Ethanol Impairs NRF2/Antioxidant and Growth Signaling in the Intact Placenta In Vivo and in Human Trophoblasts

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. HTR-8 sv/neo Trophoblast Cultures and Ethanol Treatment
2.3. In Vivo Prenatal Alcohol Exposure Model
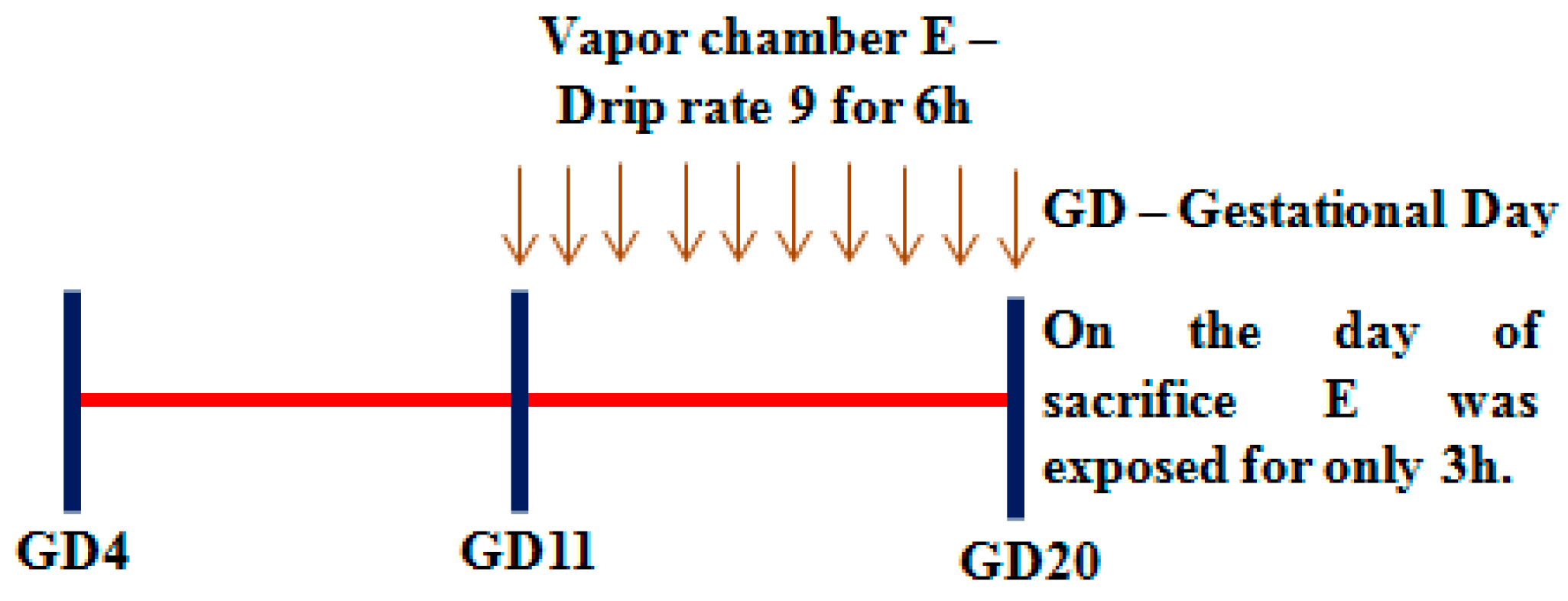
2.3.1. Rationale for the Timeline Used
2.3.2. In Vivo Prenatal Alcohol Exposure
2.4. Transfection
2.5. Luciferase Assay
2.6. RNA Extraction and Real-Time qRT-PCR Analysis
2.7. Protein Extraction
2.7.1. Whole Cell Extracts (WCE)
2.7.2. Cytosolic and Nuclear Extraction
2.8. Immunoblotting
2.9. Reactive Oxygen Species Detection With DCF-DA Using Fluorimetry
2.10. MTT Assay
2.11. Statistical Analysis
3. Results
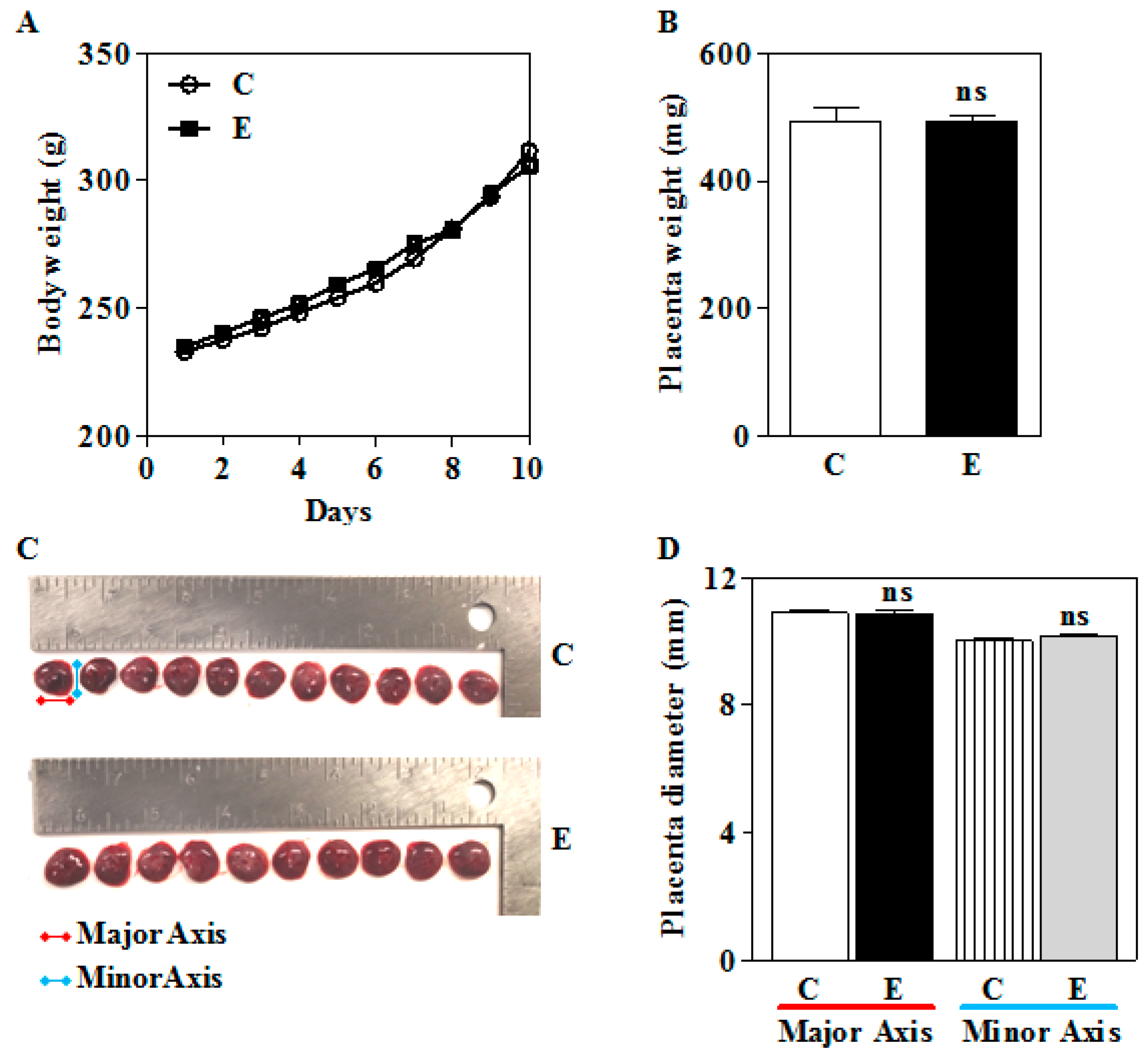
3.1. Prenatal IEV Exposure Did Not Alter the Placental and Body Weight
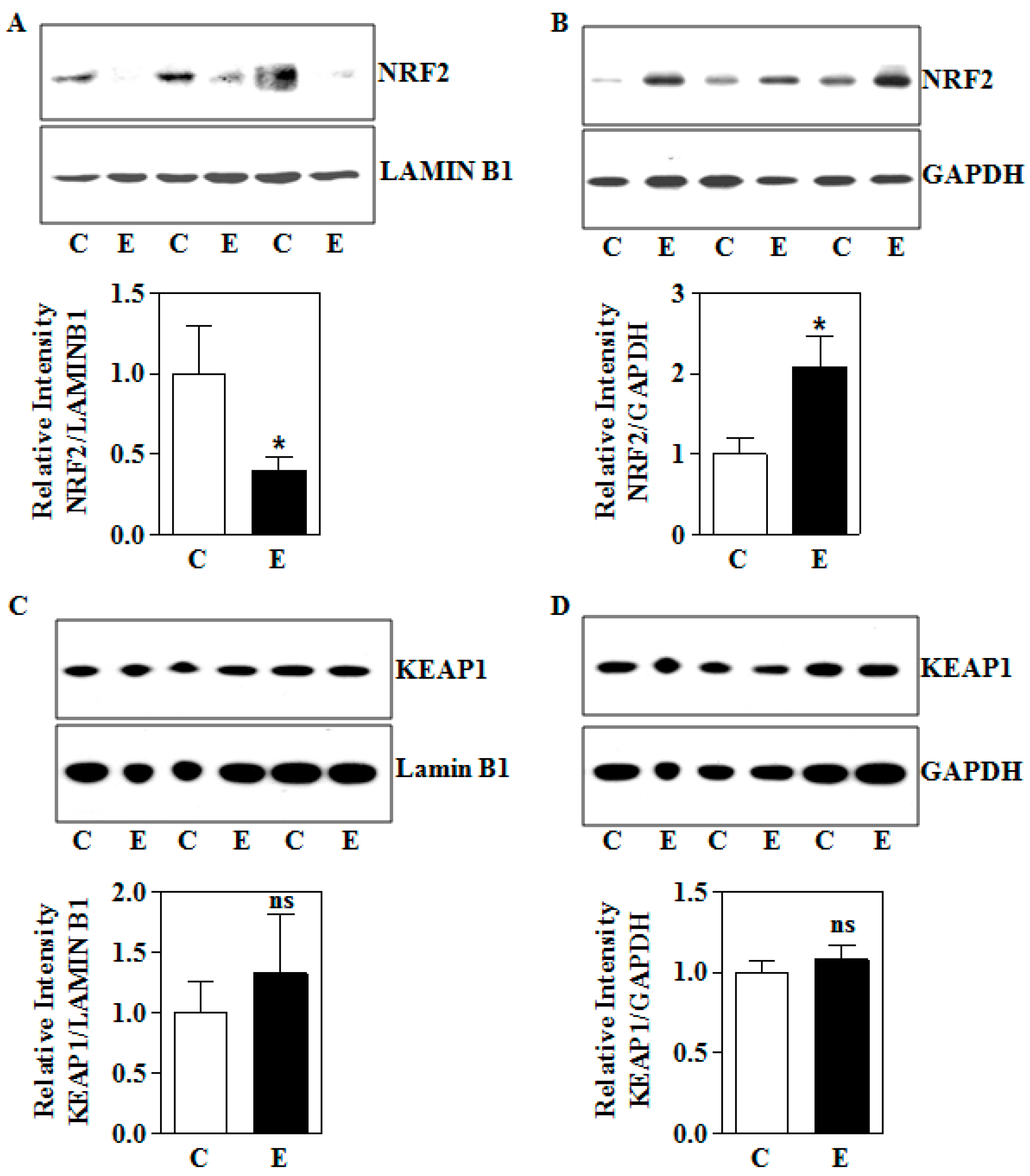
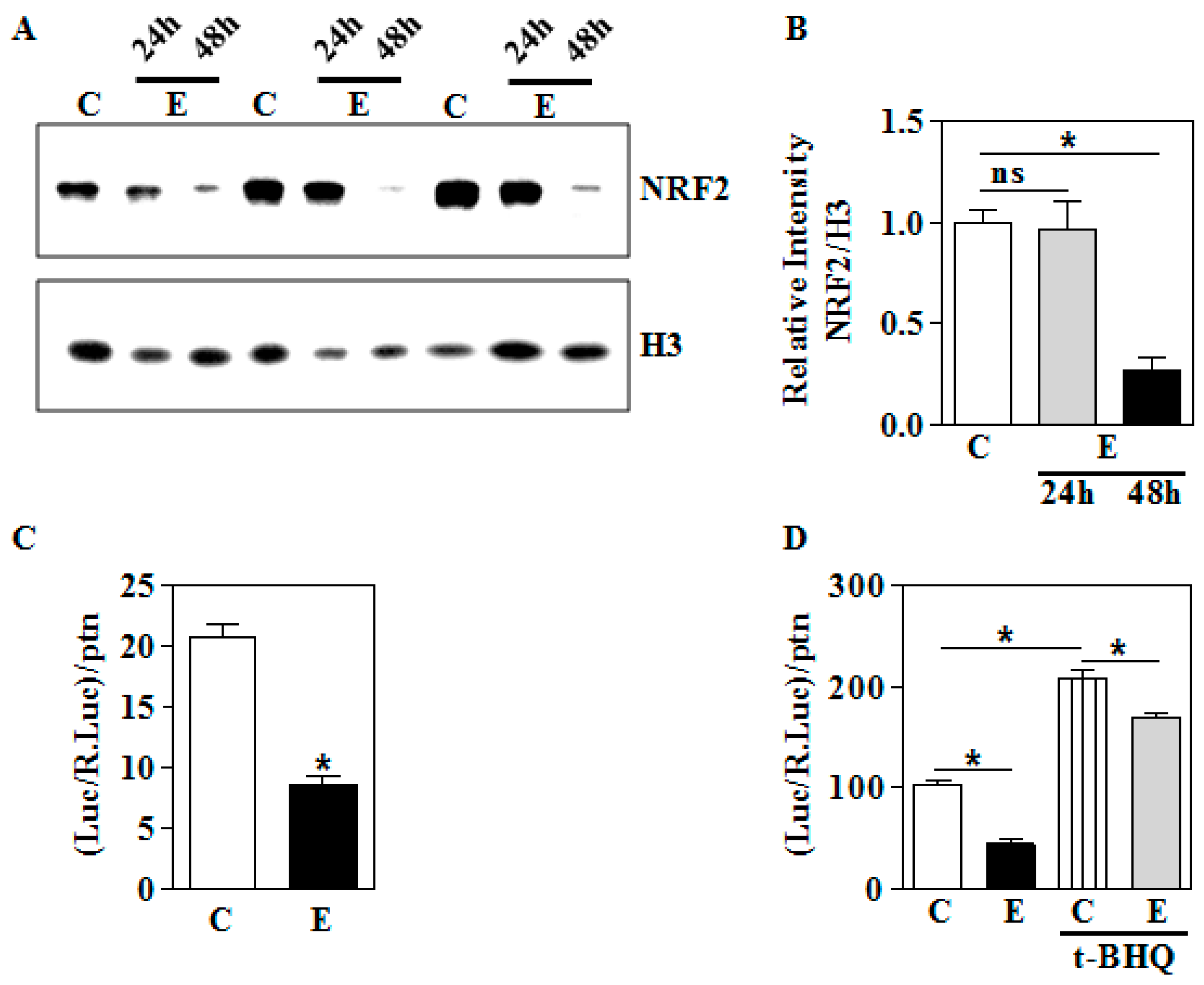
3.2. In Utero Ethanol Exposure Reduces Nuclear NRF2 Levels
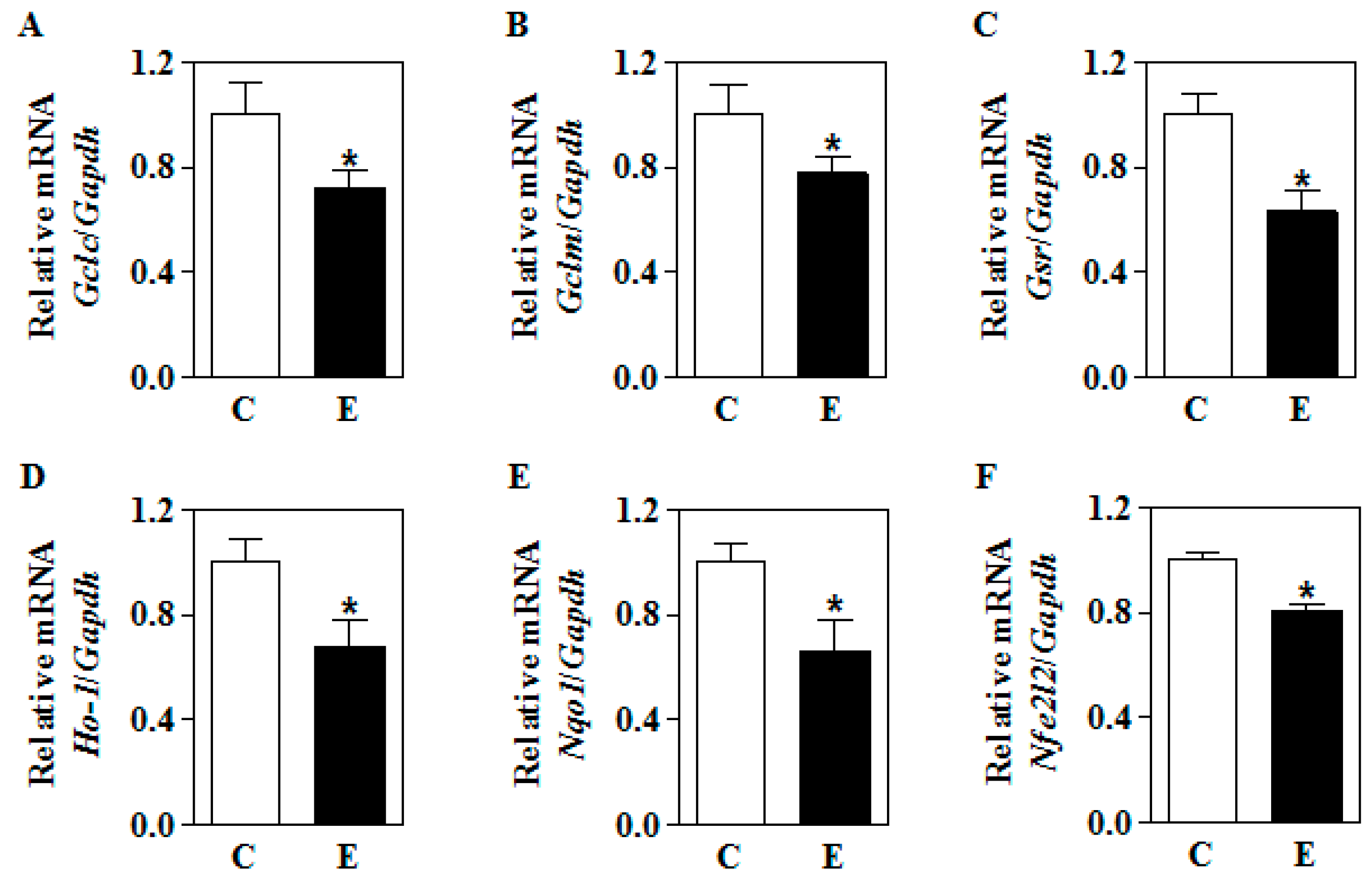
3.3. In Utero Ethanol Represses NRF2-Mediated Transcriptional Activation in the Placenta
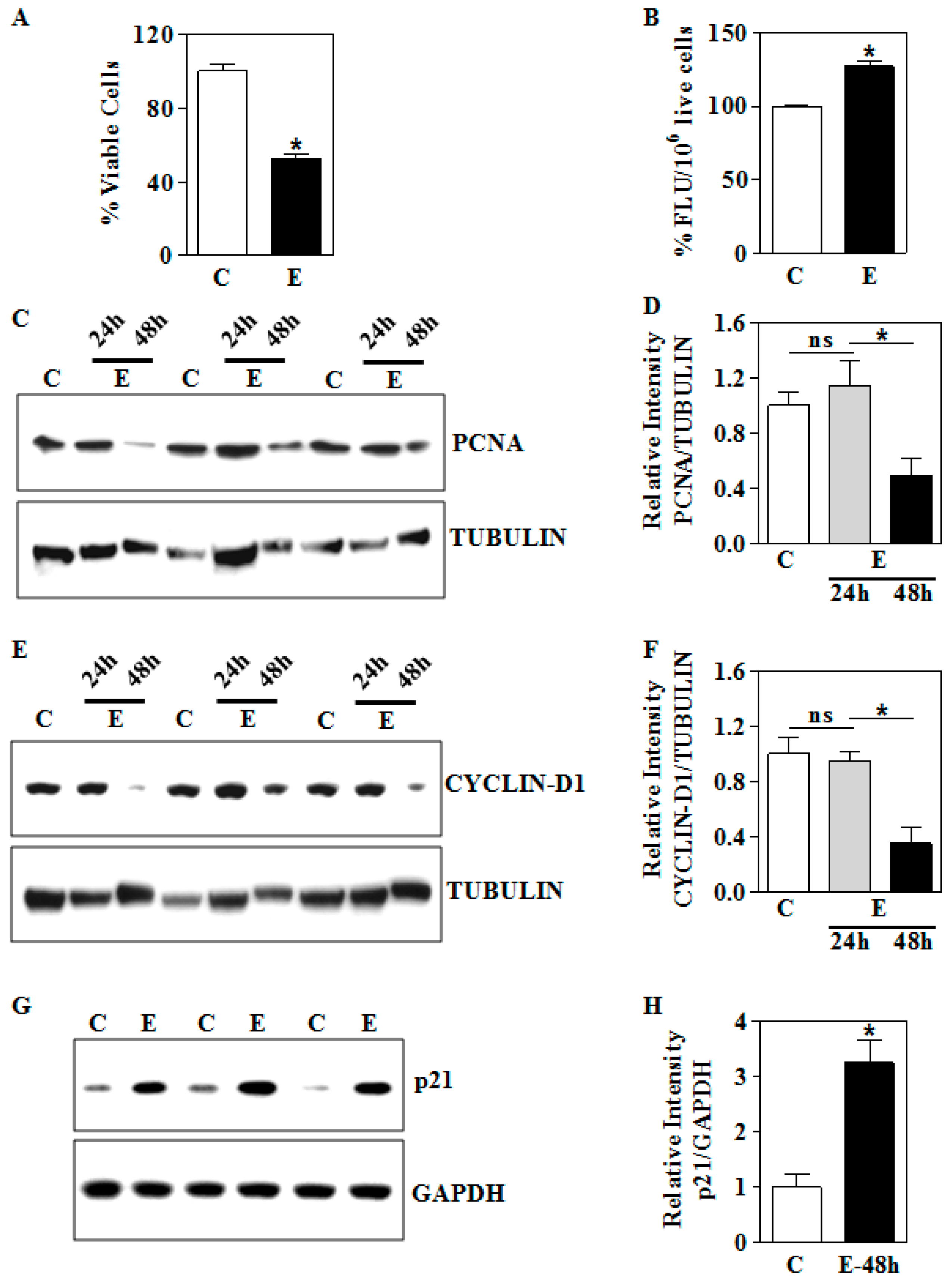
3.4. In Vitro E Impairs Nuclear Nrf2 Levels and its Transactivation Function in Human Trophoblasts
3.5. In Vitro E Inhibits Cell Viability in Association with Dysregulation of PCNA, Cyclin D1, and p21 in Human Trophoblasts
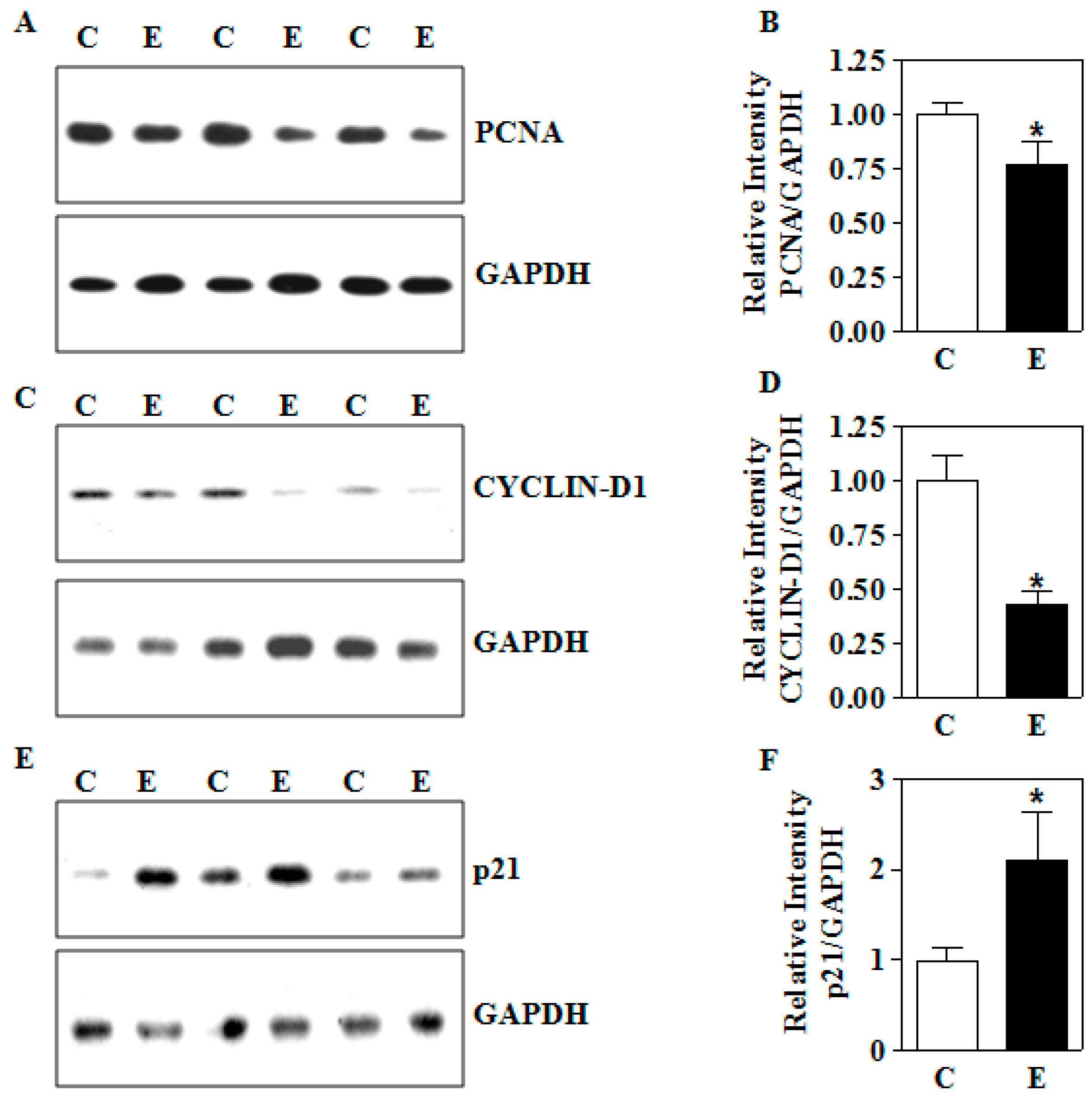
3.6. In Utero E Affects the Expression of Growth Perpetuating Factors in Placenta
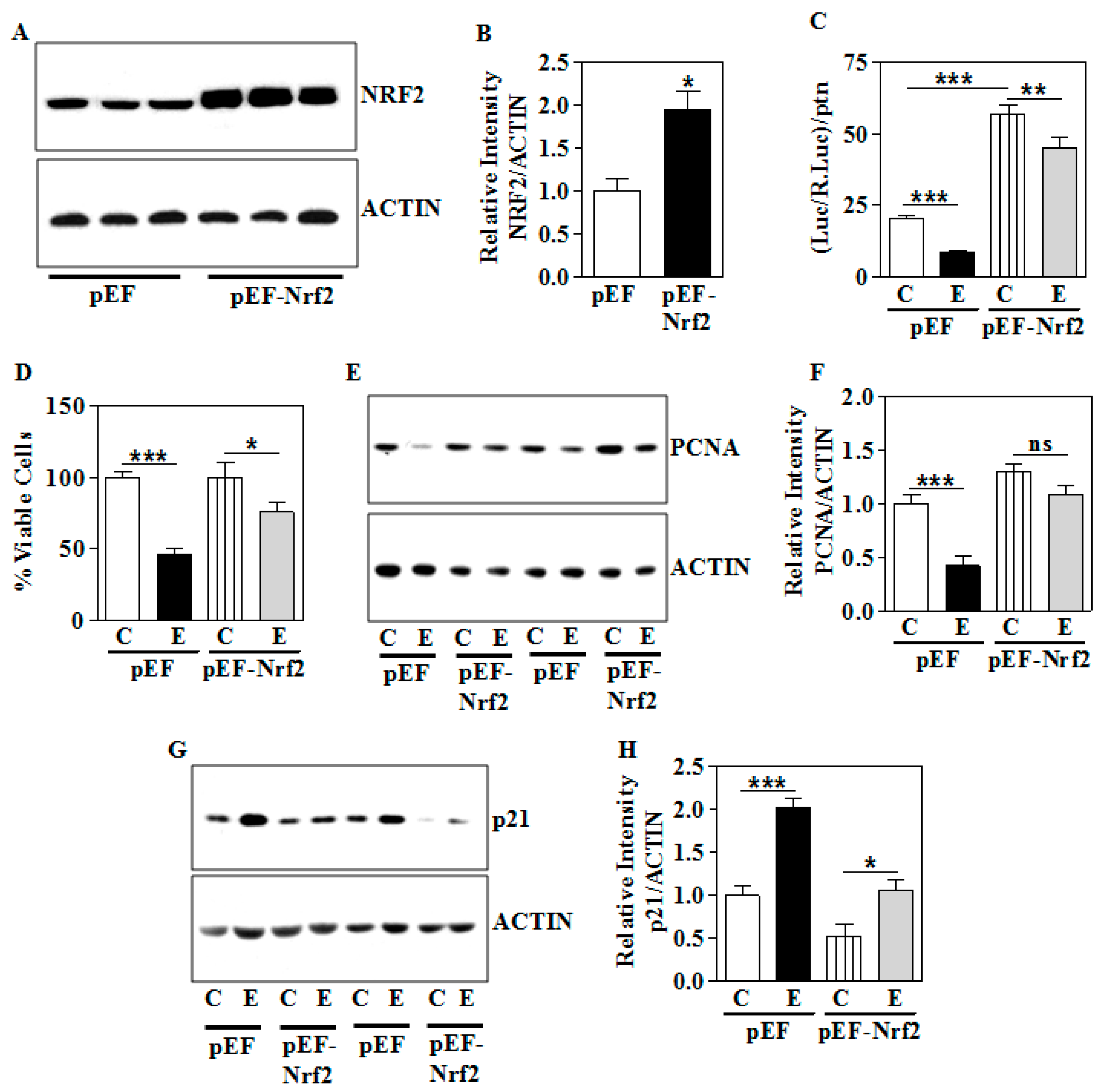
3.7. Genetic Overexpression of Nrf2 Partially Ameliorates the E-Induced Growth Suppression Phenotype
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burd, J.; Blair, J.; Dropps, K. Prenatal Alcohol Exposure, Blood Alcohol Concentrations and Alcohol Elimination Rates for the Mother, Fetus and Newborn. J. Perinatol. 2012, 32, 652–659. [Google Scholar] [CrossRef]
- Bailey, B.A.; Sokol, J. Pregnancy and Alcohol Use: Evidence and Recommendations for Prenatal Care. J. Clin. Gynecol. Obstet. 2008, 51, 436–444. [Google Scholar] [CrossRef]
- Jones, K.L.; Smith, D.L.; Ulleland, C.W.; Streissguth, A.P. Pattern of Malformation in Offspring of Chronic Alcoholics. Lancet 1973, 2, 999–1001. [Google Scholar] [CrossRef]
- Sulik, K.K. Fetal Alcohol Spectrum Disorder: Pathogenesis and Mechanisms. Handb. Clin. Neurol. 2014, 125, 463–475. [Google Scholar] [PubMed]
- Clarren, S.K.; Smith, D.W. The Fetal Alcohol Syndrome. N. Engl. J. Med. 1978, 298, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L. The Effects of Alcohol on Fetal Development. Birth Defects Res. 2011, 93, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.I.; Schenker, S. The Effects of Maternal Alcohol Consumption on The Viability and Visceral Development of The Newborn Rat. Res. Commun. Chem. Pathol. Pharmacol. 1977, 16, 15–32. [Google Scholar]
- Randall, C.L.; Taylor, W.J. Prenatal Ethanol Exposure in Mice: Teratogenic Effects. Teratology 1979, 19, 305–311. [Google Scholar] [CrossRef]
- Patwardhan, R.V.; Schenker, S.; Henderson, G.I.; Abou-Mourad, N.N.; Hoyumpa, A.M. Short-Term and Long-Term Ethanol Administration Inhibits the Placental Uptake and Transport of Valine in Rats. J. Lab. Clin. Med. 1981, 98, 251–262. [Google Scholar]
- Henderson, G.I.; Patwardhan, R.V.; McLeroy, S.; Schenker, S. Inhibition of Placental Amino Acid Uptake in Rats Following Acute and Chronic Ethanol Exposure. Alcohol. Clin. Exp. Res. 1982, 6, 495–505. [Google Scholar] [CrossRef]
- Aliyu, M.H.; Lynch, O.; Nana, P.N.; Alio, A.P.; Wilson, R.E.; Marty, P.J.; Zoorob, R.; Salihu, H.M. Alcohol Consumption During Pregnancy and Risk of Placental Abruption and Placenta Previa. Matern. Child Health J. 2011, 15, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Gundogan, F.; Gilligan, J.; Qi, W.; Chen, E.; Naram, R.; de la Monte, S.M. Dose Effect of Gestational Ethanol Exposure on Placentation and Fetal Growth. Placenta 2015, 36, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Gundogan, F.; Elwood, G.; Mark, P.; Feijoo, A.; Longato, L.; Tong, M.; de la Monte, S.M. Ethanol-Induced Oxidative Stress and Mitochondrial Dysfunction in Rat Placenta: Relevance to Pregnancy Loss. Alcohol. Clin. Exp. Res. 2010, 34, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, R.H.; Ramirez, R.J.; Tsirka, A.E.; Kloesz, J.L.; McLaughlin, M.K.; Gruetzmacher, E.M.; Devaskar, S.U. Uteroplacental Insufficiency Lowers the Threshold towards Hypoxia-Induced Cerebral Apoptosis in Growth-Retarded Fetal Rats. Brain Res. 2001, 895, 186–193. [Google Scholar] [CrossRef]
- Basilious, A.; Yager, J.; Fehlings, M.G. Neurological Outcomes of Animal Models of Uterine Artery Ligation and Relevance to Human Intrauterine Growth Restriction: A systematic review. Dev. Med. Child Neurol. 2015, 57, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Goeden, N.; Velasquez, J.; Arnold, K.A.; Chan, Y.; Lund, B.T.; Anderson, G.M.; Bonnin, A. Maternal Inflammation Disrupts Fetal Neurodevelopment Via Increased Placental Output of Serotonin to the Fetal Brain. J. Neurosci. 2016, 36, 6041–6049. [Google Scholar] [CrossRef]
- Kodomari, I.; Wada, E.; Nakamura, S.; Wada, K. Maternal Supply of BDNF to Mouse Fetal Brain Through the Placenta. Neurochem. Int. 2009, 54, 95–98. [Google Scholar] [CrossRef]
- Brunton, P.J.; Russell, J.A. Endocrine Induced Changes in Brain Function During Pregnancy. Brain Res. 2010, 1364, 198–215. [Google Scholar] [CrossRef]
- Burton, G.J.; Fowden, A.L. Review: The Placenta and Developmental Programming: Balancing Fetal Nutrient Demands with Maternal Resource Allocation. Placenta 2012, 33, 23–27. [Google Scholar] [CrossRef]
- Narasimhan, M.; Mahimainathan, L.; Rathinam, M.L.; Riar, A.K.; Henderson, G.I. Overexpression of Nrf2 Protects Cerebral Cortical Neurons from Ethanol-Induced Apoptotic Death. Mol. Pharmacol. 2011, 80, 988–999. [Google Scholar] [CrossRef]
- Patel, D.; Rathinam, M.; Jarvis, C.; Mahimainathan, L.; Henderson, G.; Narasimhan, M. Role for Cystathionine γ Lyase (CSE) in an Ethanol (E)-Induced Lesion in Fetal Brain GSH Homeostasis. Int. J. Mol. Sci. 2018, 19, 1537. [Google Scholar] [CrossRef] [PubMed]
- Bosco, C.; Preedy, V.R.; Watson, R.R. Alcohol and Xenobiotics in Placenta Damage. Compr. Handb. Alcohol Relat. Pathol. 2005, 2, 921–935. [Google Scholar]
- Rosenberg, M.J.; Wolff, C.R.; El-Emawy, A.; Staples, M.C.; Perrone-Bizzozero, N.I.; Savage, D.D. Effects of Moderate Drinking During Pregnancy on Placental Gene Expression. Alcohol 2010, 44, 673–690. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M.; Wands, J.R. Mitochondrial DNA Damage and Impaired Mitochondrial Function Contribute to Apoptosis of Insulin-Stimulated Ethanol-Exposed Neuronal Cells. Alcohol. Clin. Exp. Res. 2001, 25, 898–906. [Google Scholar] [CrossRef]
- Lyall, F. Development of The Utero-Placental Circulation: The Role of Carbon Monoxide and Nitric Oxide in Trophoblast Invasion and Spiral Artery Transformation. Microsc. Res. Tech. 2003, 60, 402–411. [Google Scholar] [CrossRef]
- Schneider, H. Oxygenation of the Placental-Fetal Unit in Humans. Respir. Physiol. Neurobiol. 2011, 178, 51–58. [Google Scholar] [CrossRef]
- Myatt, L. Review: Reactive Oxygen and Nitrogen Species and Functional Adaptation of the Placenta. Placenta 2010, 31, 66–69. [Google Scholar] [CrossRef]
- Baczyk, D.; Audette, M.C.; Coyaud, E.; Raught, B.; Kingdom, J.C. Spatiotemporal Distribution of Small Ubiquitin-Like Modifiers During Human Placental Development and in Response to Oxidative and Inflammatory Stress. J. Physiol. 2018, 596, 1587–1600. [Google Scholar] [CrossRef]
- Kay, H.H.; Grindle, K.M.; Magness, R.R. Ethanol Exposure Induces Oxidative Stress and Impairs Nitric Oxide Availability in the Human Placental Villi: A Possible Mechanism of Toxicity. Am. J. Obstet. Gynecol. 2000, 182, 682–688. [Google Scholar] [CrossRef]
- Devi, B.G.; Henderson, G.I.; Frosto, T.A.; Schenker, S. Effect of Ethanol on Mitochondria Morphologic and Biochemical Integrity of The Fetal Rat Hepatocytes in Culture. Hepatology 1993, 19, 648–659. [Google Scholar] [CrossRef]
- Chen, J.; Robinson, N.C.; Schenker, S.; Frosto, T.A.; Henderson, G.I. Formation of Hydroxynonenal Adducts with Cytochrome c Oxidase in Rats Following Short-Term Ethanol Intake. Hepatology 1999, 29, 1792–1798. [Google Scholar] [CrossRef]
- Chen, J.; Petersen, D.R.; Schenker, S.; Henderson, G.I. Formation of Malondialdehyde Adducts in Livers of Rats Exposed to Ethanol: Role in Ethanol-Mediated Inhibition of Cytochrome c Oxidase. Alcohol. Clin. Exp. Res. 2000, 24, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, P.; Cederbaum, A.I. Nrf2 is Increased by CYP2E1 in Rodent Liver and Hepg2 Cells and Protects Against Oxidative Stress Caused by CYP2E1. Hepatology 2006, 43, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Sulik, K.K.; Chen, S.Y. Nrf2-Mediated Transcriptional Induction of Antioxidant Response in Mouse Embryos Exposed to Ethanol In vivo: Implications for The Prevention of Fetal Alcohol Spectrum Disorders. Antioxid. Redox Signal. 2008, 10, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, C.K.; Lavoie, H.A.; Dipette, D.J.; Singh, U.S. Resveratrol Restores Nrf2 Level and Prevents Ethanol-Induced Toxic Effects in The Cerebellum of a Rodent Model of Fetal Alcohol Spectrum Disorders. Mol. Pharmacol. 2011, 80, 446–457. [Google Scholar] [CrossRef]
- Bass, T.; Volpe, J.J. Ethanol in Clinically Relevant Concentrations Enhances Expression of Oligodendroglial Differentiation but Has No Effect on Astrocytic Differentiation or DNA Synthesis in Primary Cultures. Dev. Neurosci. 1989, 11, 52–64. [Google Scholar] [CrossRef]
- Riar, A.K.; Narasimhan, M.; Rathinam, M.L.; Vedpathak, D.; Mummidi, S.; Henderson, G.I.; Mahimainathan, L. Ethanol-Induced Transcriptional Activation of Programmed Cell Death 4 (Pdcd4) Is Mediated by GSK-3β Signaling in Rat Cortical Neuroblasts. PLoS ONE 2014, 9, e98080. [Google Scholar] [CrossRef]
- Delbridge, L.M.; Connell, P.J.; Harris, P.J.; Morgan, T.O. Ethanol Effects on Cardiomyocyte Contractility. Clin. Sci. 2000, 98, 401–407. [Google Scholar] [CrossRef]
- Toivari, M.; Mäki, T.; Suutarla, S.; Eklund, K.K. Ethanol Inhibits Ige-Induced Degranulation and Cytokine Production in Cultured Mouse and Human Mast Cells. Life Sci. 2000, 67, 2795–2806. [Google Scholar] [CrossRef]
- Narasimhan, M.; Rathinam, M.; Riar, A.; Patel, D.; Mummidi, S.; Yang, H.S.; Colburn, N.H.; Henderson, G.I.; Mahimainathan, L. Programmed Cell Death 4 (PDCD4): A Novel Player in Ethanol-Mediated Suppression of Protein Translation in Primary Cortical Neurons and Developing Cerebral Cortex. Alcohol. Clin. Exp. Res. 2013, 37, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Riar, A.K.; Narasimhan, M.; Rathinam, M.L.; Henderson, G.I.; Mahimainathan, L. Ethanol Induces Cytostasis of Cortical Basal Progenitors. J. Biomed. Sci. 2016, 23, 6. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.H.; Hawley, T.S.; Hawley, R.G.; MacDougall, J.R.; Kerbel, R.S.; Khoo, N.; Lala, P.K. Establishment and Characterization of First Trimester Human Trophoblast Cells with Extended Lifespan. Exp. Cell Res. 1993, 206, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Kilburn, B.A.; Wang, J.; Duniec-Dmuchowski, Z.M.; Leach, R.E.; Romero, R.; Armant, D.R. Extracellular Matrix Composition and Hypoxia Regulate the Expression of HLA-G and Integrins in a Human Trophoblast Cell Line. Biol. Reprod. 2000, 62, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Patten, A.R.; Fontaine, C.J.; Christie, B.R. A Comparison of the Different Animal Models of Fetal Alcohol Spectrum Disorders and their Use in Studying Complex Behaviors. Front. Pediatr. 2014, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.; Barone, S., Jr. Critical Periods of Vulnerability for the Developing Nervous System: Evidence from Humans and Animal Models. Environ. Health Perspect. 2000, 108, 511–533. [Google Scholar]
- Wong, C.T.; Wais, J.; Crawford, D.A. Prenatal Exposure to Common Environmental Factors Affects Brain Lipids and Increases Risk of Developing Autism Spectrum Disorders. Eur. J. Neurosci. 2015, 42, 2742–2760. [Google Scholar] [CrossRef]
- Adamson, S.L.; Lu, Y.; Whiteley, K.J.; Holmyard, D.; Hemberger, M.; Pfarrer, C.; Cross, J.C. Interactions Between Trophoblast Cells and the Maternal and Fetal Circulation in the Mouse Placenta. Dev. Biol. 2002, 250, 358–373. [Google Scholar] [CrossRef]
- Gilpin, N.W.; Richardson, H.N.; Cole, M.; Koob, G.F. Vapor Inhalation of Alcohol in Rats. Curr. Protoc. Neurosci. 2008, 9, 9–29. [Google Scholar]
- Morton, R.A.; Diaz, M.R.; Topper, L.A.; Valenzuela, C.F. Construction of Vapor Chambers used to Expose Mice to Alcohol During the Equivalent of all Three Trimesters of Human Development. J. Vis. Exp. 2014, 89, 51839. [Google Scholar] [CrossRef]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of Novel MicroRNAs in Post-Transcriptional Control of Nrf2 Expression and Redox Homeostasis in Neuronal, SH-SY5Y Cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef]
- Narasimhan, M.; Riar, A.K.; Rathinam, M.L.; Vedpathak, D.; Henderson, G.; Mahimainathan, L. Hydrogen Peroxide Responsive Mir153 Targets Nrf2/ARE Cytoprotection in Paraquat Induced Dopaminergic Neurotoxicity. Toxicol. Lett. 2014, 228, 179–191. [Google Scholar] [CrossRef]
- Dimauro, I.; Pearson, T.; Caporossi, D.; Jackson, M.J. A Simple Protocol for the Subcellular Fractionation of Skeletal Muscle Cells and Tissue. BMC Res. Notes 2012, 5, 513. [Google Scholar] [CrossRef]
- Morabito, C.; Guarnieri, S.; Catizone, A.; Schiraldi, C.; Ricci, G.; Mariggiò, M.A. Transient Increases in Intracellular Calcium and Reactive Oxygen Species Levels in Tcam-2 Cells Exposed to Microgravity. Sci. Rep. 2017, 7, 15648. [Google Scholar] [CrossRef]
- Kay, H.H.; Tsoi, S.; Grindle, K.; Magness, R.R. Markers of Oxidative Stress in Placental Villi Exposed to Ethanol. J. Soc. Gynecol. Investig. 2006, 13, 118–121. [Google Scholar] [CrossRef]
- Rodrigo, S.; Rodríguez, L.; Otero, P.; Panadero, M.I.; García, A.; Barbas, C.; Roglans, N.; Ramos, S.; Goya, L.; Laguna, J.C.; et al. Fructose During Pregnancy Provokes Fetal Oxidative Stress: The Key Role of The Placental Heme Oxygenase-1. Mol. Nutr. Food Res. 2016, 60, 2700–2711. [Google Scholar] [CrossRef]
- Zhao, F.; Lei, F.; Yan, X.; Zhang, S.; Wang, W.; Zheng, Y. Protective Effects of Hydrogen Sulfide against Cigarette Smoke Exposure-Induced Placental Oxidative Damage by Alleviating Redox Imbalance Via Nrf2 Pathway in Rats. Cell. Physiol. Biochem. 2018, 48, 1815–1828. [Google Scholar] [CrossRef]
- Lee, H.M.; Choi, K.C. Cigarette Smoke Extract and Isoprene Resulted in the Induction of Apoptosis and Autophagy in Human Placenta Choriocarcinoma JEG-3 Cells. Environ. Toxicol. 2018, 33, 178–190. [Google Scholar] [CrossRef]
- Gu, H.; Li, H.; Zhang, L.; Luan, H.; Huang, T.; Wang, L.; Fan, Y.; Zhang, Y.; Liu, X.; Wang, W.; et al. Diagnostic Role of MicroRNA Expression Profile in the Serum of Pregnant Women with Fetuses with Neural Tube Defects. J. Neurochem. 2012, 122, 641–649. [Google Scholar] [CrossRef]
- Erlebacher, A. Immunology of the Maternal-Fetal Interface. Annu. Rev. Immunol. 2013, 31, 387–411. [Google Scholar] [CrossRef]
- Genbacev, O.; Joslin, R.; Damsky, C.H.; Polliotti, B.M.; Fisher, S.J. Hypoxia Alters Early Gestation Human Cytotrophoblast Differentiation/Invasion In vitro and Models the Placental Defects that Occur in Preeclampsia. J. Clin. Investig. 1996, 97, 540–550. [Google Scholar] [CrossRef]
- Rosario, G.X.; Konno, T.; Soares, M.J. Maternal Hypoxia Activates Endovascular Trophoblast Cell Invasion. Dev. Biol. 2008, 314, 362–375. [Google Scholar] [CrossRef]
- Sourisseau, T.; Georgiadis, A.; Tsapara, A.; Ali, R.R.; Pestell, R.; Matter, K.; Balda, M.S. Regulation of PCNA and Cyclin D1 Expression and Epithelial Morphogenesis by the ZO-1-Regulated Transcription Factor ZONAB/DbpA. Mol. Cell. Biol. 2006, 26, 2387–2398. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Hua, X.; Wang, X.; Chen, L. Exposure of Pregnant Mice to Triclosan Impairs Placental Development and Nutrient Transport. Sci. Rep. 2017, 7, 44803. [Google Scholar] [CrossRef]
- Banu, S.K.; Stanley, J.A.; Sivakumar, K.K.; Taylor, R.J.; Arosh, J.A.; Burghardt, R.C. Editor’s Highlight: Exposure to CrVI during Early Pregnancy Increases Oxidative Stress and Disrupts the Expression of Antioxidant Proteins in Placental Compartments. Toxicol. Sci. 2017, 155, 497–511. [Google Scholar] [CrossRef]
- Xu, B.; Hua, J.; Zhang, Y.; Jiang, X.; Zhang, H.; Ma, T.; Zheng, W.; Sun, R.; Shen, W.; Sha, J.; et al. Proliferating Cell Nuclear Antigen (PCNA) Regulates Primordial Follicle Assembly by Promoting Apoptosis of Oocytes in Fetal and Neonatal Mouse Ovaries. PLoS ONE 2011, 6, e16046. [Google Scholar] [CrossRef]
- Abbas, T.; Sivaprasad, U.; Terai, K.; Amador, V.; Pagano, M.; Dutta, A. PCNA-Dependent Regulation of P21 Ubiquitylation and Degradation Via the CRL4Cdt2 Ubiquitin Ligase Complex. Genes Dev. 2008, 22, 2496–2506. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell. 2008, 14, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, G.; Ma, C.; Bower, K.A.; Xu, M.; Fan, Z.; Shi, X.; Ke, Z.J.; Luo, J. Overexpression of Glycogen Synthase Kinase 3beta Sensitizes Neuronal Cells to Ethanol Toxicity. J. Neurosci. Res. 2009, 87, 2793–2802. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Assay ID | Fluorophore |
|---|---|---|
| Gclc | Rn00689046_m1 | FAM |
| Gclm | Rn00568900_m1 | FAM |
| Gsr | Rn01482159_m1 | FAM |
| Ho-1 | Rn01536933_m1 | FAM |
| Nqo1 | Rn00566528_m1 | FAM |
| Nfe2l2 | Rn00477784_m1 | FAM |
| Gapdh | Rn01775763_g1 | VIC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shanmugam, S.; Patel, D.; Wolpert, J.M.; Keshvani, C.; Liu, X.; Bergeson, S.E.; Kidambi, S.; Mahimainathan, L.; Henderson, G.I.; Narasimhan, M. Ethanol Impairs NRF2/Antioxidant and Growth Signaling in the Intact Placenta In Vivo and in Human Trophoblasts. Biomolecules 2019, 9, 669. https://doi.org/10.3390/biom9110669
Shanmugam S, Patel D, Wolpert JM, Keshvani C, Liu X, Bergeson SE, Kidambi S, Mahimainathan L, Henderson GI, Narasimhan M. Ethanol Impairs NRF2/Antioxidant and Growth Signaling in the Intact Placenta In Vivo and in Human Trophoblasts. Biomolecules. 2019; 9(11):669. https://doi.org/10.3390/biom9110669
Chicago/Turabian StyleShanmugam, Sambantham, Dhyanesh Patel, John M. Wolpert, Caezaan Keshvani, Xiaobo Liu, Susan E. Bergeson, Srivatsan Kidambi, Lenin Mahimainathan, George I. Henderson, and Madhusudhanan Narasimhan. 2019. "Ethanol Impairs NRF2/Antioxidant and Growth Signaling in the Intact Placenta In Vivo and in Human Trophoblasts" Biomolecules 9, no. 11: 669. https://doi.org/10.3390/biom9110669
APA StyleShanmugam, S., Patel, D., Wolpert, J. M., Keshvani, C., Liu, X., Bergeson, S. E., Kidambi, S., Mahimainathan, L., Henderson, G. I., & Narasimhan, M. (2019). Ethanol Impairs NRF2/Antioxidant and Growth Signaling in the Intact Placenta In Vivo and in Human Trophoblasts. Biomolecules, 9(11), 669. https://doi.org/10.3390/biom9110669