Pharmacology Study of the Multiple Angiogenesis Inhibitor RC28-E on Anti-Fibrosis in a Chemically Induced Lung Injury Model

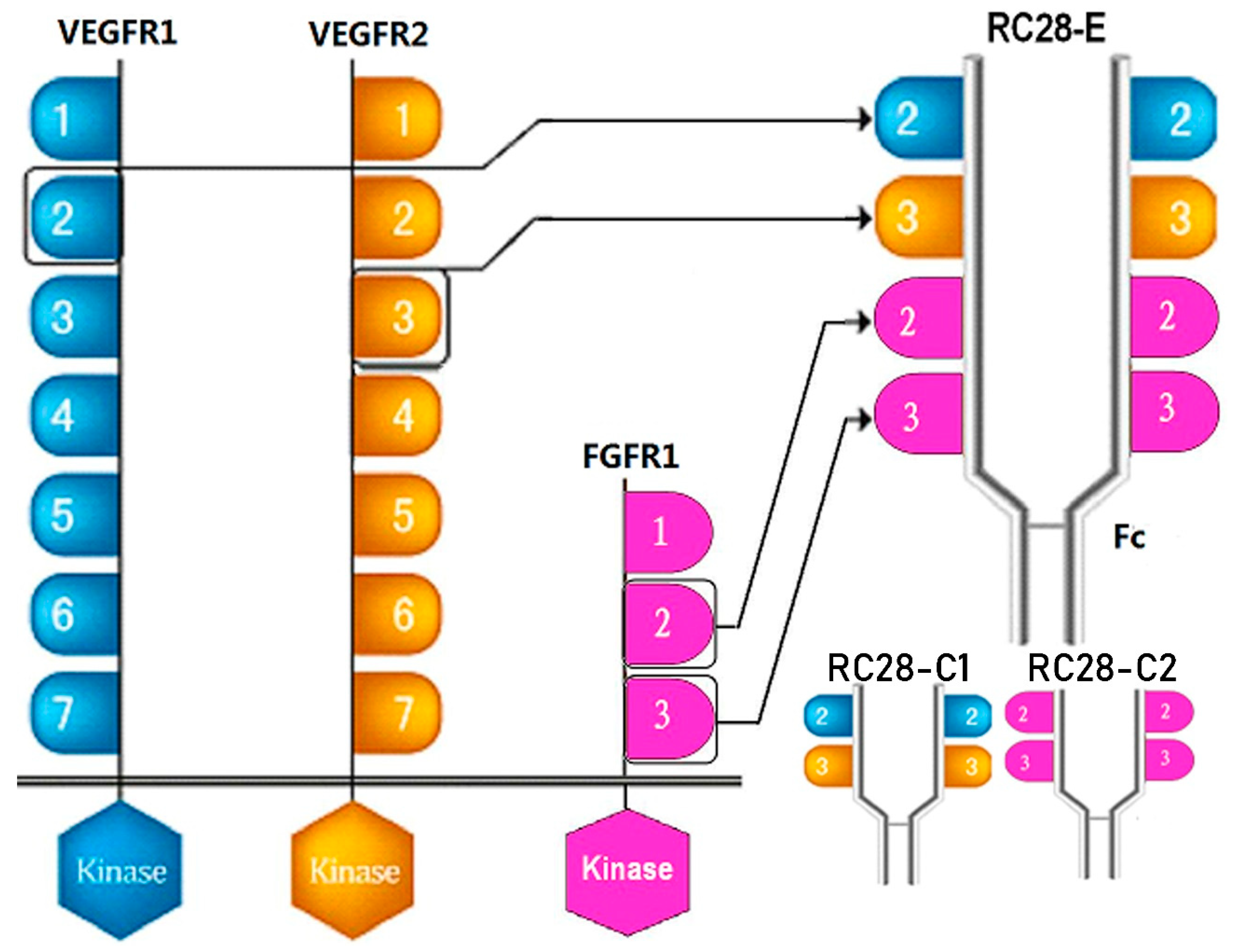{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Animals
2.3. Bleomycin-Induced Mouse Pulmonary Fibrosis Model and Treatment
2.4. Isolation of Primary Murine Fibroblasts
2.5. Cell Culture
2.6. Histopathological Analysis
2.7. Detecting Hydroxyproline (HYP) in Lung Tissues by Alkaline Hydrolysis
2.8. Immunohistochemistry
2.9. Cytometric Bead Analysis
2.10. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (Q-PCR)
2.11. Western Blot Analysis
2.12. Immunofluorescence Staining
2.13. Cell Proliferation Assay
2.14. Transwell Migration Assay
2.15. Statistical Analysis
3. Results
3.1. Bleomycin-Induced Pulmonary Fibrosis Formation
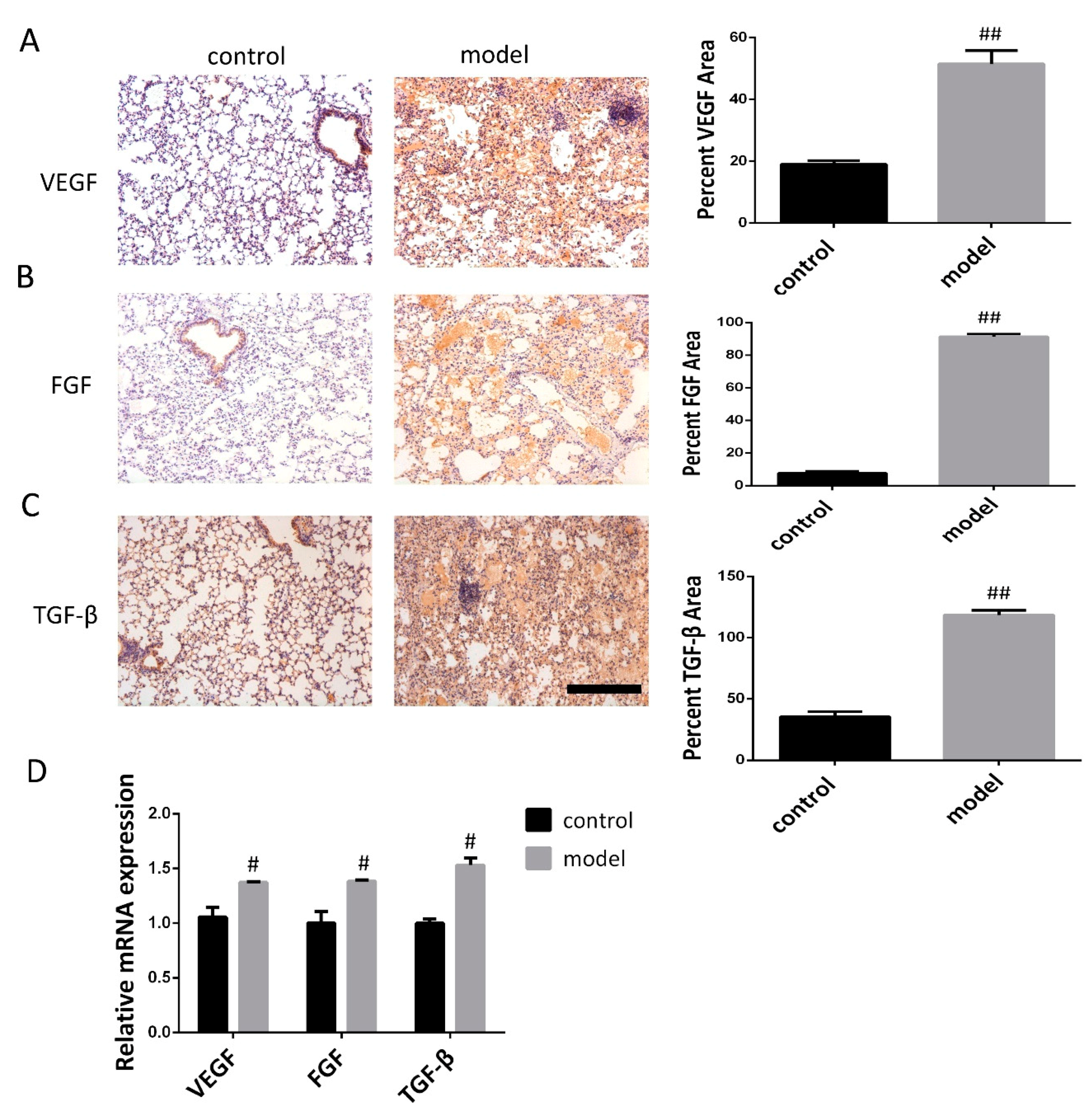
3.2. Increased VEGF, FGF, and TGF-β Expression in the Lung Following Bleomycin-Induced Pulmonary Fibrosis Formation
3.3. Effects of RC28-E on the Expression of VEGF, FGF, and TGF-β During Bleomycin-Induced Fibrosis Formation
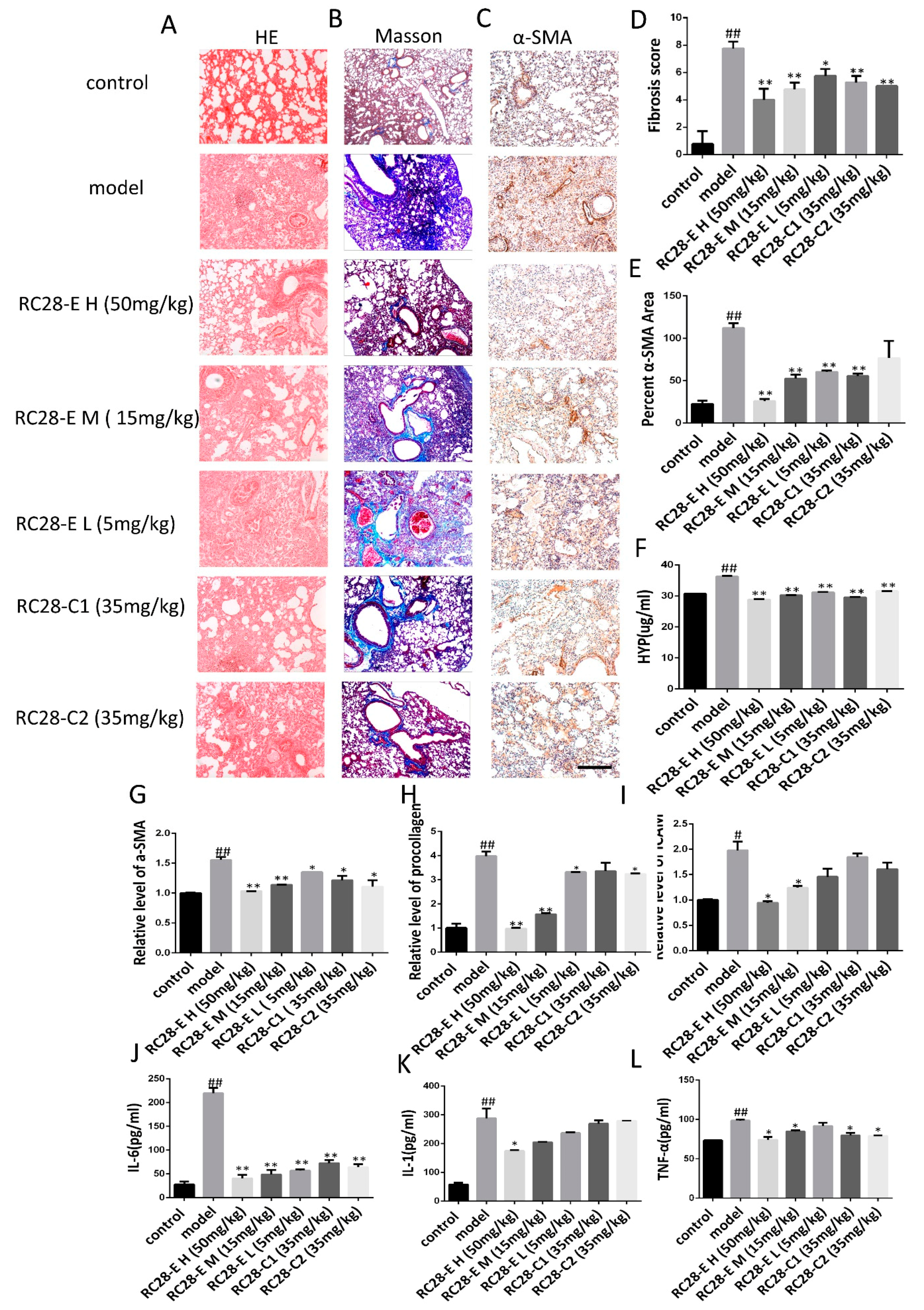
3.4. Effects of RC28-E on Lung Fibrosis Induced by Bleomycin in Mice
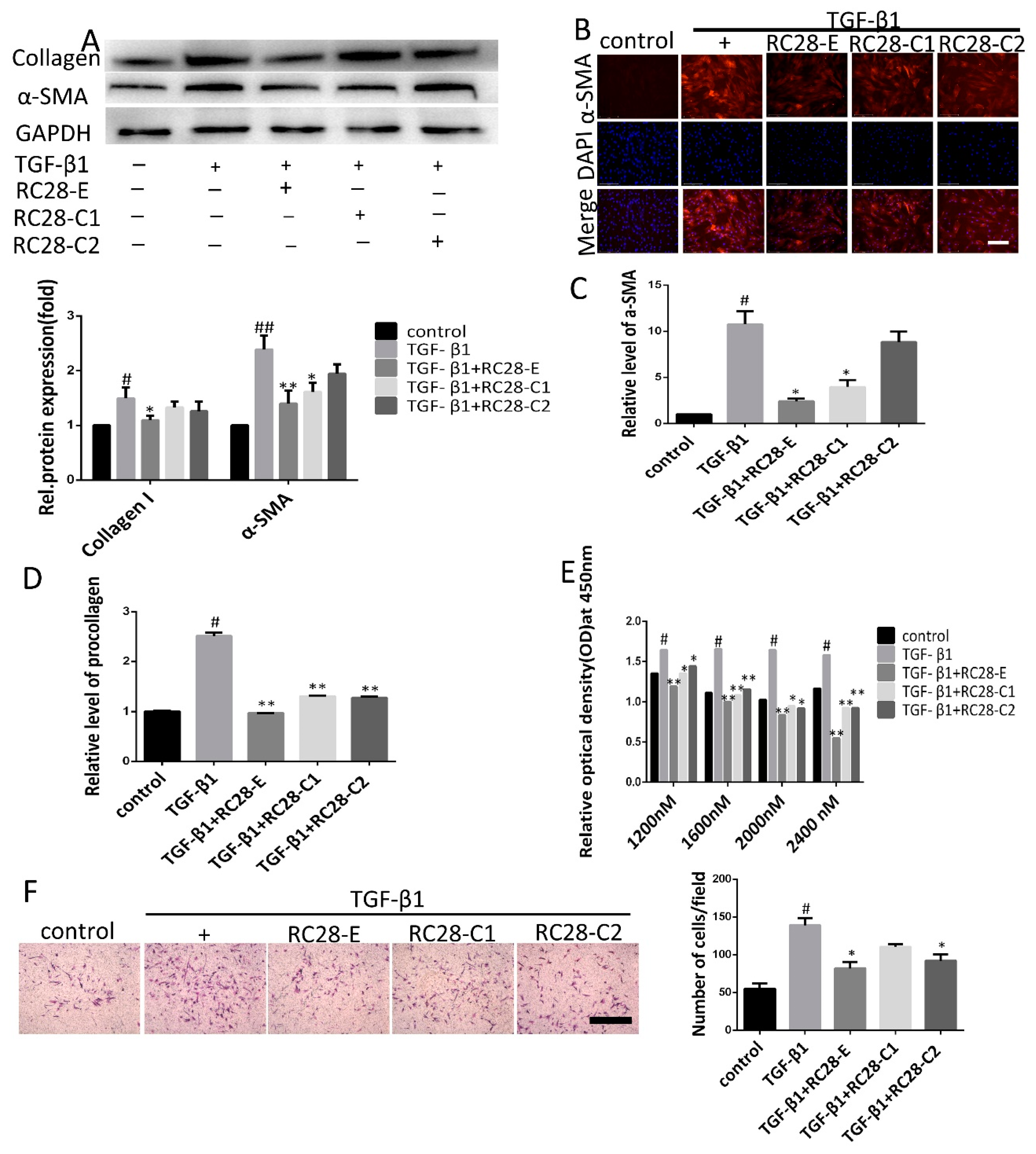
3.5. RC28-E Inhibits TGF-β1-Induced Fibroblast Activation In Vitro
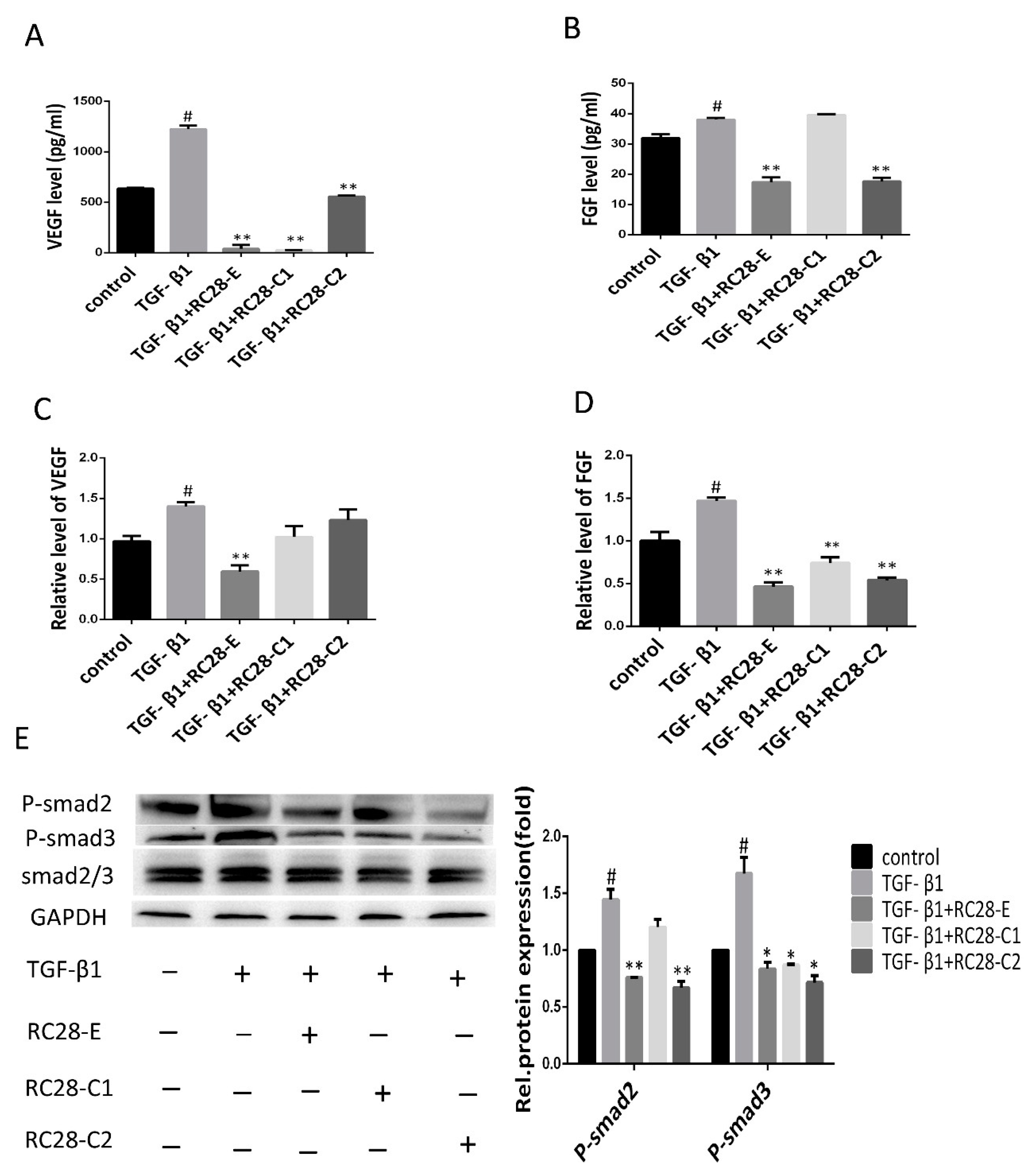
3.6. RC28-E Downregulated Protein Levels of VEGF and FGF and Inhibited the Phosphorylation Activity of the Downstream Signaling Molecules Smad in Human Skin Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zeisberg, M.; Kalluri, R. Cellular mechanisms of tissue fibrosis: 1. common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C216–C225. [Google Scholar] [CrossRef] [PubMed]
- Salem, C.; Slim, R.; Fathallah, N. Fibrosis—A common pathway to organ injury and failure. New Engl. J. Med. 2015, 373, 95–96. [Google Scholar] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Kaissling, B.; Lehir, M.; Kriz, W. Renal epithelial injury and fibrosis. Biochim. Biophys. Acta 2013, 1832, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chen, J.-K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGFβ-dependent renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar] [CrossRef]
- Mezzano, S.A.; Ruiz-Ortega, M.; Egido, J. Angiotensin II and renal fibrosis. Hypertension 2001, 38, 635–638. [Google Scholar] [CrossRef]
- Saylor, P.J.; Escudier, B.; Michaelson, M.D. Importance of fibroblast growth factor receptor in neovascularization and tumor escape from antiangiogenic therapy. Clin. Genitourin. Cancer 2012, 10, 77–83. [Google Scholar] [CrossRef]
- Sun, X.T.; Ding, Y.T.; Yan, X.G.; Wu, L.Y.; Li, Q.; Cheng, N.; Qiu, Y.D.; Zhang, M.Y. Angiogenic synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in an in vitro quantitative microcarrier-based three-dimensional fibrin angiogenesis system. World J. Gastroenterol. 2004, 10, 2524–2528. [Google Scholar] [CrossRef]
- Giavazzi, R.; Sennino, B.; Coltrini, D.; Garofalo, A.; Dossi, R.; Ronca, R.; Tosatti, M.P.M.; Presta, M. Distinct role of fibroblast growth factor-2 and vascular endothelial growth factor on tumor growth and angiogenesis. Am. J. Pathol. 2003, 162, 1913–1926. [Google Scholar] [CrossRef]
- Jhappan, C.; Stahle, C.; Harkins, R.N.; Fausto, N.; Smith, G.H.; Merlino, G.T. TGFcalpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell 1990, 61, 1137–1146. [Google Scholar] [CrossRef]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagni, A.; Wilgenbus, P.; Impagnatiello, M.A.; Cotton, M.; Christofori, G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res. 2000, 60, 7163–7169. [Google Scholar]
- Ogata, T.; Miyauchi, T.; Irukayama-Tomobe, Y.; Takanashi, M.; Goto, K.; Yamaguchi, I. The peroxisome proliferatoractivated receptor alpha activator fenofibrate inhibits endothelin-1-induced cardiac fibroblast proliferation. J. Cardiovasc Pharm. 2004, 44, S279–S282. [Google Scholar] [CrossRef] [PubMed]
- Strhutz, F.; Zeisberg, M.; Ziyadeh, F.N.; Yang, C.Q.; Kalluri, R.; Müller, G.A.; Neilson, E.G.; Renziehausen, A.; Sisic, Z. Role of basic fibroblast growt factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002, 61, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Wang, F.; Jin, C.; Huang, X.; Miller, D.L.; Basilico, C.; McKeehan, W.L. Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis. Am. J. Pathol. 2003, 163, 1653–1662. [Google Scholar] [CrossRef]
- Triantafylla, M.; Massa, H.F.; Dardabounis, D.; Gatzioufas, Z.; Kozobolis, V.; Ioannakis, K.; Perente, I.; Panos, G.D. Ranibizumab for the treatment of degenerative ocular conditions. Clin. Ophthalmol. 2014, 8, 1187–1198. [Google Scholar]
- Blick, S.K.; Keating, G.M.; Wagstaff, A.J. Ranibizumab. Drugs 2007, 67, 1199–1206. [Google Scholar] [CrossRef]
- Yilmaz, A.; Karatay, M.; Yildirim, T.; Celik, H.; Sertbas, I.; Erdem, Y.; Caydere, M.; Isi, H.S.; Bayar, M.A. Prevention of epidural fibrosis using ranibizumab in a postlaminectomy rat model. Turk. Neurosurg. 2017, 27, 119–123. [Google Scholar] [CrossRef]
- Denizli, N.; Demirci, R.; Duranay, M.; Yilmaz, B. New insight into treatment of diseases of glomerular endothelium. Turk. Nephrol. Dial. Transplant. J. 2011, 20, 220–226. [Google Scholar] [CrossRef]
- Huang, Y.; Feng, H.; Kan, T.; Huang, B.; Zhang, M.; Li, Y.; Shi, C.; Wu, M.; Luo, Y.; Yang, J.; et al. Bevacizumab attenuates hepatic fibrosis in rats by inhibiting activation of hepatic stellate cells. PLoS ONE 2013, 8, e73492. [Google Scholar] [CrossRef]
- Kahook, M.Y. Bleb morphology and vascularity after trabecu-lectomy with intravitreal ranibizumab: A pilot study. Am. J. Ophthalmol. 2010, 150, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Nilforushan, N.; Yadgari, M.; Kish, S.K.; Nassiri, N. Subcon-junctival bevacizumab versus mitomycin C adjunctive to trabeculectomy. Am. J. Ophthalmol. 2012, 153, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Gunther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.-Y.L.; Kim, J.H.; Park, C.K. VEGF induces TGF-β1 expres-sion and myofibroblast transformation after glaucoma surgery. Am. J. Pathol. 2013, 182, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.T. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ. Res. 2008, 102, 637–652. [Google Scholar] [CrossRef]
- Barratt, S.L.; Flower, V.A.; Pauling, J.D. VEGF (vascular endothelial growth factor) and fibrotic lung disease. Int. J. Mol. Sci. 2018, 19, 1269. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T. Molecular mechanisms of pulmonary fibrogenesis and its progression to lung cancer: A review. Int. J. Mol. Sci. 2019, 20, 1461. [Google Scholar] [CrossRef]
- Jiang, J.; Xu, K.; Wang, L.; Xin, W.; Zhao, G.; Huang, M.; Li, S.; Luan, X.; Fang, J. Pharmacology study of a chimeric decoy receptor trap fusion protein on retina neovascularization by dual blockage of VEGF and FGF-2. Eur. J. Pharm. Sci. 2018, 121, 251–259. [Google Scholar] [CrossRef]
- Li, D.; Xie, K.; Zhang, L.; Yao, X.; Li, H.; Xu, Q.; Wang, X.; Jiang, J.; Fang, J. Dual blockade of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (FGF-2) exhibits potent anti-angiogenic effects. Cancer Lett. 2016, 377, 164–173. [Google Scholar] [CrossRef]
- Yang, Q.H.; Zhang, Y.; Jiang, J.; Wu, M.M.; Han, Q.; Bo, Q.Y.; Yu, G.-W.; Ru, Y.S.; Liu, X.; Huang, M.; et al. Protective effects of a novel drug RC28-E blocking both VEGF and FGF2 on early diabetic rat retina. Int. J. Ophthalmol. 2018, 11, 935–944. [Google Scholar] [CrossRef]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1998, 41, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yang, J.J.; Shi, K.H. Wnt signaling pathway in cardiac fibrosis: New insights and directions. Metabolism 2016, 65, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Pintucci, G.; Seghezzi, G.; Hyman, K.; Galloway, A.C.; Mignatti, P. VEGF, a prosurvival factor, acts in concert with TGF-1 to induce endothelial cell apoptosis. Proc. Natl. Acad. Sci. USA 2006, 103, 17260–17265. [Google Scholar] [CrossRef] [PubMed]
- Mishra, L.; Derynck, R.; Mishra, B. Transforming growth factor-beta signaling in stem cells and cancer. Science 2005, 310, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.A.; Leof, E.B. TGF-beta signaling: A tale of two responses. J. Cell Biochem. 2007, 102, 593–608. [Google Scholar] [CrossRef]
- Hoshino, M.; Takahashi, M.; Aoike, N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J. Allergy Clin. Immunol. 2001, 107, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Hamada, N.; Kuwano, K.; Yamada, M.; Hagimoto, N.; Hiasa, K.; Egashira, K.; Nakashima, N.; Maeyama, T.; Yoshimi, M.; Nakanishi, Y. Anti-vascular endothelial growth factor gene therapy attenuates lung injury andfibrosis in mice. J. Immunol. 2005, 175, 1224–1231. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Wang, X.; Ma, W.; Han, S.; Meng, Z.; Zhao, L.; Yin, Y.; Wang, Y.; Li, J. TGF-β participates choroid neovascularization through Smad2/3-VEGF/TNF-α signaling in mice with Laser-induced wet age-related macular degeneration. Sci. Rep. 2017, 7, 9672. [Google Scholar] [CrossRef]
- Mishra, A.; Mishra, S.C.; Tripathi, A.M. Clinical correlation of molecular (VEGF, FGF, PDGF, c-Myc, c-Kit, Ras, p53) expression in juvenile nasopharyngeal angiofibroma. Eur. Arch. Oto-Rhino-L 2018, 275, 2719–2726. [Google Scholar] [CrossRef]
- Tang, B.; Vu, M.; Booker, T.; Santner, S.J.; Miller, F.R.; Anver, M.R.; Wakefield, L.M. TGF-beta switches fromtumor suppressor to prometastatic factor in a model of breast cancer progression. J. Clin. Investig. 2003, 112, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix. Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.R.; Chung, A.C.K.; Zhou, L.; Wang, X.J.; Lan, H.Y. Latent TGF-β1 protects against crescentic glornerulonephritis. J. Am. Soc. Nephrol. 2008, 19, 233–242. [Google Scholar] [CrossRef]
- Xu, F.Y.; Liu, C.W.; Zhou, D.D.; Zhang, L. TGF-/SMAD pathway and its regulation in hepatic fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.S.; Shi, W.; Wang, Y.L.; Chen, H.; Bringas, P.; Datto, M.B.; Frederick, J.P.; Wang, X.F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol.-Lung C 2002, 282, L585–L593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.M.; Zhou, S.; Li, C.J.; Liao, J.; Xiao, J.; Wang, Q.M.; Lian, G.Y.; Li, J.; Huang, X.R.; To, K.F.; et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018, 93, 173–187. [Google Scholar] [CrossRef]
- Andrianifahanana, M.; Wilkes, M.C.; Gupta, S.K.; Rahimi, R.A.; Repellin, C.E.; Edens, M.; Wittenberger, J.; Yin, X.; Maidl, E.; Becker, J.; et al. Profibrotic TGFβ responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases. FASEB J. 2013, 27, 4444–4454. [Google Scholar] [CrossRef]
- Li, C.M.; Khosla, J.; Pagan, I.; Hoyle, P.; Sannes, P.L. TGF-beta1 and fibroblast growth factor-1 modify fibroblast growth factor-2 production in type II cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1038–L1046. [Google Scholar] [CrossRef]
- Kucich, U.; Rosenbloom, J.C.; Herrick, D.J.; Abrams, W.R.; Hamilton, A.D.; Sebti, S.M.; Rosenbloom, J. Signaling events required for transforming growth factor-beta stimulation of connective tissue growth factor expression by cultured human lung fibroblasts. Arch. Biochem. Biophys. 2001, 395, 103–112. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Roper, J.; Richardson, M.P.; Wang, W.V.; Richard, L.G.; Chen, W.; Coffee, E.M.; Sinnamon, M.J.; Lee, L.; Chen, P.C.; Bronson, R.T.; et al. The dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a genetically engineered mouse model of PIK3CA wild-type colorectal cancer. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kou, X.; Sun, Y.; Li, S.; Bian, W.; Liu, Z.; Zhang, D.; Jiang, J. Pharmacology Study of the Multiple Angiogenesis Inhibitor RC28-E on Anti-Fibrosis in a Chemically Induced Lung Injury Model. Biomolecules 2019, 9, 644. https://doi.org/10.3390/biom9110644
Kou X, Sun Y, Li S, Bian W, Liu Z, Zhang D, Jiang J. Pharmacology Study of the Multiple Angiogenesis Inhibitor RC28-E on Anti-Fibrosis in a Chemically Induced Lung Injury Model. Biomolecules. 2019; 9(11):644. https://doi.org/10.3390/biom9110644
Chicago/Turabian StyleKou, Xiangying, Yeying Sun, Shenjun Li, Weihua Bian, Zhihao Liu, Daolai Zhang, and Jing Jiang. 2019. "Pharmacology Study of the Multiple Angiogenesis Inhibitor RC28-E on Anti-Fibrosis in a Chemically Induced Lung Injury Model" Biomolecules 9, no. 11: 644. https://doi.org/10.3390/biom9110644
APA StyleKou, X., Sun, Y., Li, S., Bian, W., Liu, Z., Zhang, D., & Jiang, J. (2019). Pharmacology Study of the Multiple Angiogenesis Inhibitor RC28-E on Anti-Fibrosis in a Chemically Induced Lung Injury Model. Biomolecules, 9(11), 644. https://doi.org/10.3390/biom9110644