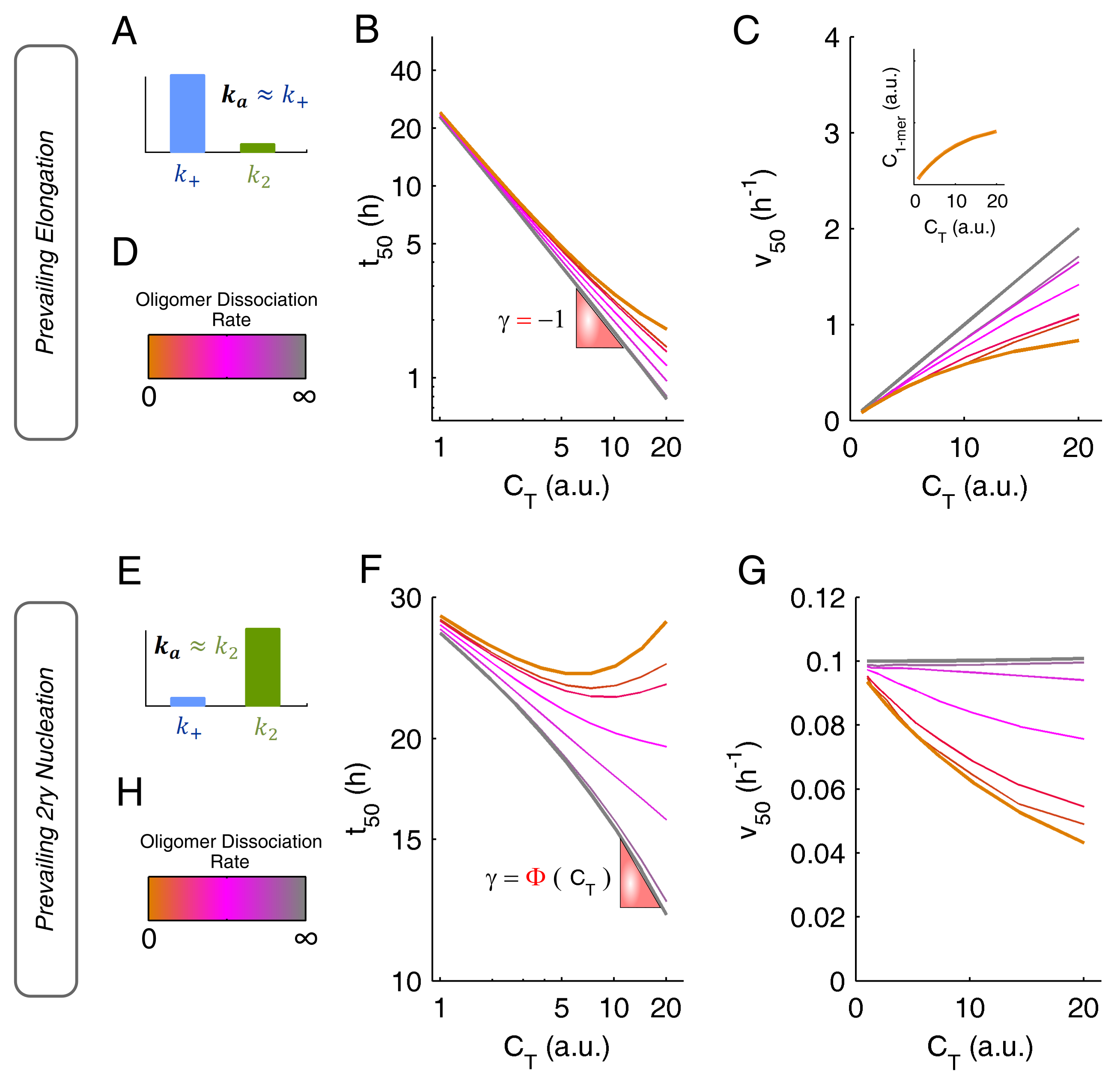
3.4. Systematization of Concepts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Model Equations
Appendix A.1. Closed-Form Model Equations
Appendix A.2. Oligomerization Equilibrium of Insulin
Appendix A.3. Oligomerization Equilibrium of Ataxin-3
Appendix B. Numerical Methods
Appendix B.1. Scaling Laws
Appendix B.2. Discretized Population Balance
References
- David, B.; Hayer-Hartl, M.; Hartl, F.U. In Vivo Aspects of Protein Folding and Quality Control. Science 2016, 353, aac4354. [Google Scholar]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Wienkers, L.C.; Heath, T.G. Predicting In Vivo Drug Interactions from In Vitro Drug Discovery Data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Kundel, F.; Tosatto, L.; Whiten, D.R.; Wirthensohn, D.C.; Horrocks, M.H.; Klenerman, D. Shedding Light on Aberrant Interactions: A Review of Modern Tools for Studying Protein Aggregates. FEBS J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Uversky, V.N. Uversky. Structural, Morphological, and Functional Diversity of Amyloid Oligomers. FEBS Lett. 2015, 589, 2640–2648. [Google Scholar] [CrossRef] [PubMed]
- Young, L.M.; Ashcroft, A.E.; Radford, S.E. Small Molecule Probes of Protein Aggregation. Curr. Opin. Chem. Biol. 2017, 39, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an Understanding of Amyloid-β Oligomers: Characterization, Toxicity Mechanisms, and Inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Chemical Kinetics for Drug Discovery to Combat Protein Aggregation Diseases. Trends Pharmacol. Sci. 2014, 35, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.; Villar-Alvarez, E.; Taboada, P.; Rocha, F.A.; Damas, A.M.; Martins, P.M. What Can the Kinetics of Amyloid Fibril Formation Tell about Off-Pathway Aggregation? J. Biol. Chem. 2016, 291, 2018–2032. [Google Scholar] [CrossRef] [PubMed]
- Padrick, S.B.; Miranker, A.D. Islet Amyloid: Phase Partitioning and Secondary Nucleation Are Central to the Mechanism of Fibrillogenesis. Biochemistry 2002, 41, 4694–4703. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.; Rocha, F.A.; Damas, A.M.; Martins, P.M. A Generic Crystallization-Like Model That Describes the Kinetics of Amyloid Fibril Formation. J. Biol. Chem. 2012, 287, 30585–30594. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.; Dobson, C.M.; Linse, S.; Knowles, T.P. Differences in Nucleation Behavior Underlie the Contrasting Aggregation Kinetics of the Aβ40 and Aβ42 Peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Kirkegaard, J.B.; Arosio, P.; Michaels, T.C.; Vendruscolo, M.; Dobson, C.M.; Linse, S.; Knowles, T.P. Molecular Mechanisms of Protein Aggregation from Global Fitting of Kinetic Models. Nat. Protoc. 2016, 11, 252–272. [Google Scholar] [CrossRef] [PubMed]
- Eden, K.; Morris, R.; Gillam, J.; MacPhee, C.E.; Allen, R.J. Competition between Primary Nucleation and Autocatalysis in Amyloid Fibril Self-Assembly. Biophys. J. 2015, 108, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.J.; Eden, K.; Yarwood, R.; Jourdain, L.; Allen, R.J.; MacPhee, C.E. Mechanistic and Environmental Control of the Prevalence and Lifetime of Amyloid Oligomers. Nat. Commun. 2013, 4, 1891. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Almeida, B.; Fraga, J.S.; Taboada, P.; Martins, P.M.; Macedo-Ribeiro, S. Distribution of Amyloid-Like and Oligomeric Species from Protein Aggregation Kinetics. Angew. Chem. Int. Ed. 2017, 56, 14042–14045. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.V.; Dovidchenko, N.V.; Galzitskaya, O.V. What is Responsible for Atypical Dependence of the Rate of Amyloid Formation on Protein Concentration: Fibril-Catalyzed Initiation of New Fibrils or Competition with Oligomers? J. Phys. Chem. Lett. 2018, 9, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Sun, Z.; Hayden, E.Y.; Teplow, D.B.; Lyubchenko, Y.L. Nanoscale Dynamics of Amyloid β-42 Oligomers as Revealed by High-Speed Atomic Force Microscopy. ACS Nano 2017, 11, 12202–12209. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.; Frokjaer, S.; Brange, J.; Uversky, V.N.; Fink, A.L. Probing the Mechanism of Insulin Fibril Formation with Insulin Mutants. Biochemistry 2001, 40, 8397–8409. [Google Scholar] [CrossRef] [PubMed]
- Bocian, W.; Sitkowski, J.; Tarnowska, A.; Bednarek, E.; Kawȩcki, R.; Koźmiński, W.; Kozerski, L. Direct Insight into Insulin Aggregation by 2D NMR Complemented by PFGSE NMR. Proteins Struct. Funct. Bioinf. 2008, 71, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, B.; Groenning, M.; Roessle, M.; Kastrup, J.S.; Van De Weert, M.; Flink, J.M.; Frokjaer, S.; Gajhede, M.; Svergun, D.I. A Helical Structural Nucleus is the Primary Elongating Unit of Insulin Amyloid Fibrils. PLoS Biol. 2007, 5, e134. [Google Scholar] [CrossRef] [PubMed]
- Scarff, C.A.; Almeida, B.; Fraga, J.; Macedo-Ribeiro, S.; Radford, S.E.; Ashcroft, A.E. Examination of Ataxin-3 (Atx-3) Aggregation by Structural Mass Spectrometry Techniques: A Rationale for Expedited Aggregation Upon Polyglutamine (polyQ) Expansion. Mol. Cell. Proteom. 2015, 14, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Ellisdon, A.M.; Thomas, B.; Bottomley, S.P. The Two-Stage Pathway of Ataxin-3 Fibrillogenesis Involves a Polyglutamine-Independent Step. J. Biol. Chem. 2006, 281, 16888–16896. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W. A Constrained Regularization Method for Inverting Data Represented by Linear Algebraic or Integral Equations. Comput. Phys. Comm. 1982, 27, 213–227. [Google Scholar] [CrossRef]
- Fodera, V.; Librizzi, F.; Groenning, M.; Van De Weert, M.; Leone, M. Secondary Nucleation and Accessible Surface in Insulin Amyloid Fibril Formation. J. Phys. Chem. B 2008, 112, 3853–3858. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.L.; Lomont, J.P.; Tu, L.H.; Raleigh, D.P.; Zanni, M.T. A Free Energy Barrier Caused by the Refolding of an Oligomeric Intermediate Controls the Lag Time of Amyloid Formation by hIAPP. J. Am. Chem. Soc. 2017, 139, 16748–16758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraman, M.; Kodali, R.; Sahoo, B.; Thakur, A.K.; Mayasundari, A.; Mishra, R.; Peterson, C.B.; Wetzel, R. Slow Amyloid Nucleation Via α-Helix-Rich Oligomeric Intermediates in Short Polyglutamine-Containing Huntingtin Fragments. J. Mol. Biol. 2012, 415, 881–899. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Bernacki, J.P.; Murphy, R.M. Model Discrimination and Mechanistic Interpretation of Kinetic Data in Protein Aggregation Studies. Biophys. J. 2009, 96, 2871–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditlev, J.A.; Mayer, B.J.; Loew, L.M. There Is More Than One Way to Model an Elephant. Experiment-Driven Modeling of the Actin Cytoskeleton. Biophys. J. 2013, 104, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fersht, A.R. Mechanism of Initiation of Aggregation of P53 Revealed by Φ-Value Analysis. Proc. Natl. Acad. Sci. USA 2015, 112, 2437–2442. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, D.J.; Wenger, A.; Sundin, E.; Wesén, E.; Westerlund, F.; Esbjörner, E.K. Binding of Thioflavin-T to Amyloid Fibrils Leads to Fluorescence Self-Quenching and Fibril Compaction. Biochemistry 2017, 56, 2170–2174. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Lin, T.Y.; Chang, D.; Guo, Z. Thioflavin-T as an Amyloid Dye: Fibril Quantification, Optimal Concentration and Effect on Aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Ansaloni, A.; Mezzenga, R.; Lashuel, H.A.; Dietler, G. Novel Mechanistic Insight into the Molecular Basis of Amyloid Polymorphism and Secondary Nucleation During Amyloid Formation. J. Mol. Biol. 2013, 425, 1765–1781. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Peters, B. Solute Precipitate Nucleation: A Review of Theory and Simulation Advances. In Advances in Chemical Physics: Volume 155; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 97–160. [Google Scholar]
- Lomakin, A.; Chung, D.S.; Benedek, G.B.; Kirschner, D.A.; Teplow, D.B. On the Nucleation and Growth of Amyloid Beta-Protein Fibrils: Detection of Nuclei and Quantitation of Rate Constants. Proc. Natl. Acad. Sci. USA 1996, 93, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Parmar, A.S.; Gottschall, P.E.; Muschol, M. Pre-Assembled Clusters Distort Crystal Nucleation Kinetics in Supersaturated Lysozyme Solutions. Biophys. Chem. 2007, 129, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.; Barbosa, S.; Taboada, P.; Rocha, F.A.; Damas, A.M.; Martins, P.M. The Nucleation of Protein Crystals as a Race against Time with On- and Off-Pathways. J. Appl. Cryst. 2017, 50, 1056–1065. [Google Scholar] [CrossRef]
- Yan, L.M.; Velkova, A.; Tatarek-Nossol, M.; Rammes, G.; Sibaev, A.; Andreetto, E.; Kracklauer, M.; Bakou, M.; Malideli, E.; Göke, B.; et al. Selectively N-Methylated Soluble IAPP Mimics as Potent IAPP Receptor Agonists and Nanomolar Inhibitors of Cytotoxic Self-Assembly of Both IAPP and Aβ40. Angew. Chem. Int. Ed. 2013, 52, 10378–10383. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Kumar, S.; Rhoades, E.; Miranker, A.D. Conformational Switching within Dynamic Oligomers Underpins Toxic Gain-of-Function by Diabetes-Associated Amyloid. Nat. Commun. 2018, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C.; Sani, M.A.; Ding, F.; Kakinen, A.; Javed, I.; Separovic, F.; Davis, T.P.; Mezzenga, R. Implications of Peptide Assemblies in Amyloid Diseases. Chem. Soc. Rev. 2017, 46, 6492–6531. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-Human Assessment of PRX002, an Anti—α-Synuclein Monoclonal Antibody, in Healthy Volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.M. True and Apparent Inhibition of Amyloid Fibril Formation. Prion 2013, 7, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.R.; Douglass, A.; Vale, R.D.; Weissman, J.S. Mechanism of Prion Propagation: Amyloid Growth Occurs by Monomer Addition. PLoS Biol. 2004, 2, e321. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, A.; Sárkány, Z.; Fraga, J.S.; Taboada, P.; Macedo-Ribeiro, S.; Martins, P.M. Probing the Occurrence of Soluble Oligomers through Amyloid Aggregation Scaling Laws. Biomolecules 2018, 8, 108. https://doi.org/10.3390/biom8040108
Silva A, Sárkány Z, Fraga JS, Taboada P, Macedo-Ribeiro S, Martins PM. Probing the Occurrence of Soluble Oligomers through Amyloid Aggregation Scaling Laws. Biomolecules. 2018; 8(4):108. https://doi.org/10.3390/biom8040108
Chicago/Turabian StyleSilva, Alexandra, Zsuzsa Sárkány, Joana S. Fraga, Pablo Taboada, Sandra Macedo-Ribeiro, and Pedro M. Martins. 2018. "Probing the Occurrence of Soluble Oligomers through Amyloid Aggregation Scaling Laws" Biomolecules 8, no. 4: 108. https://doi.org/10.3390/biom8040108
APA StyleSilva, A., Sárkány, Z., Fraga, J. S., Taboada, P., Macedo-Ribeiro, S., & Martins, P. M. (2018). Probing the Occurrence of Soluble Oligomers through Amyloid Aggregation Scaling Laws. Biomolecules, 8(4), 108. https://doi.org/10.3390/biom8040108