Analytical Considerations of Stable Isotope Labelling in Lipidomics


Abstract
:
1. Introduction
2. Fundamentals
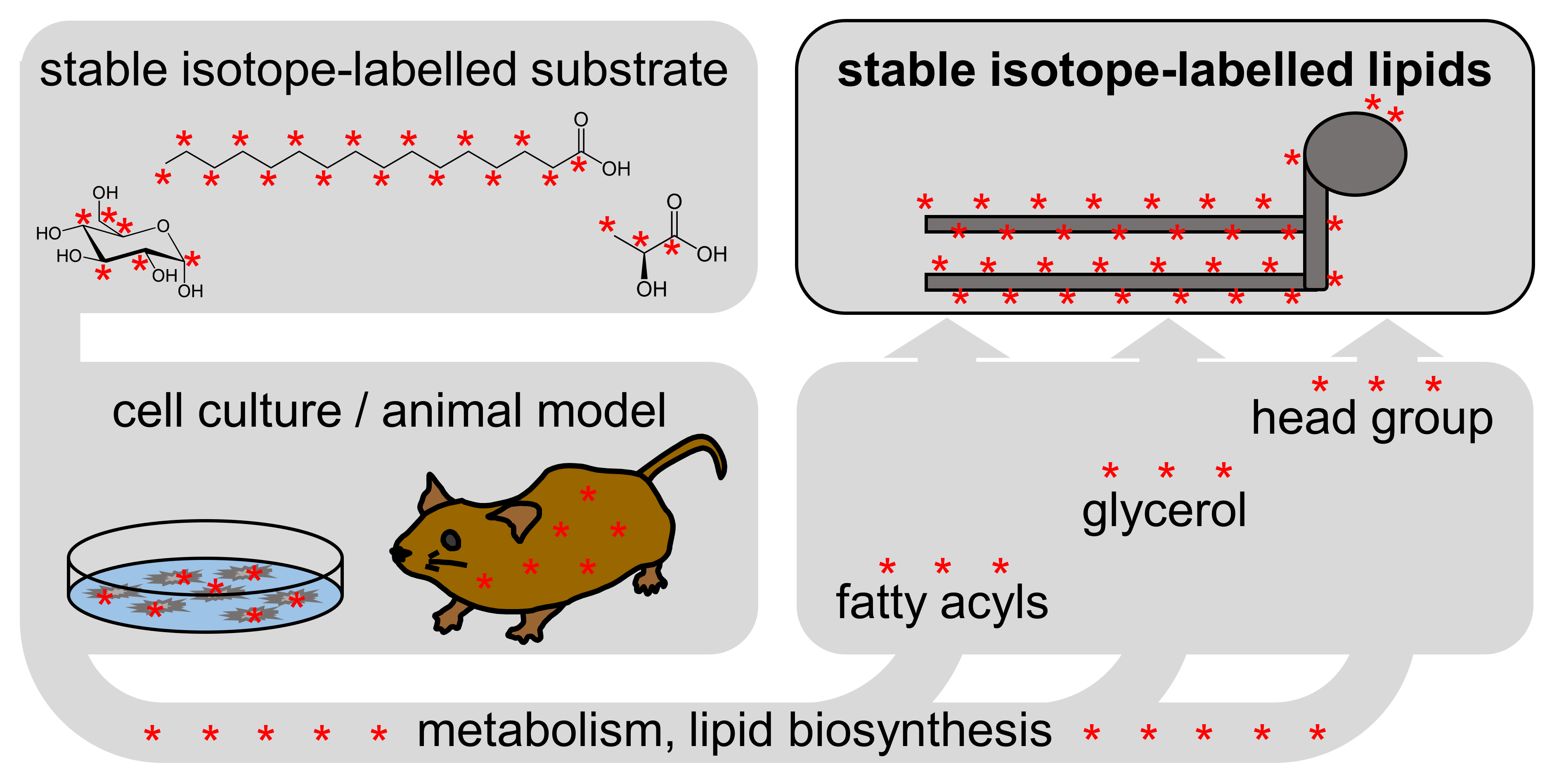
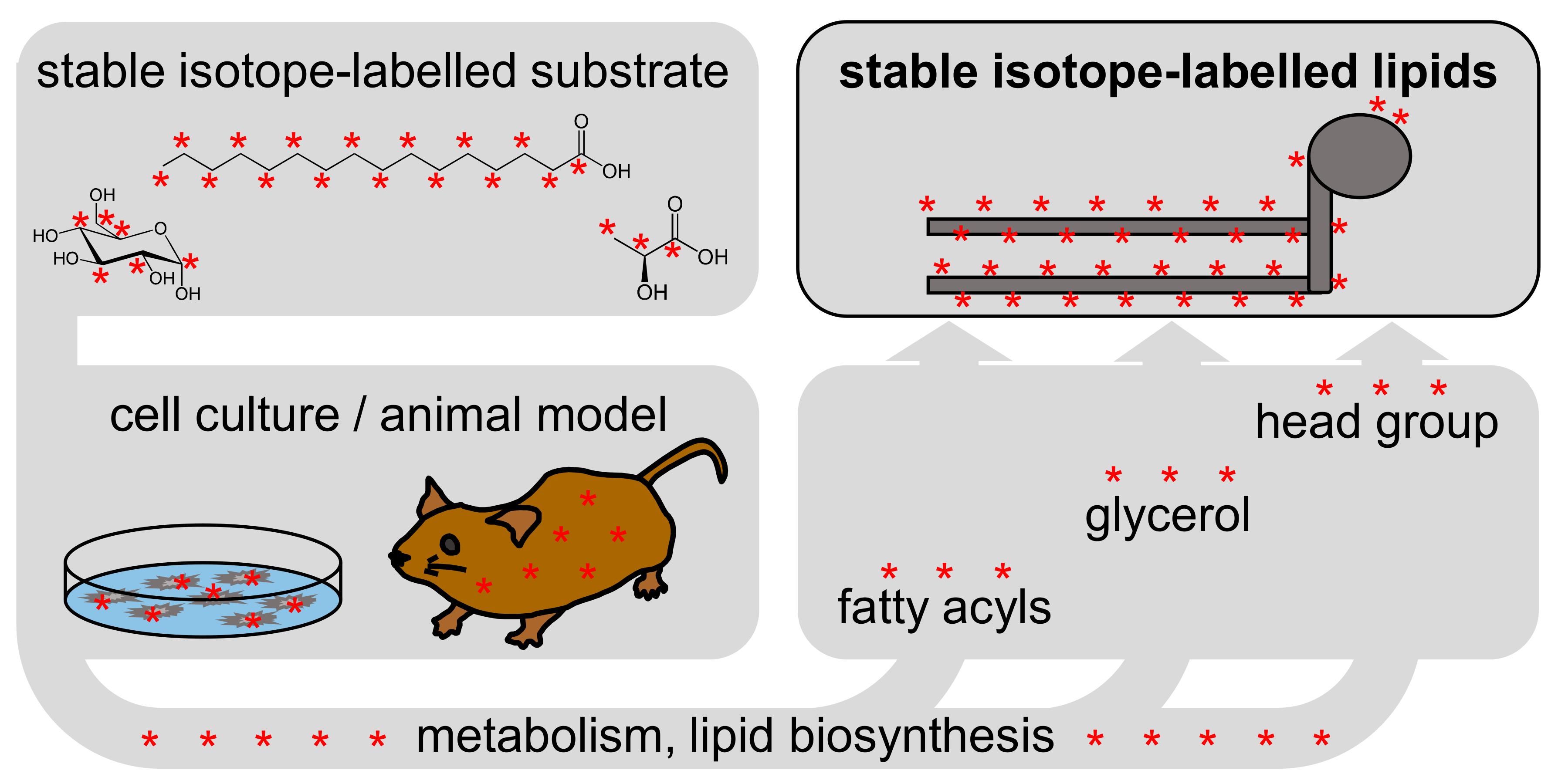
2.1. Stable Isotope-Labelled Compounds in Lipidomics
2.2. Tracers and Tracees
3. Analytical Considerations
3.1. Choice of Tracer
3.2. Sample Preparation
3.3. Sample Introduction
3.4. Instrumental Considerations
4. Data Analysis
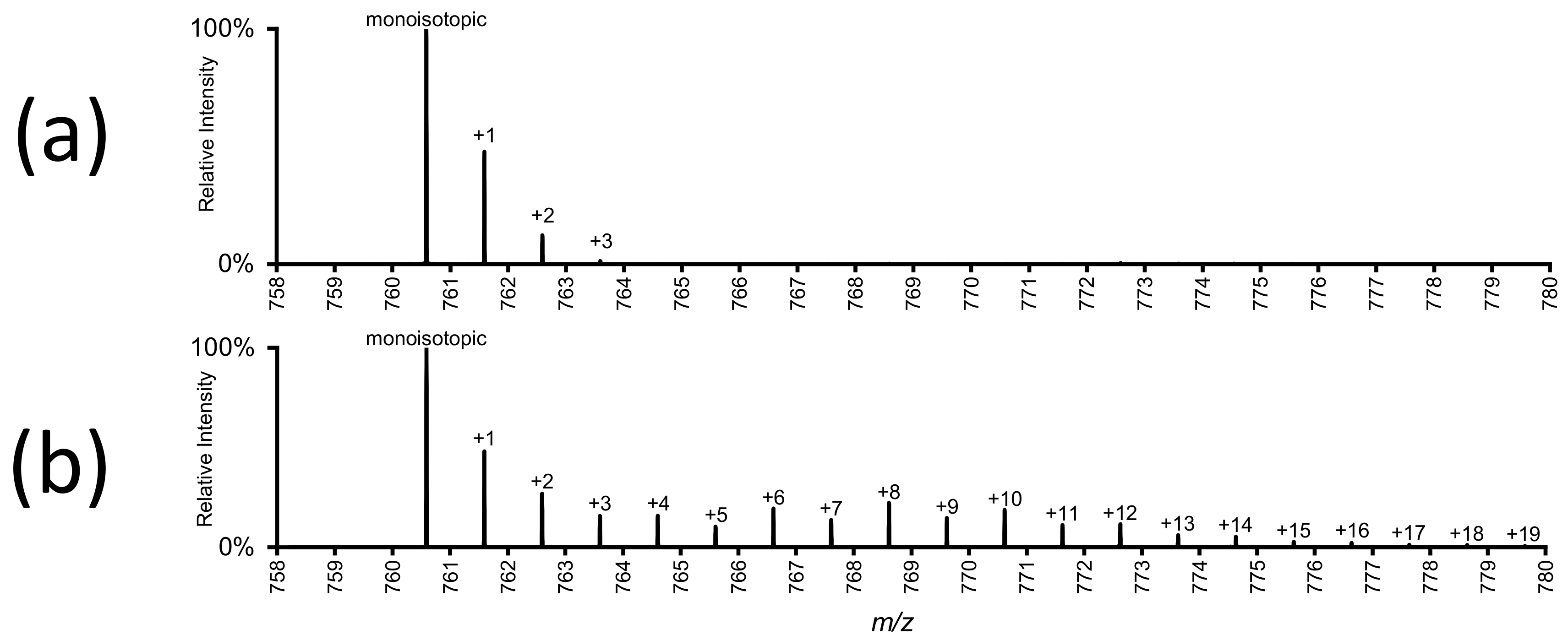
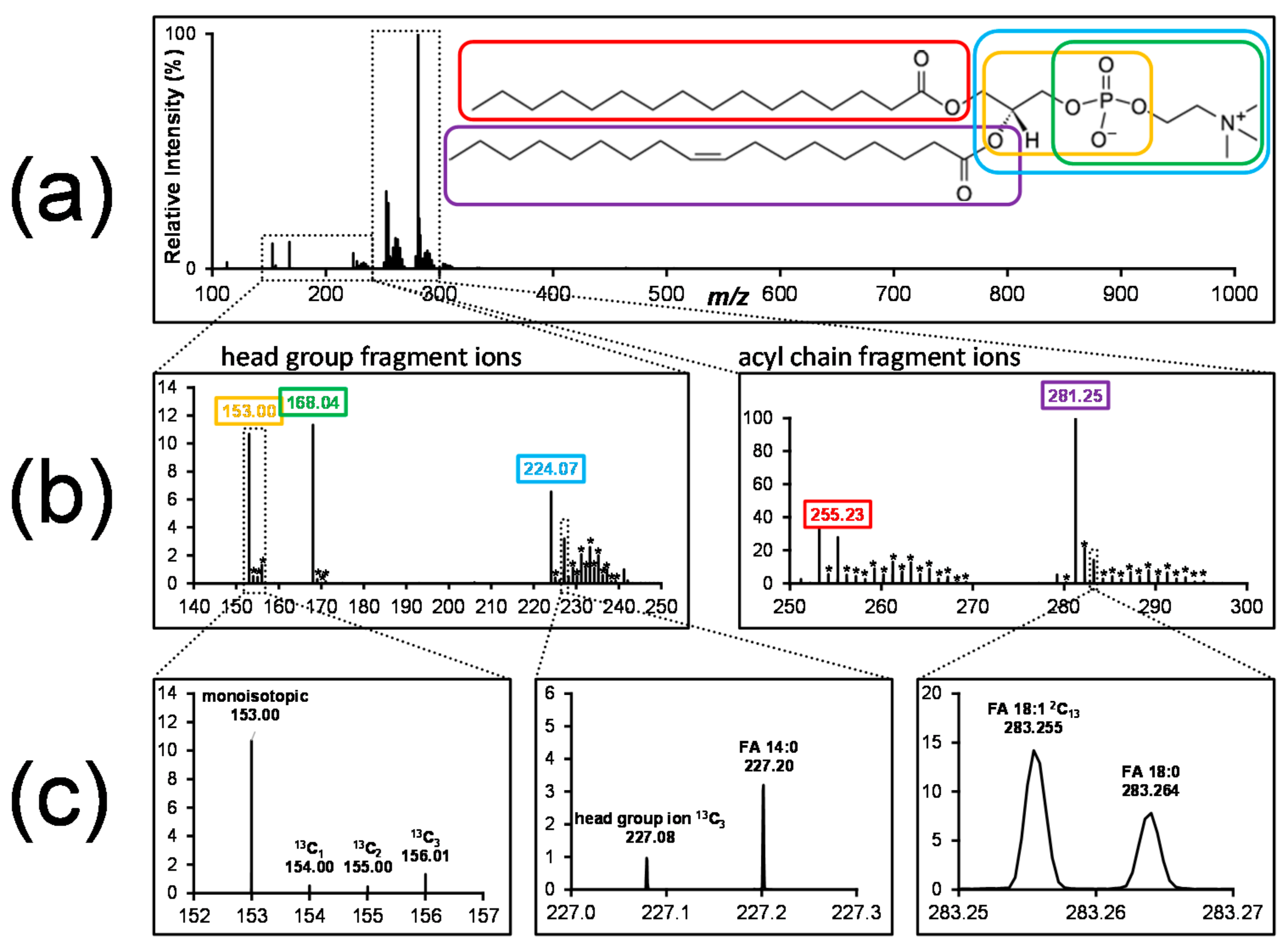
4.1. Information Content
4.2. Isotope Correction
4.3. Flux Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wenk, M.R. The emerging field of lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, M.; Géloën, A.; Record, M.; Vance, D.; Spener, F. Lipidomics is emerging. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2003, 1634, 61. [Google Scholar] [CrossRef]
- Han, X.; Gross, R.W. Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: A bridge to lipidomics. J. Lipid Res. 2003, 44, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Athenstaedt, K.; Daum, G. Phosphatidic acid, a key intermediate in lipid metabolism. Eur. J. Biochem. 1999, 266, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef] [PubMed]
- Schoenheimer, R. The investigation of intermediary metabolism with the aid of heavy hydrogen. Bull. N. Y. Acad. Med. 1937, 13, 272–295. [Google Scholar] [PubMed]
- Ecker, J.; Liebisch, G. Application of stable isotopes to investigate the metabolism of fatty acids, glycerophospholipid and sphingolipid species. Prog. Lipid Res. 2014, 54, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, C.; Han, X. Selection of internal standards for accurate quantification of complex lipid species in biological extracts by electrospray ionization mass spectrometry-What, how and why? Mass Spectrom. Rev. 2017, 36, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.; Schwaiger, M.; Volejnik, P.; Koellensperger, G. 13C-labelled yeast as internal standard for LC–MS/MS and LC high resolution MS based amino acid quantification in human plasma. J. Pharm. Biomed. Anal. 2018, 155, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Ogundare, M.; Williams, C.M.; Wang, Y. On the future of “omics”: Lipidomics. J. Inherit. Metab. Dis. 2011, 34, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Rampler, E.; Coman, C.; Hermann, G.; Sickmann, A.; Ahrends, R.; Koellensperger, G. LILY-lipidome isotope labeling of yeast: In vivo synthesis of 13C labeled reference lipids for quantification by mass spectrometry. Analyst 2017, 142, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Rampler, E.; Criscuolo, A.; Zeller, M.; El Abiead, Y.; Schoeny, H.; Hermann, G.; Sokol, E.; Cook, K.; Peake, D.A.; Delanghe, B.; et al. A Novel Lipidomics Workflow for Improved Human Plasma Identification and Quantification Using RPLC-MSn Methods and Isotope Dilution Strategies. Anal. Chem. 2018, 90, 6494–6501. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-Y.; Suh, S.-H.; Lee, I.-K.; Wolfe, R.R. Applications of stable, nonradioactive isotope tracers in in vivo human metabolic research. Exp. Mol. Med. 2016, 48, e203. [Google Scholar] [CrossRef] [PubMed]
- Batista Silva, W.; Daloso, D.M.; Fernie, A.R.; Nunes-Nesi, A.; Araújo, W.L. Can stable isotope mass spectrometry replace radiolabelled approaches in metabolic studies? Plant Sci. 2016, 249, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Hiller, K.; Metallo, C.M.; Kelleher, J.K.; Stephanopoulos, G. Nontargeted Elucidation of Metabolic Pathways Using Stable-Isotope Tracers and Mass Spectrometry. Anal. Chem. 2010, 82, 6621–6628. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Freund, D.M.; Hegeman, A.D. Recent advances in stable isotope-enabled mass spectrometry-based plant metabolomics. Curr. Opin. Biotechnol. 2017, 43, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, D.G.; Cardasis, H.L.; Stout, S.J.; Wang, S.-P.; Mendoza, V.; Castro-Perez, J.M.; Miller, P.L.; Murphy, B.A.; Cumiskey, A.-M.; Cleary, M.A.; et al. Use of [13C18] Oleic Acid and Mass Isotopomer Distribution Analysis to Study Synthesis of Plasma Triglycerides In Vivo: Analytical and Experimental Considerations. Anal. Chem. 2013, 85, 6287–6294. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Anal. Chem. 2007, 79, 7554–7559. [Google Scholar] [CrossRef] [PubMed]
- Mairinger, T.; Steiger, M.; Nocon, J.; Mattanovich, D.; Koellensperger, G.; Hann, S. Gas Chromatography-Quadrupole Time-of-Flight Mass Spectrometry-Based Determination of Isotopologue and Tandem Mass Isotopomer Fractions of Primary Metabolites for 13C-Metabolic Flux Analysis. Anal. Chem. 2015, 87, 11792–11802. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K.; Neese, R.A. Mass isotopomer distribution analysis at eight years: Theoretical, analytic, and experimental considerations. Am. J. Physiol. 1999, 276, E1146–E1170. [Google Scholar] [CrossRef] [PubMed]
- IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. Available online: https://goldbook.iupac.org/html/I/I03351.html.
- IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. Available online: https://goldbook.iupac.org/html/I/I03352.html.
- Castro, B.M.; Prieto, M.; Silva, L.C. Ceramide: A simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 2014, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Bhujwalla, Z.M.; Glunde, K.; Lane, A.N.; Fan, T.W.-M.; Xie, Z.; Moseley, H.N.B.; Higashi, R.M. Isotopomer analysis of lipid biosynthesis by high resolution mass spectrometry and NMR. Anal. Chim. Acta 2009, 651, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Kamphorst, J.J.; Rabinowitz, J.D.; Shlomi, T. Fatty Acid Labeling from Glutamine in Hypoxia Can be Explained by Isotope Exchange without Net Reductive Isocitrate Dehydrogenase (IDH) Flux. J. Biol. Chem. 2013, 288, 31363–31369. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Lang, W.; Geisler, J.G.; Wang, P.; Petrounia, I.; Mai, S.; Smith, C.; Askari, H.; Struble, G.T.; Williams, R.; et al. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and -2. J. Lipid Res. 2012, 53, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Fan, T.W.M.; Deng, P.; Higashi, R.M.; Lane, A.N.; Le, A.-T.; Scott, T.L.; Sun, Q.; Warmoes, M.O.; Yang, Y. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat. Commun. 2017, 8, 1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leithner, K.; Triebl, A.; Trötzmüller, M.; Hinteregger, B.; Leko, P.; Wieser, B.I.; Grasmann, G.; Bertsch, A.L.; Züllig, T.; Stacher, E.; et al. The glycerol backbone of phospholipids derives from noncarbohydrate precursors in starved lung cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, 6225–6230. [Google Scholar] [CrossRef] [PubMed]
- Herath, K.B.; Zhong, W.; Yang, J.; Mahsut, A.; Rohm, R.J.; Shah, V.; Castro-Perez, J.; Zhou, H.; Attygalle, A.B.; Kang, L.; et al. Determination of low levels of 2H-labeling using high-resolution mass spectrometry: Application in studies of lipid flux and beyond. Rapid Commun. Mass Spectrom. 2014, 28, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.J.; Hellerstein, M.K. Thematic review series: Patient-Oriented Research. Recent advances in liver triacylglycerol and fatty acid metabolism using stable isotope labeling techniques. J. Lipid Res. 2006, 47, 1651–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, B.W.; Mittendorfer, B.; Elias, N.; Satyanarayana, R.; Klein, S. Use of stable isotopically labeled tracers to measure very low density lipoprotein-triglyceride turnover. J. Lipid Res. 2002, 43, 223–233. [Google Scholar] [PubMed]
- Tigas, S.K.; Sunehag, A.L.; Haymond, M.W. Impact of Duration of Infusion and Choice of Isotope Label on Isotope Recycling in Glucose Homeostasis. Diabetes 2002, 51, 3170–3175. [Google Scholar] [CrossRef] [PubMed]
- Horning, M.A.; Colberg, S.R.; Casazza, G.A.; Brooks, G.A. Recycling of Deuterium from Dideuterated Glucose during Moderate Exercise. Ann. Clin. Biochem. Int. J. Biochem. Lab. Med. 2000, 37, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Han, X. Sample Preparation. In Lipidomics: Comprehensive Mass Spectrometry of Lipids; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 281–304. [Google Scholar]
- Sarafian, M.H.; Gaudin, M.; Lewis, M.R.; Martin, F.; Holmes, E.; Nicholson, J.K.; Dumas, M. Objective Set of Criteria for Optimization of Sample Preparation Procedures for Ultra-High Throughput Untargeted Blood Plasma Lipid Profiling by Ultra Performance Liquid Chromatography–Mass Spectrometry. Anal. Chem. 2014, 86, 5766–5774. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Bligh, E.; Dyer, W. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1978, 37, 1978. [Google Scholar]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lofgren, L.; Stahlman, M.; Forsberg, G.-B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Fauland, A.; Trötzmüller, M.; Eberl, A.; Afiuni-Zadeh, S.; Köfeler, H.; Guo, X.; Lankmayr, E. An improved SPE method for fractionation and identification of phospholipids. J. Sep. Sci. 2013, 36, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Wewer, V.; Dörmann, P.; Hölzl, G. Analysis and Quantification of Plant Membrane Lipids by Thin-Layer Chromatography and Gas Chromatography; Munnik, T., Heilmann, I., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1009, pp. 69–78. ISBN 978-1-62703-400-5. [Google Scholar]
- Wei, F.; Wang, X.; Ma, H.; Lv, X.; Dong, X.; Chen, H. Rapid profiling and quantification of phospholipid molecular species in human plasma based on chemical derivatization coupled with electrospray ionization tandem mass spectrometry. Anal. Chim. Acta 2018, 1024, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.; Reid, G.E. Chemical Derivatization and Ultrahigh Resolution and Accurate Mass Spectrometry Strategies for “Shotgun” Lipidome Analysis. Acc. Chem. Res. 2016, 49, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Holčapek, M.; Liebisch, G.; Ekroos, K. Lipidomic Analysis. Anal. Chem. 2018, 90, 4249–4257. [Google Scholar] [CrossRef] [PubMed]
- Schwudke, D.; Schuhmann, K.; Herzog, R.; Bornstein, S.R.; Shevchenko, A. Shotgun lipidomics on high resolution mass spectrometers. Cold Spring Harb. Perspect. Biol. 2011, 3, a004614. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, K.; Srzentić, K.; Nagornov, K.O.; Thomas, H.; Gutmann, T.; Coskun, Ü.; Tsybin, Y.O.; Shevchenko, A. Monitoring Membrane Lipidome Turnover by Metabolic 15N Labeling and Shotgun Ultra-High-Resolution Orbitrap Fourier Transform Mass Spectrometry. Anal. Chem. 2017, 89, 12857–12865. [Google Scholar] [CrossRef] [PubMed]
- Boumann, H.A.; Damen, M.J.A.; Versluis, C.; Heck, A.J.R.; de Kruijff, B.; de Kroon, A.I.P.M. The Two Biosynthetic Routes Leading to Phosphatidylcholine in Yeast Produce Different Sets of Molecular Species. Evidence for Lipid Remodeling. Biochemistry 2003, 42, 3054–3059. [Google Scholar] [CrossRef] [PubMed]
- Turowski, M.; Yamakawa, N.; Meller, J.; Kimata, K.; Ikegami, T.; Hosoya, K.; Tanaka, N.; Thornton, E.R. Deuterium Isotope Effects on Hydrophobic Interactions: The Importance of Dispersion Interactions in the Hydrophobic Phase. J. Am. Chem. Soc. 2003, 125, 13836–13849. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Zhang, Z.-P.; Kellogg, G.E.; Karnes, H.T. Evaluation of deuterium isotope effects in normal-phase LC-MS-MS separations using a molecular modeling approach. J. Chromatogr. Sci. 2004, 42, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cyronak, M.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response? J. Pharm. Biomed. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Dennis, E.A. High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography-mass spectrometry. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Strawford, A.; Antelo, F.; Christiansen, M.; Hellerstein, M.K. Adipose tissue triglyceride turnover, de novo lipogenesis, and cell proliferation in humans measured with 2H2O. Am. J. Physiol. Metab. 2004, 286, E577–E588. [Google Scholar] [CrossRef]
- Tumanov, S.; Bulusu, V.; Kamphorst, J.J. Analysis of Fatty Acid Metabolism Using Stable Isotope Tracers and Mass Spectrometry. Methods Enzymol. 2015, 561, 197–217. [Google Scholar] [PubMed]
- Lagerstedt, S.A.; Hinrichs, D.R.; Batt, S.M.; Magera, M.J.; Rinaldo, P.; McConnell, J.P. Quantitative Determination of Plasma C8–C26 Total Fatty Acids for the Biochemical Diagnosis of Nutritional and Metabolic Disorders. Mol. Genet. Metab. 2001, 73, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Gagné, S.; Crane, S.; Huang, Z.; Li, C.S.; Bateman, K.P.; Lévesque, J.-F. Rapid measurement of deuterium-labeled long-chain fatty acids in plasma by HPLC-ESI-MS. J. Lipid Res. 2007, 48, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Fan, J.; Lu, W.; White, E.; Rabinowitz, J.D. Liquid Chromatography-High Resolution Mass Spectrometry Analysis of Fatty Acid Metabolism. Anal. Chem. 2011, 83, 9114–9122. [Google Scholar] [CrossRef] [PubMed]
- Persson, X.-M.T.; Błachnio-Zabielska, A.U.; Jensen, M.D. Rapid measurement of plasma free fatty acid concentration and isotopic enrichment using LC/MS. J. Lipid Res. 2010, 51, 2761–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dushianthan, A.; Cusack, R.; Grocott, M.P.W.; Postle, A.D. Abnormal liver phosphatidylcholine synthesis revealed in patients with acute respiratory distress syndrome. J. Lipid Res. 2018, 59, 1034–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elahee Doomun, S.; Loke, S.; O’Callaghan, S.; Callahan, D. A Simple Method for Measuring Carbon-13 Fatty Acid Enrichment in the Major Lipid Classes of Microalgae Using GC-MS. Metabolites 2016, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Kainu, V.; Hermansson, M.; Somerharju, P. Electrospray Ionization Mass Spectrometry and Exogenous Heavy Isotope-labeled Lipid Species Provide Detailed Information on Aminophospholipid Acyl Chain Remodeling. J. Biol. Chem. 2008, 283, 3676–3687. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hoene, M.; Zhao, X.; Chen, S.; Wei, H.; Häring, H.-U.; Lin, X.; Zeng, Z.; Weigert, C.; Lehmann, R.; Xu, G. Stable Isotope-Assisted Lipidomics Combined with Nontargeted Isotopomer Filtering, a Tool to Unravel the Complex Dynamics of Lipid Metabolism. Anal. Chem. 2013, 85, 4651–4657. [Google Scholar] [CrossRef] [PubMed]
- Holčapek, M.; Jirásko, R.; Lísa, M. Recent developments in liquid chromatography-mass spectrometry and related techniques. J. Chromatogr. A 2012, 1259, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Trötzmüller, M.; Triebl, A.; Ajsic, A.; Hartler, J.; Köfeler, H.; Regittnig, W. Determination of the Isotopic Enrichment of 13C- and 2H-Labeled Tracers of Glucose Using High-Resolution Mass Spectrometry: Application to Dual- and Triple-Tracer Studies. Anal. Chem. 2017, 89, 12252–12260. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Brüschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Vosegaard, T.; Guo, Z. Applications of nuclear magnetic resonance in lipid analyses: An emerging powerful tool for lipidomics studies. Prog. Lipid Res. 2017, 68, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Castro-Perez, J.; Previs, S.F.; McLaren, D.G.; Shah, V.; Herath, K.; Bhat, G.; Johns, D.G.; Wang, S.-P.; Mitnaul, L.; Jensen, K.; et al. In vivo D2O labeling to quantify static and dynamic changes in cholesterol and cholesterol esters by high resolution LC/MS. J. Lipid Res. 2011, 52, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Pulfer, M.; Murphy, R.C. Electrospray Mass Spectrometry of Phospholipids. Mass Spectrom. Rev. 2003, 22, 332–364. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.-F.; Turk, J. Electrospray ionization with low-energy collisionally activated dissociation tandem mass spectrometry of glycerophospholipids: Mechanisms of fragmentation and structural characterization. J. Chromatogr. B 2009, 877, 2673–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X. Fragmentation Patterns of Glycerophospholipids. In Lipidomics: Comprehensive Mass Spectrometry of Lipids; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 173–200. [Google Scholar]
- Skotland, T.; Ekroos, K.; Kavaliauskiene, S.; Bergan, J.; Kauhanen, D.; Lintonen, T.; Sandvig, K. Determining the Turnover of Glycosphingolipid Species by Stable-Isotope Tracer Lipidomics. J. Mol. Biol. 2016, 428, 4856–4866. [Google Scholar] [CrossRef] [PubMed]
- De Laeter, J.R.; Böhlke, J.K.; de Bièvre, P.; Hidaka, H.; Peiser, H.S.; Rosman, K.J.R.; Taylor, P.D.P. Atomic Weight of the Elements: Review 2000. Pure Appl. Chem. 2003, 75, 683–800. [Google Scholar] [CrossRef]
- Su, X.; Lu, W.; Rabinowitz, J.D. Metabolite Spectral Accuracy on Orbitraps. Anal. Chem. 2017, 89, 5940–5948. [Google Scholar] [CrossRef] [PubMed]
- Millard, P.; Letisse, F.; Sokol, S.; Portais, J.-C. IsoCor: Correcting MS data in isotope labeling experiments. Bioinformatics 2012, 28, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, Y.-J.; Cho, K.; Nikolskiy, I.; Crawford, P.A.; Patti, G.J. X13CMS: Global Tracking of Isotopic Labels in Untargeted Metabolomics. Anal. Chem. 2014, 86, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Jungreuthmayer, C.; Neubauer, S.; Mairinger, T.; Zanghellini, J.; Hann, S. ICT: Isotope correction toolbox. Bioinformatics 2015, 32, btv514. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Zeeshan, S.; Huber, C.; Hensel, M.; Schomburg, D.; Münch, R.; Eisenreich, W.; Dandekar, T. Software LS-MIDA for efficient mass isotopomer distribution analysis in metabolic modelling. BMC Bioinform. 2013, 14, 218. [Google Scholar] [CrossRef] [PubMed]
- Niedenführ, S.; ten Pierick, A.; van Dam, P.T.N.; Suarez-Mendez, C.A.; Nöh, K.; Wahl, S.A. Natural isotope correction of MS/MS measurements for metabolomics and 13C fluxomics. Biotechnol. Bioeng. 2016, 113, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. 13C metabolic flux analysis: Optimal design of isotopic labeling experiments. Curr. Opin. Biotechnol. 2013, 24, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Crown, S.B.; Antoniewicz, M.R. Publishing 13C metabolic flux analysis studies: A review and future perspectives. Metab. Eng. 2013, 20, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Crown, S.B.; Ahn, W.; Antoniewicz, M.R. Rational design of 13C-labeling experiments for metabolic flux analysis in mammalian cells. BMC Syst. Biol. 2012, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. Methods and advances in metabolic flux analysis: A mini-review. J. Ind. Microbiol. Biotechnol. 2015, 42, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med. 2018, 50, 19. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.K. Quantifying plant phenotypes with isotopic labeling & metabolic flux analysis. Curr. Opin. Biotechnol. 2016, 37, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, N.; Fendt, S.-M.; Rühl, M.; Sauer, U. 13C-based metabolic flux analysis. Nat. Protoc. 2009, 4, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Niedenführ, S.; Wiechert, W.; Nöh, K. How to measure metabolic fluxes: A taxonomic guide for 13C fluxomics. Curr. Opin. Biotechnol. 2015, 34, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W.; Nöh, K. Isotopically non-stationary metabolic flux analysis: Complex yet highly informative. Curr. Opin. Biotechnol. 2013, 24, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.K.; Bates, P.D.; Tjellström, H. Tracking the metabolic pulse of plant lipid production with isotopic labeling and flux analyses: Past, present and future. Prog. Lipid Res. 2015, 58, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, J.K.; Nickol, G.B. Isotopomer Spectral Analysis: Utilizing Nonlinear Models in Isotopic Flux Studies. Methods Enzymol. 2015, 561, 303–330. [Google Scholar] [PubMed]
- Zamboni, N.; Fischer, E.; Sauer, U. FiatFlux—A software for metabolic flux analysis from 13C-glucose experiments. BMC Bioinform. 2005, 6, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quek, L.-E.; Wittmann, C.; Nielsen, L.K.; Krömer, J.O. OpenFLUX: Efficient modelling software for 13C-based metabolic flux analysis. Microb. Cell Fact. 2009, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W.; Möllney, M.; Petersen, S.; de Graaf, A.A. A Universal Framework for 13C Metabolic Flux Analysis. Metab. Eng. 2001, 3, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Kajihata, S.; Furusawa, C.; Matsuda, F.; Shimizu, H. OpenMebius: An Open Source Software for Isotopically Nonstationary 13C-Based Metabolic Flux Analysis. BioMed Res. Int. 2014, 2014, 627014. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D. INCA: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Agilent MassHunter VistaFlux for Qualitative Flux Analysis. Agilent Technologies, Inc., 2016. Available online: https://www.agilent.com/cs/library/technicaloverviews/public/5991-6756EN.pdf.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Triebl, A.; Wenk, M.R. Analytical Considerations of Stable Isotope Labelling in Lipidomics. Biomolecules 2018, 8, 151. https://doi.org/10.3390/biom8040151
Triebl A, Wenk MR. Analytical Considerations of Stable Isotope Labelling in Lipidomics. Biomolecules. 2018; 8(4):151. https://doi.org/10.3390/biom8040151
Chicago/Turabian StyleTriebl, Alexander, and Markus R. Wenk. 2018. "Analytical Considerations of Stable Isotope Labelling in Lipidomics" Biomolecules 8, no. 4: 151. https://doi.org/10.3390/biom8040151
APA StyleTriebl, A., & Wenk, M. R. (2018). Analytical Considerations of Stable Isotope Labelling in Lipidomics. Biomolecules, 8(4), 151. https://doi.org/10.3390/biom8040151