Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements


 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Role of ROS in Cancer Progression
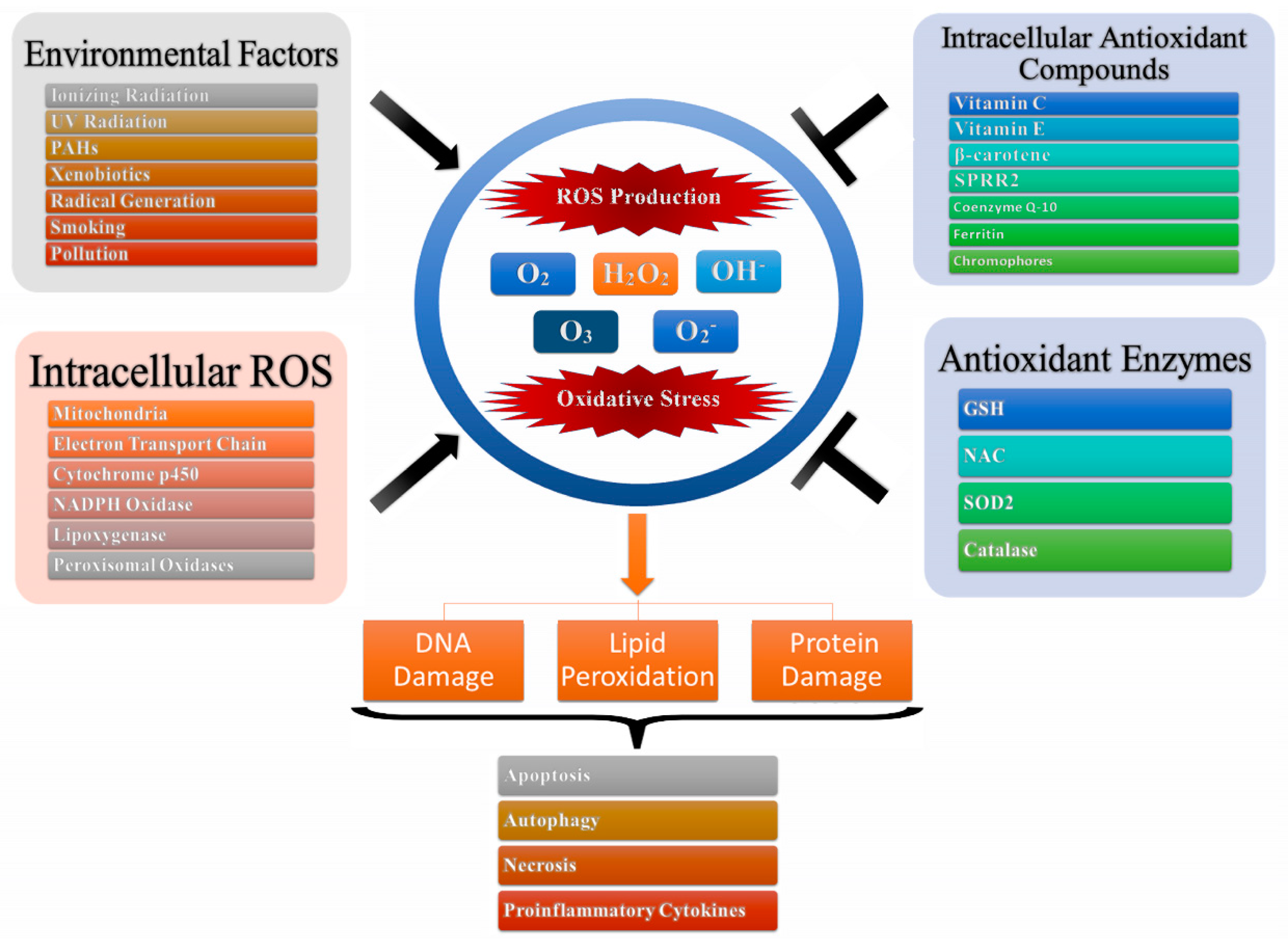
2.1. ROS-Mediated Induction of Oxidative Stress
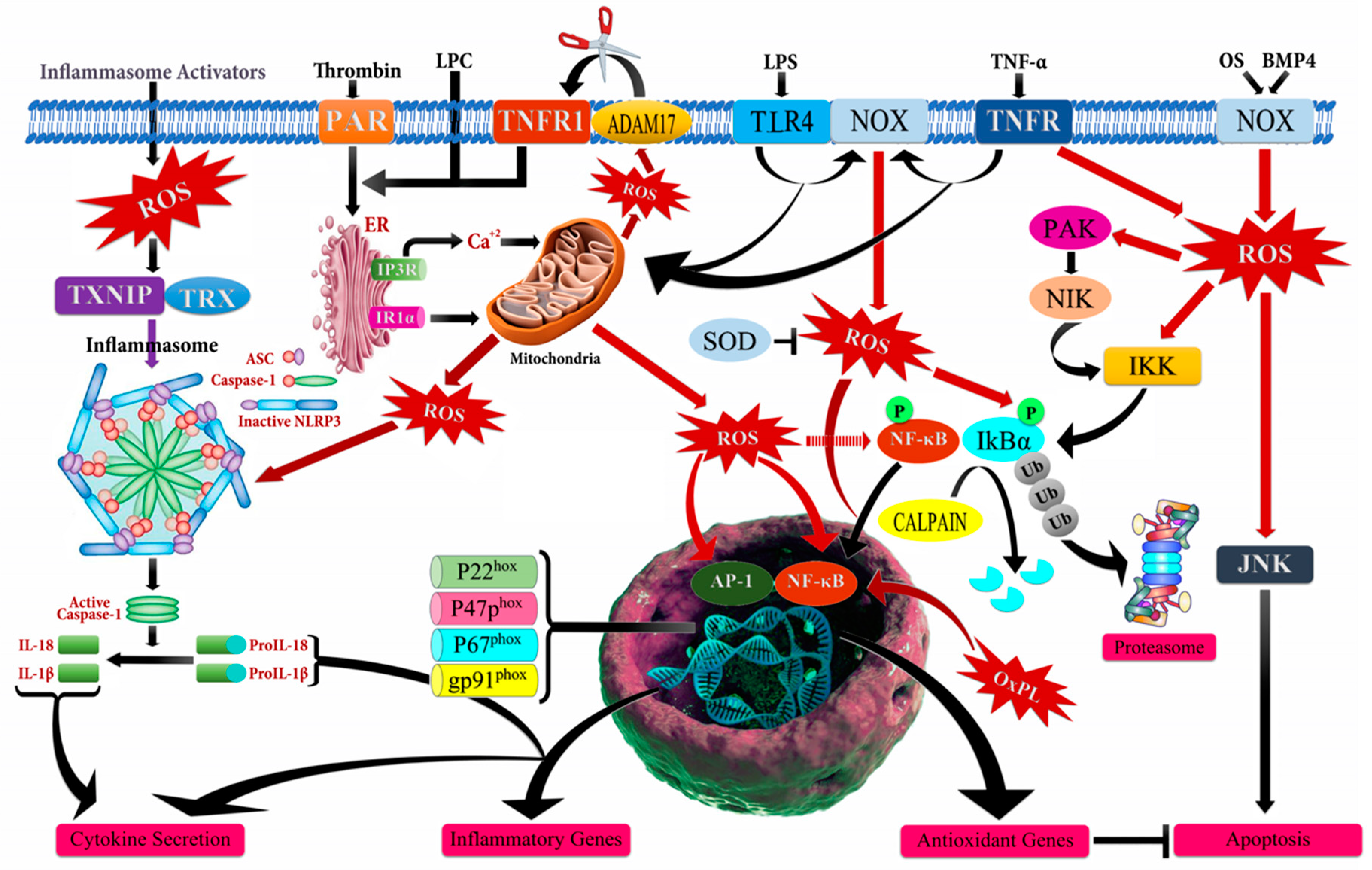
2.2. Inflammatory Markers and ROS
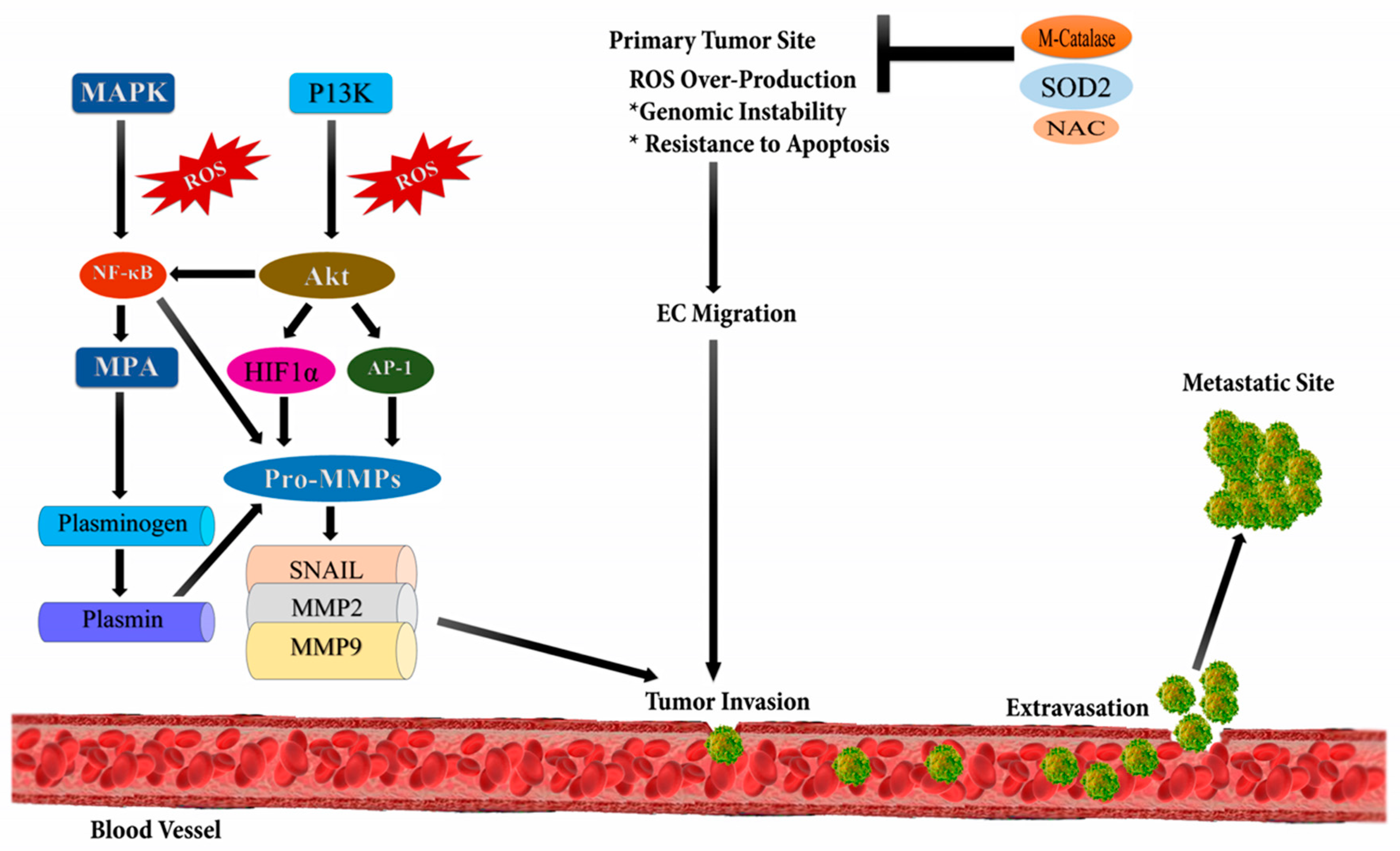
2.3. Cancer Metastasis and ROS
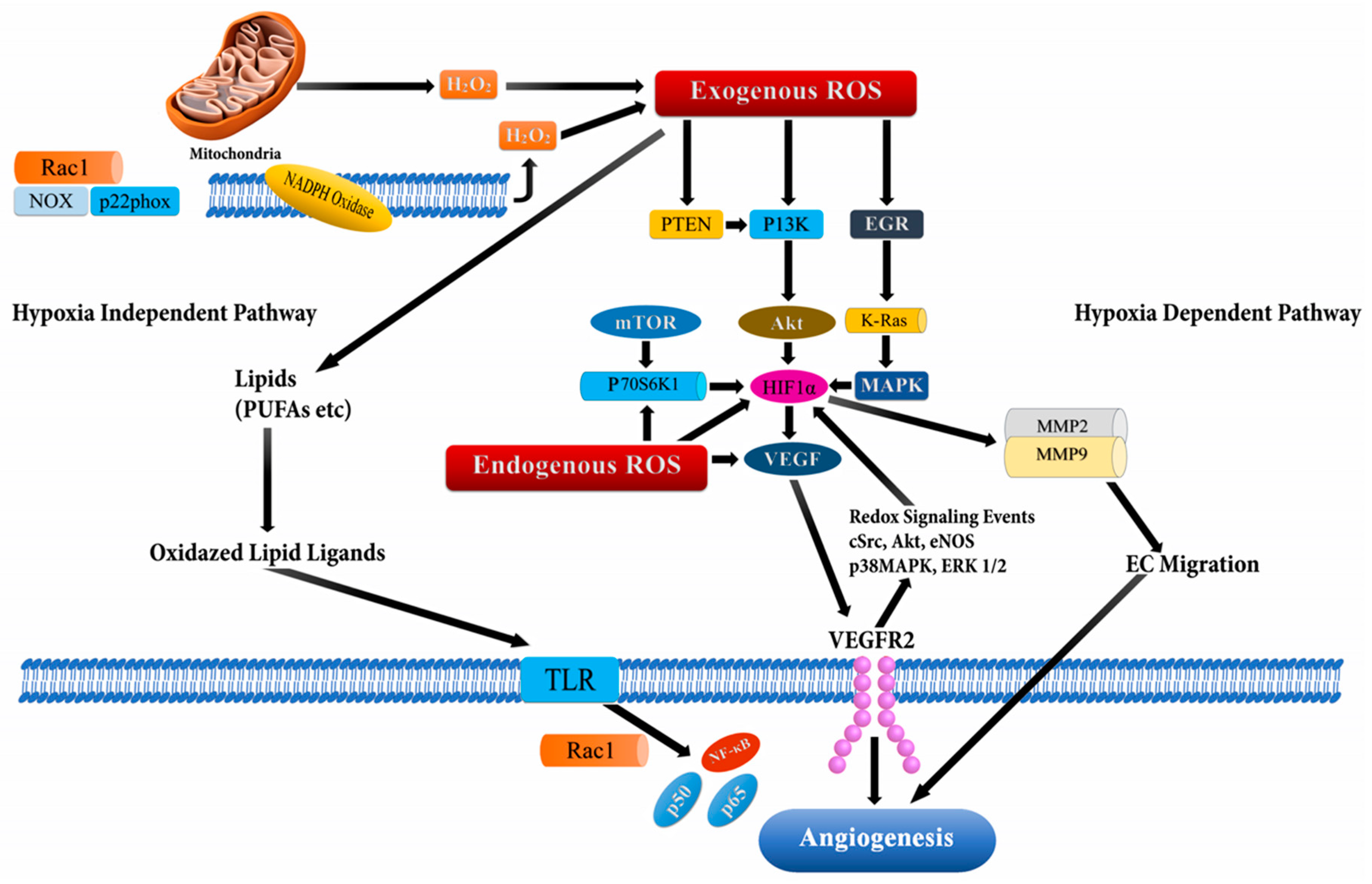
2.4. Angiogenesis and ROS
3. Role of ROS in Cancer Cell Killing
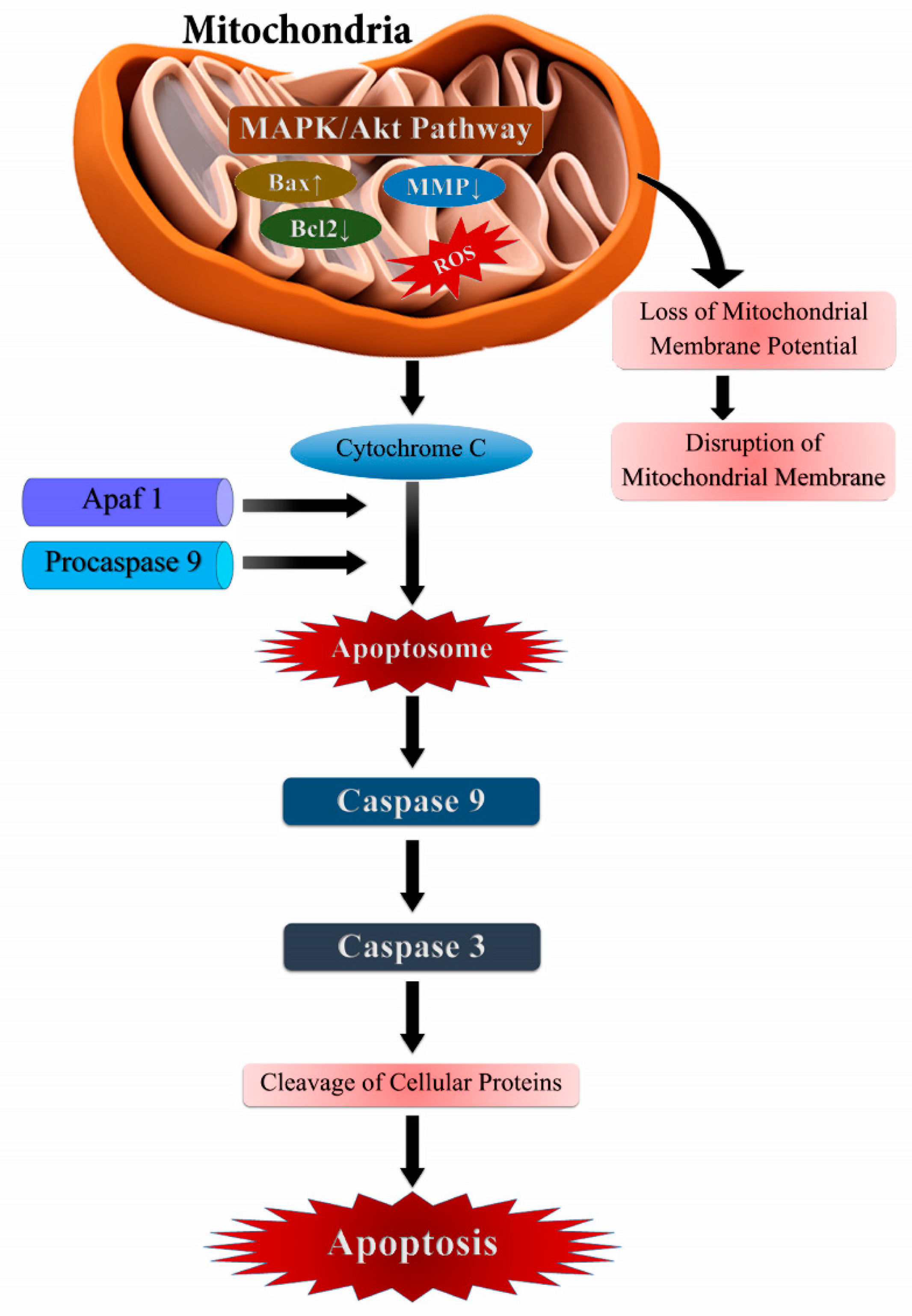
3.1. Cellular Apoptosis and ROS
3.2. Autophagy and ROS
3.3. Anticancer Therapy and ROS
3.4. Inhibition of Antioxidant System in Cancer Cells
3.5. Production of ROS Directly in Cancer Cells
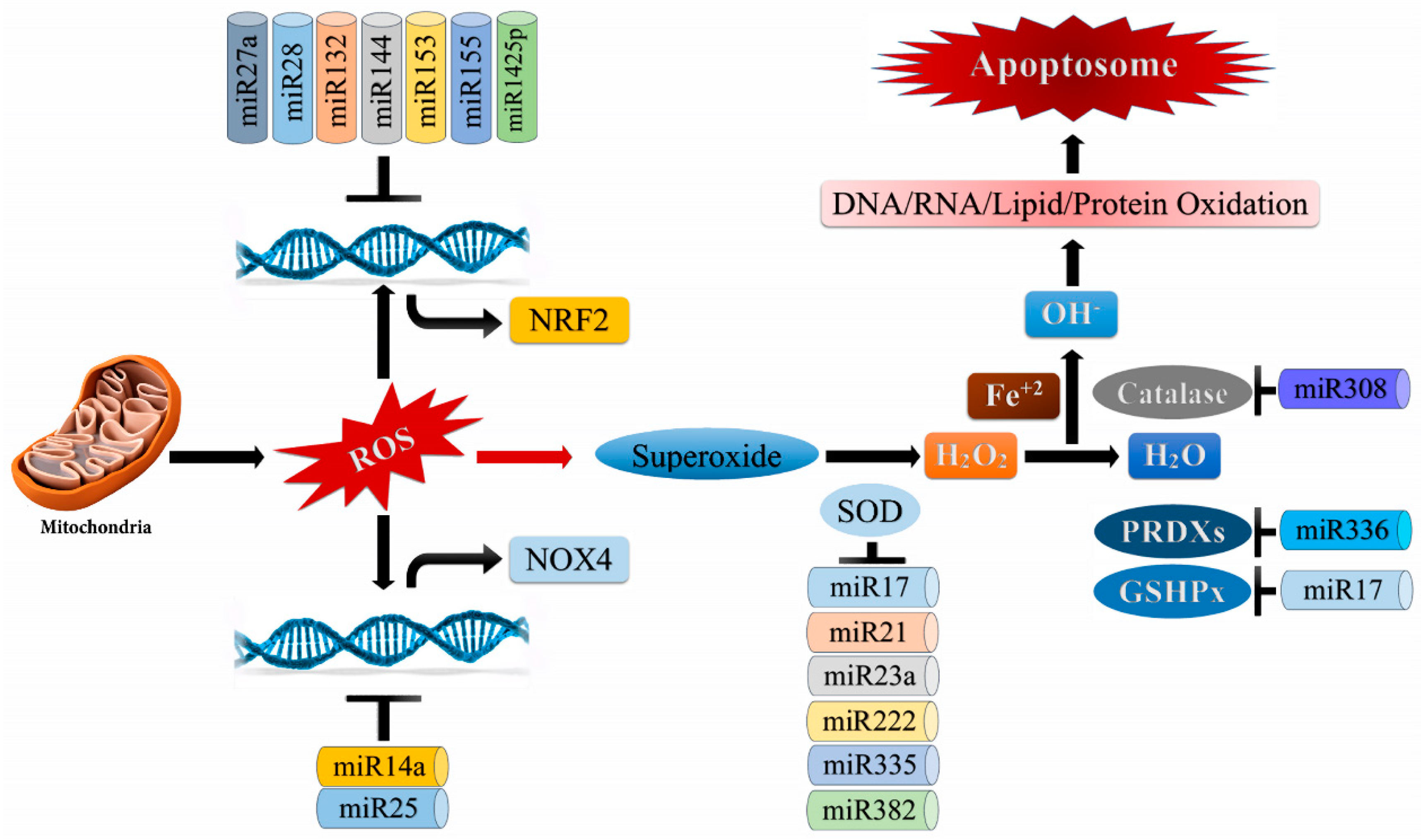
3.6. miRNAs and ROS
4. ROS: A Double-Edged Sword
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- The Lancet. GLOBOCAN 2018: Counting the toll of cancer. Lancet 2018, 392. [Google Scholar] [CrossRef]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Nourazarian, A.R.; Kangari, P.; Salmaninejad, A. Roles of oxidative stress in the development and progression of breast cancer. Asian Pac. J. Cancer Prev. 2014, 15, 4745–4751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ahn, K.S.; Kim, C.; Shanmugam, M.K.; Siveen, K.S.; Arfuso, F.; Samym, R.P.; Deivasigamanim, A.; Lim, L.H.K.; Wang, L.; et al. Nimbolide-induced oxidative stress abrogates STAT3 signaling cascade and inhibits tumor growth in transgenic adenocarcinoma of mouse prostate model. Antioxid. Redox Signal. 2016, 24, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Puar, Y.; Shanmugam, M.; Fan, L.; Arfuso, F.; Sethi, G.; Tergaonkar, V. Evidence for the involvement of the master transcription factor NF-κB in cancer initiation and progression. Biomedicines 2018, 6, 82. [Google Scholar] [CrossRef]
- Rodrigues, C.; Pimpão, C.; Mósca, A.F.; Coxixo, A.S.; Lopes, D.; da Silva, I.V.; Pedersen, P.A.; Antunes, F.; Soveral, G. Human Aquaporin-5 Facilitates Hydrogen Peroxide Permeation Affecting Adaption to Oxidative Stress and Cancer Cell Migration. Cancers 2019, 11, 932. [Google Scholar] [CrossRef]
- Liang, W.; Zhang, Y.; Song, L.; Li, Z. 2, 3′4, 4′, 5-Pentachlorobiphenyl induces hepatocellular carcinoma cell proliferation through pyruvate kinase M2-dependent glycolysis. Toxicol. Lett. 2019, 313, 108–119. [Google Scholar] [CrossRef]
- Sarkar, R.; Kishida, S.; Kishida, M.; Nakamura, N.; Kibe, T.; Karmalar, D.; Chaudhuri, C.R.; Barui, A. Effect of cigarette smoke extract on mitochondrial heme-metabolism: An in vitro model of oral cancer progression. Toxicol. In Vitro 2019, 60, 336–346. [Google Scholar] [CrossRef]
- Bousquet, P.A.; Meltzer, S.; Sønstevold, L.; Esbensen, Y.; Dueland, S.; Flatmark, K.; Sitter, B.; Bathen, T.F.; Seierstad, T.; Redalen, K.R.; et al. Markers of mitochondrial metabolism in tumor hypoxia, systemic inflammation, and adverse outcome of rectal cancer. Transl. Oncol. 2019, 12, 76–83. [Google Scholar] [CrossRef]
- Echizen, K.; Oshima, H.; Nakayama, M.; Oshima, M. The inflammatory microenvironment that promotes gastrointestinal cancer development and invasion. Adv. Biol. Regul. 2018, 68, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Zanotto-Filho, A.; Rajamanickam, S.; Loranc, E.; Masamsetti, V.P.; Gorthi, A.; Romero, J.C.; Tonapi, S.; Gonçalves, R.M.; Reddick, R.L.; Benavides, R.; et al. Sorafenib improves alkylating therapy by blocking induced inflammation, invasion and angiogenesis in breast cancer cells. Cancer Lett. 2018, 425, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Reactive oxygen species induce epithelial--mesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx--2/Snail signaling pathways in MCF--7 cells. Mol. Med. Rep. 2019, 20, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Ren, Y.; Zhu, C. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res. Ther. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.F.; Yang, X.L.; Zhao, Y.; Tian, Q.; Chen, M.T.; Zhao, Y.Y.; Jin, W. Loss of TMEM126A promotes extracellular matrix remodeling, epithelial-to-mesenchymal transition, and breast cancer metastasis by regulating mitochondrial retrograde signaling. Cancer Lett. 2019, 440, 189–201. [Google Scholar] [CrossRef]
- Deng, X.; Feng, N.; Zheng, M.; Ye, X.; Lin, H.; Yu, X.; Gan, Z.; Fang, Z.; Zhang, H.; Gao, M.; et al. PM2.5 exposure-induced autophagy is mediated by lncRNA loc146880 which also promotes the migration and invasion of lung cancer cells. Biochimica Biophysica Acta Gen. Subj. 2017, 1861, 112–125. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, T.; Ke, T.; Huang, X.; Ke, D.; Wang, Q.; Li, H. Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. Br. J. Cancer 2014, 110, 1034–1044. [Google Scholar] [CrossRef]
- Kansestani, A.N.; Mansouri, K.; Hemmati, S.; Zare, M.E.; Moatafaei, A. High Glucose-reduced Apoptosis in Human Breast Cancer Cells Is Mediated by Activation of NF-κB. Iran. J. Allergy Asthma Immunol. 2019, 18, 153–162. [Google Scholar] [CrossRef]
- Zhu, D.; Shen, Z.; Liu, J.; Chen, J.; Liu, Y.; Hu, C.; Li, Z.; Li, Y. The ROS-mediated activation of STAT-3/VEGF signaling is involved in the 27-hydroxycholesterol-induced angiogenesis in human breast cancer cells. Toxicol. Lett. 2016, 264, 79–86. [Google Scholar] [CrossRef]
- Gerschmann, R.; Gilbert, D.; Nye, S.W.; Dwyer, P.; Fenn, W.O. Oxygen poisoning and X-irradiation: A mechanism in common.1954. Nutrition 2001, 17, 162. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Lenaz, G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol. 2012, 942, 93–136. [Google Scholar] [PubMed]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Gonda, T.A.; Tu, S.; Wang, T.C. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle 2009, 8, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Antony, S.; Meitzler, J.L.; Doroshow, J.H. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014, 345, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyap, D.; Tuli, H.S.; Sak, K.; Garg, V.K.; Goel, N.; Punia, S.; Chaudhary, A. Role of Reactive Oxygen Species in Cancer Progression. Curr. Pharmacol. Rep. 2019, 5, doi. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Azad, N.; Rojanasakul, Y.; Vallyathan, V. Inflammation and lung cancer: Roles of reactive oxygen/nitrogen species. J. Toxicol. Environ. Health B Crit. Rev. 2008, 11, 1–15. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017, 15. [Google Scholar] [CrossRef]
- Li, F.; Sethi, G. Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochimica Biophysica Acta 2010, 1805, 167–180. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Chua, A.W.L.; Hay, H.S.; Rajendran, P.; Shanmugam, M.K.; Li, F.; Bist, P.; Koay, E.S.; Lim, L.H.; Kumar, A.P.; Sethi, G. Butein downregulates chemokine receptor CXCR4 expression and function through suppression of NF-κB activation in breast and pancreatic tumor cells. Biochem. Pharmacol. 2010, 80, 1553–1562. [Google Scholar] [CrossRef]
- Nair, A.S.; Shishodia, S.; Ahn, K.S.; Kunnumakkara, A.B.; Sethi, G.; Aggarwal, B.B. Deguelin, an Akt inhibitor, suppresses IκBα kinase activation leading to suppression of NF-κB-regulated gene expression, potentiation of apoptosis, and inhibition of cellular invasion. J. Immunol. 2006, 177, 5612–5622. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.S.; Sethi, G.; Chaturvedi, M.M.; Aggarwal, B.B. Simvastatin, 3--hydroxy--3--methylglutaryl coenzyme A reductase inhibitor, suppresses osteoclastogenesis induced by receptor activator of nuclear factor--κB ligand through modulation of NF--κB pathway. Int. J. Cancer 2008, 123, 1733–1740. [Google Scholar] [CrossRef]
- Manna, S.K.; Aggarwal, R.S.; Sethi, G.; Aggarwal, B.B.; Ramesh, G.T. Morin (3, 5, 7, 2′, 4′-pentahydroxyflavone) abolishes nuclear factor-κB activation induced by various carcinogens and inflammatory stimuli, leading to suppression of nuclear factor-κB–regulated gene expression and up-regulation of apoptosis. Clin. Cancer Res. 2007, 13, 2290–2297. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Onco. 2013, 18, 43–73. [Google Scholar] [CrossRef] [Green Version]
- Brooks, S.A.; Lomax-Browne, H.J.; Carter, T.M.; Kinch, C.E.; Hall, D.M. Molecular interactions in cancer cell metastasis. Acta Histochemica 2010, 112, 3–25. [Google Scholar] [CrossRef]
- Guerrini, L.; Garcia-Rico, E.; Alvarez-Puebla, R. The Role of Nanoscience in Cancer Diagnosis. In Handbook of Nanomaterials for Cancer Theranostics; Conde, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 177–197. [Google Scholar]
- Chitty, J.L.; Filipe, E.C.; Lucas, M.C.; Herrmann, D.; Cox, T.R.; Timpson, P. Recent advances in understanding the complexities of metastasis. F1000Research 2018. [Google Scholar] [CrossRef]
- Liao, Z.; Chua, D.; Tan, N.S. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef]
- Kamiya, T.; Goto, A.; Kurokawa, E.; Hara, H.; Adachi, T. Cross talk mechanism among EMT, ROS, and histone acetylation in phorbol ester-treated human breast cancer MCF-7 cells. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.H.; Dier, U.; Melendez, J.A.; Hempel, N. Regulation of MMP-1 expression in response to hypoxia is dependent on the intracellular redox status of metastatic bladder cancer cells. Biochim. Biophys. Acta 2015, 1852, 2593–2602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Liu, Z.; Hu, X. Inhibiting cancer metastasis via targeting NAPDH oxidase 4. Biochem. Pharmacol. 2013, 86, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagati, A.; Moparthy, S.; Fink, E.E.; Bianchi-Smiraglia, A.; Yun, D.H.; Kolesnikova, M.; Udartseva, O.O.; Wolff, D.W.; Roll, M.V.; Lipchick, B.C.; et al. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene 2019, 38, 3585–3597. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38. [Google Scholar] [CrossRef] [Green Version]
- Aydin, E.; Johansson, J.; Nazir, F.H.; Hellstrand, K.; Martner, A. Role of NOX2-derived reactive oxygen species in NK cell–mediated control of murine melanoma metastasis. Cancer Immunol. Res. 2017, 5, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef]
- Paccione, R.J.; Miyazaki, H.; Patel, V.; Waseem, A.; Gutkind, J.S.; Zehner, Z.E.; Yeudall, W.A. Keratin down-regulation in vimentin-positive cancer cells is reversible by vimentin RNA interference, which inhibits growth and motility. Mol. Cancer Ther. 2008, 7, 2894–2903. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.O.; Gu, J.M.; Kim, M.S.; Kim, H.S.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: Methylation of the E-cadherin promoter. Gastroenterology 2008, 135, 2128–2140. [Google Scholar] [CrossRef]
- Senger, D.R.; Brown, L.F.; Claffey, K.P.; Dvorak, H.F. Vascular permeability factor, tumor angiogenesis and stroma generation. Invasion Metastasis 1994, 14, 385–394. [Google Scholar]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sa Junior, P.L.; Camara, D.A.D.; Porcacchia, A.S.; Fonseca, P.M.M.; Jorge, S.D.; Araldi, R.P.; Ferreira, A.K. The Roles of ROS in Cancer Heterogeneity and Therapy. Oxid. Med. Cell. Longev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Schito, L. Hypoxia-Dependent Angiogenesis and Lymphangiogenesis in Cancer. Adv. Exp. Med. Biol. 2019, 1136, 71–85. [Google Scholar] [PubMed]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4. [Google Scholar] [CrossRef] [Green Version]
- Rezatabar, S.; Karimian, A.; Rameshknia, V.; Parsian, H.; Majidinia, M.; Kopi, T.A.; Bishayee, A.; Sadeghinia, A.; Yousefi, M.; Monirialamdari, M.; et al. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Khromova, N.V.; Kopnin, P.B.; Stepanova, E.V.; Agapova, L.S.; Kopnin, B.P. p53 hot-spot mutants increase tumor vascularization via ROS-mediated activation of the HIF1/VEGF-A pathway. Cancer Lett. 2009, 276, 143–151. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, Y.; Shi, M.; Zhang, Q.; Wang, Q.; Chen, X. Deferoxamine promotes MDA-MB-231 cell migration and invasion through increased ROS-dependent HIF-1alpha accumulation. Cell. Physiol. Biochem. 2014, 33, 1036–1046. [Google Scholar] [CrossRef]
- Han, X.; Sun, S.; Zhao, M.; Cheng, X.; Chen, G.; Lin, S.; Guan, Y.; Yu, X. Celastrol stimulates hypoxia-inducible factor-1 activity in tumor cells by initiating the ROS/Akt/p70S6K signaling pathway and enhancing hypoxia-inducible factor-1alpha protein synthesis. PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Liu, L.Z.; Hu, X.W.; Xia, C.; He, J.; Zhou, Q.; Shi, X.; Fang, J.; Jiang, B.H. Reactive oxygen species regulate epidermal growth factor-induced vascular endothelial growth factor and hypoxia-inducible factor-1alpha expression through activation of AKT and P70S6K1 in human ovarian cancer cells. Free Radic. Biol. Med. 2006, 41, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Brewer, T.F.; Garcia, F.J.; Onak, C.S.; Carroll, K.S.; Chang, C.J. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu. Rev. Biochem. 2015, 84, 765–790. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, F.; Wu, Z.; Hu, X.; Zhang, J.; Gao, Z.; Han, X.; Qin, J.; Li, C.; Wang, Y. The PI3K/Akt/GSK-3beta/ROS/eIF2B pathway promotes breast cancer growth and metastasis via suppression of NK cell cytotoxicity and tumor cell susceptibility. Cancer Biol. Med. 2019, 16, 38–54. [Google Scholar] [PubMed] [Green Version]
- Govindarajan, B.; Sligh, J.E.; Vincent, B.J.; Li, M.; Canter, J.A.; Nickoloff, B.J.; Rodenburg, R.J.; Smeitink, J.A.; Oberley, L.; Zhang, Y.; et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Invest. 2007, 117, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoni, E.; Fiaschi, T.; Ramponi, G.; Chiarugi, P. Redox regulation of anoikis resistance of metastatic prostate cancer cells: Key role for Src and EGFR-mediated pro-survival signals. Oncogene 2009, 28, 2074–2086. [Google Scholar] [CrossRef] [Green Version]
- Aydin, E.; Hallner, A.; Grauers Wiktorin, H.; Staffas, A.; Hellstrand, K.; Martner, A. NOX2 inhibition reduces oxidative stress and prolongs survival in murine KRAS-induced myeloproliferative disease. Oncogene 2019, 38, 1534–1543. [Google Scholar] [CrossRef] [Green Version]
- Bino, L.; Vesela, I.; Papezikova, I.; Prochazkova, J.; Vasicek, O.; Stefkova, K.; Kucera, J.; Hanackova, M.; Kubala, L.; Pachernik, J. The depletion of p38alpha kinase upregulates NADPH oxidase 2/NOX2/gp91 expression and the production of superoxide in mouse embryonic stem cells. Arch. Biochem. Biophys. 2019, 671, 18–26. [Google Scholar] [CrossRef]
- Komatsu, D.; Kato, M.; Nakayama, J.; Miyagawa, S.; Kamata, T. NADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expression. Oncogene 2008, 27, 4724–4732. [Google Scholar] [CrossRef] [Green Version]
- De Bessa, T.C.; Pagano, A.; Moretti, A.I.S.; Oliveira, P.V.S.; Mendonca, S.A.; Kovacic, H.; Laurindo, F.R.M. Subverted regulation of Nox1 NADPH oxidase-dependent oxidant generation by protein disulfide isomerase A1 in colon carcinoma cells with overactivated KRas. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kim, K.E.; Koh, G.Y.; Ho, Y.S.; Lee, K.J. Hydrogen peroxide produced by angiopoietin-1 mediates angiogenesis. Cancer Res. 2006, 66, 6167–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; De Marco, P.; Cirillo, F.; Cappello, A.R.; Dolce, V.; Belfiore, A.; et al. Copper activates HIF-1alpha/GPER/VEGF signalling in cancer cells. Oncotarget 2015, 6, 34158–34177. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liu, L.Z.; Jiang, Y.; Zhu, Y.; Guo, N.L.; Barnett, J.; Rojanasakul, Y.; Agani, F.; Jiang, B.H. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol. Sci. 2012, 125, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Coso, S.; Harrison, I.; Harrison, C.B.; Vinh, A.; Sobey, C.G.; Drummond, G.R.; Williams, E.D.; Selemidis, S. NADPH oxidases as regulators of tumor angiogenesis: Current and emerging concepts. Antioxid. Redox Signal. 2012, 16, 1229–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, H.; Wartenberg, M. Reactive oxygen species as signaling molecules in cardiovascular differentiation of embryonic stem cells and tumor-induced angiogenesis. Antioxid. Redox Signal. 2005, 7, 1423–1434. [Google Scholar] [CrossRef]
- Harrison, I.P.; Vinh, A.; Johnson, I.R.D.; Luong, R.; Drummond, G.R.; Sobey, C.G.; Tiganis, T.; Williams, E.D.; JJ, O.L.; Brooks, D.A.; et al. NOX2 oxidase expressed in endosomes promotes cell proliferation and prostate tumour development. Oncotarget 2018, 9, 35378–35393. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Uchida, T.; Yoshie, T.; Mizote, Y.; Ishikawa, F.; Katsuyama, M.; Shibanuma, M. A mitochondrial ROS pathway controls matrix metalloproteinase 9 levels and invasive properties in RAS-activated cancer cells. FEBS J. 2019, 286, 459–478. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Wang, Q.; Li, C.; Wang, Y.; Chen, X. VEGF stimulated the angiogenesis by promoting the mitochondrial functions. Oncotarget 2017, 8, 77020–77027. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, S.; Nakagawa, Y.; Kitagishi, Y.; Nakanishi, A.; Murai, T. Reactive Oxygen Species, Superoxide Dimutases, and PTEN-p53-AKT-MDM2 Signaling Loop Network in Mesenchymal Stem/Stromal Cells Regulation. Cells 2018, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Fu, Q.; Xu, B.; Zhou, H.; Gao, J.; Shao, X.; Xiong, J.; Gu, Q.; Wen, S.; Li, F.; et al. Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ROS-mediated AKT activation. Int. J. Cancer 2018, 142, 1786–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, H.P.; Rodrigues, E.G.; Amorim Reis, A.K.C.; Longo, L.S., Jr.; Ogata, F.T.; Moretti, A.I.S.; da Costa, P.E.; Teodoro, A.C.S.; Toledo, M.S.; Stern, A. Nitric oxide and interactions with reactive oxygen species in the development of melanoma, breast, and colon cancer: A redox signaling perspective. Nitric Oxide 2019, 89, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yue, G.G.; Chan, A.M.; Tsui, S.K.; Fung, K.P.; Sun, H.; Pu, J.; Lau, C.B. Eriocalyxin B, a novel autophagy inducer, exerts anti-tumor activity through the suppression of Akt/mTOR/p70S6K signaling pathway in breast cancer. Biochem. Pharmacol. 2017, 142, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Tania, M.; Zhang, D.Z.; Chen, H.C. Antioxidant enzymes and cancer. Chin. J. Cancer Res. 2010, 22, 87–92. [Google Scholar] [CrossRef]
- Woo, C.C.; Hsu, A.; Kumar, A.P.; Sethi, G.; Tan, K.H.B. Thymoquinone inhibits tumor growth and induces apoptosis in a breast cancer xenograft mouse model: The role of p38 MAPK and ROS. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Wang, L.; Deivasigamni, A.; Looi, C.Y.; Karthikeyan, C.; Trivedi, P.; Chinnathambi, A.; Alharbi, S.A.; Arfuso, F.; Dharmarajan, A.; et al. A novel benzimidazole derivative, MBIC inhibits tumor growth and promotes apoptosis via activation of ROS-dependent JNK signaling pathway in hepatocellular carcinoma. Oncotarget 2017, 8, 12831–12842. [Google Scholar] [CrossRef]
- Kim, C.; Lee, S.G.; Yang, W.M.; Arfuso, F.; Um, J.Y.; Kumar, A.P.; Bian, J.; Sethi, G.; Ahn, K.S. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018, 431, 123–141. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Park, K.R.; Nam, D.; Yun, H.M.; Lee, S.G.; Jang, H.J.; Sethi, G.; Cho, S.K.; Ahn, K.S. β-Caryophyllene oxide inhibits growth and induces apoptosis through the suppression of PI3K/AKT/mTOR/S6K1 pathways and ROS-mediated MAPKs activation. Cancer Lett. 2011, 312, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A.; Loo, S.Y.; Rajendran, P.; Manu, K.A.; Perumal, E.; Li, F.; Shanmugam, M.K.; Siveen, K.S.; Park, J.I.; Ahn, K.S.; et al. An anthraquinone derivative, emodin sensitizes hepatocellular carcinoma cells to TRAIL induced apoptosis through the induction of death receptors and downregulation of cell survival proteins. Apoptosis 2013, 18, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Hirpara, J.L.; Eu, J.-Q.; Sethi, G.; Wang, L.; Goh, B.-C.; Wong, A.L. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol. 2018. [Google Scholar] [CrossRef]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef]
- Liu, L.; Ahn, K.S.; Shanmugam, M.K.; Wang, H.; Shen, H.; Arfuso, F.; Chinnathambi, A.; Alharbi, S.A.; Chang, Y.; Sethi, G.; et al. Oleuropein induces apoptosis via abrogating NF--κB activation cascade in estrogen receptor–negative breast cancer cells. J. Cell. Biochem. 2019, 120, 4504–4513. [Google Scholar] [CrossRef]
- Khan, M.A.; Chen, H.C.; Wan, X.X.; Tania, M.; Xu, A.H.; Chen, F.Z.; Zhang, D.Z. Regulatory effects of resveratrol on antioxidant enzymes: A mechanism of growth inhibition and apoptosis induction in cancer cells. Mol. Cells 2013, 35, 219–225. [Google Scholar] [CrossRef]
- Hussain, A.R.; Ahmed, M.; Ahmed, S.; Manogaran, P.; Platanias, L.C.; Alvi, S.N.; Al-Kuraya, K.S.; Uddin, S. Thymoquinone suppresses growth and induces apoptosis via generation of reactive oxygen species in primary effusion lymphoma. Free Radic. Biol. Med. 2011, 50, 978–987. [Google Scholar] [CrossRef]
- Dergarabetian, E.; Ghattass, K.; El-Sitt, S.; Al-Mismar, R.; El-Baba, C.; Itani, W.; Melhem, N.; El-Hajj, H.; Bazarbachi, A.; Schneider-Stock, R.; et al. Thymoquinone induces apoptosis in malignant T-cells via generation of ROS. Front. Biosci. 2013, 5, 706–719. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Tania, M.; Fu, S.; Fu, J. Thymoquinone, as an anticancer molecule: From basic research to clinical investigation. Oncotarget 2017, 8, 51907–51919. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, Y.K.; Abdelrazek, H.M. Cancer: Thymoquinone antioxidant/pro-oxidant effect as potential anticancer remedy. Biomed. Pharmacother. 2019, 115. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Shanmugam, M.K.; Chen, L.; Chatterjee, S.; Basha, J.; Kumar, A.P.; Kundu, T.K.; Sethi, G. Garcinol, a polyisoprenylated benzophenone modulates multiple proinflammatory signaling cascades leading to the suppression of growth and survival of head and neck carcinoma. Cancer Prev. Res. 2013, 6, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Sodrul, I.M.; Wang, C.; Chen, X.; Du, J.; Sun, H. Role of ginsenosides in reactive oxygen species-mediated anticancer therapy. Oncotarget 2018, 9, 2931–2950. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Balan, M.; Flynn, E.; Zurakowski, D.; Choueiri, T.K.; Pal, S. Activation of c-Met in cancer cells mediates growth-promoting signals against oxidative stress through Nrf2-HO-1. Oncogenesis 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Li, Y.; Lin, Z.; Zhao, M.; Xu, T.; Wang, C.; Deng, N. Silver nanoparticles induce HePG-2 cells apoptosis through ROS-mediated signaling pathways. Nanoscale Res. Lett. 2016, 11. [Google Scholar] [CrossRef] [Green Version]
- An, B.C.; Choi, Y.D.; Oh, I.J.; Kim, J.H.; Park, J.I.; Lee, S.W. GPx3-mediated redox signaling arrests the cell cycle and acts as a tumor suppressor in lung cancer cell lines. PLoS One 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhuang, Z.; Wu, T.; Lin, J.C.; Liu, Z.X.; Zhou, L.F.; Dai, T.; Lu, L.; Ju, H.Q. Nicotinamide nucleotide transhydrogenase-mediated redox homeostasis promotes tumor growth and metastasis in gastric cancer. Redox Biol. 2018, 18, 246–255. [Google Scholar] [CrossRef]
- Kim, C.; Song, H.S.; Park, H.; Kim, B. Activation of ER stress-dependent miR-216b has a critical role in Salvia miltiorrhiza ethanol-extract-induced apoptosis in U266 and U937 cells. Int. J. Mol. Sci. 2018, 19, 1240. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Cheng, L.; Jiang, Z.; Chen, K.; Zhou, C.; Sun, L.; Cao, J.; Qian, W.; Li, J.; Shan, T.; et al. Resveratrol inhibits ROS-promoted activation and glycolysis of pancreatic stellate cells via suppression of miR-21. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef]
- Kim, S.M.; Hur, D.Y.; Hong, S.W.; Kim, J.H. EBV-encoded EBNA1 regulates cell viability by modulating miR34a-NOX2-ROS signaling in gastric cancer cells. Biochem. Biophys. Res. Commun. 2017, 494, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.K.; Bhoopathi, P.; Talukdar, S.; Scheunemann, D.; Sarkar, D.; Cavenee, W.K.; Das, S.K.; Emdad, L.; Fisher, P.B. MDA-7/IL-24 regulates the miRNA processing enzyme DICER through downregulation of MITF. Proc. Natl. Acad. Sci. USA 2019, 116, 5687–5692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wang, Y.; Zhou, W.; Chen, T.; Wu, Q.; Chutturghoon, V.K.; Lin, B.; Geng, L.; Yang, Z.; Zhou, L.; et al. YAP promotes multi-drug resistance and inhibits autophagy-related cell death in hepatocellular carcinoma via the RAC1-ROS-mTOR pathway. Cancer Cell Int. 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, H.; Dong, L.; Xu, M.; Zhang, L.; Ding, W.; Zhang, J.; Lin, J.; Zhang, Y.; Qiu, B.; et al. Enhancing tumor chemotherapy and overcoming drug resistance through autophagy mediated intracellular dissolution of zinc oxide nanoparticles. Nanoscale 2019, 11, 11789–11807. [Google Scholar] [CrossRef]
- Hui, K.F.; Yeung, P.L.; Chiang, A.K. Induction of MAPK-and ROS-dependent autophagy and apoptosis in gastric carcinoma by combination of romidepsin and bortezomib. Oncotarget 2016, 7, 4454–4467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.F.; Brisson, L. Autophagy and mitophagy in cancer metabolic remodelling. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Ciccarone, F.; Castelli, S.; Ciriolo, M.R. Oxidative Stress-Driven Autophagy acROSs Onset and Therapeutic Outcome in Hepatocellular Carcinoma. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.D.; Sun, C.; Lambeth, J.D.; Marshall, F.; Amin, M.; Chung, L.; Petros, J.A.; Arnold, R.S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2005, 62, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Asoodeh, A.; Klonisch, T.; Halayko, A.J.; Kadkhoda, K.; Kroczak, T.J.; Gibson, S.B.; Booy, E.P.; Naderi--Manesh, H.; Los, M. Brevinin--2R1 semi--selectively kills cancer cells by a distinct mechanism, which involves the lysosomal--mitochondrial death pathway. J. Cell. Mol. Med. 2008, 12, 1005–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Niu, X.; Chen, Y.; Hu, Q.; Shi, G.; Wu, H.; Wang, J.; Yi, J. Emodin-induced generation of reactive oxygen species inhibits RhoA activation to sensitize gastric carcinoma cells to anoikis. Neoplasia 2008, 10, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Vargas, J.M.; Oliver-Pozo, F.J.; Dantzer, F. PARP1 and Poly (ADP-ribosyl) ation Signaling during Autophagy in Response to Nutrient Deprivation. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Boyer-Guittaut, M.; Poillet, L.; Liang, Q.; Bôle-Richard, E.; Ouyang, X.; Benavides, G.A.; Chakrama, F.-Z.; Fraichard, A.; Darley-Usmar, V.M.; Despouy, G.; et al. The role of GABARAPL1/GEC1 in autophagic flux and mitochondrial quality control in MDA-MB-436 breast cancer cells. Autophagy 2014, 10, 986–1003. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Morgan, M.J.; Choksi, S.; Liu, Z.G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 2007, 26, 675–687. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and autophagy: Interactions and molecular regulatory mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Zhao, J.; Shi, J.; Wang, M.; Qiu, S.; Hu, Y.; Xu, Y.; Cui, Y.; Liu, C. The specific inhibition of SOD1 selectively promotes apoptosis of cancer cells via regulation of the ROS signaling network. Oxid. Med. Cell. Longev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.A.; Fang, Q. Are peroxiredoxins tumor suppressors? Curr. Opin. Pharmacol. 2007, 7, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-H. MicroRNA Networks Modulate Oxidative Stress in Cancer. Int. J. Mol. Sci. 2019, 20, 4497. [Google Scholar] [CrossRef] [Green Version]
- Cadzow, M. Selection and Metabolic Disease in the Pacific. Ph.D. Thesis, University of Otago, Dunedin, New Zealand, 2018. [Google Scholar]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 reduces experimental non–small-cell lung cancer. J. Clin. Invest. 2014, 124, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Gomez, M.; Germain, D. Cross talk between SOD1 and the mitochondrial UPR in cancer and neurodegeneration. Mol. Cell. Neurosci. 2019, 98, 12–18. [Google Scholar] [CrossRef]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef]
- Mattes, K.; Vellenga, E.; Schepers, H. Differential redox-regulation and mitochondrial dynamics in normal and leukemic hematopoietic stem cells: A potential window for leukemia therapy. Crit. Rev. Oncol. Hematol. 2019, 144. [Google Scholar] [CrossRef]
- Sánchez-Álvarez, M.; Strippoli, R.; Donadelli, M.; Bazhin, A.V.; Cordani, M. Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer. Cancers 2019, 11, 1415. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Wang, B.; Yang, L.; Zhang, Y. The role of ROS-induced autophagy in hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 306–312. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37. [Google Scholar] [CrossRef]
- Ma, J.; Kavelaars, A.; Dougherty, P.M.; Heijnen, C.J. Beyond symptomatic relief for chemotherapy--induced peripheral neuropathy: Targeting the source. Cancer 2018, 124, 2289–2298. [Google Scholar] [CrossRef]
- Bartolini, D.; Torquato, P.; Piroddi, M.; Galli, F. Targeting glutathione S-transferase P and its interactome with selenium compounds in cancer therapy. Biochim. Biophys. Acta. Gen. Subj. 2019, 1863, 130–143. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Renschler, M.F. The emerging role of reactive oxygen species in cancer therapy. Eur. J. Cancer 2004, 40, 1934–1940. [Google Scholar] [CrossRef]
- Gana, C.C.; Hanssen, K.M.; Denise, M.; Flemming, C.L.; Wheatley, M.S.; Conseil, G.; Cole, S.P.; Norris, M.D.; Haber, M.; Fletcher, J.I. MRP1 modulators synergize with buthionine sulfoximine to exploit collateral sensitivity and selectively kill MRP1-expressing cancer cells. Biochem. Pharmacol. 2019, 168, 237–248. [Google Scholar] [CrossRef]
- Cilurzo, F.; Cristiano, M.; Da, M.P.; Quintieri, L.; Paolinod, D.; Pasut, G. Overcoming Cancer Cell Drug Resistance by a Folic Acid Targeted Polymeric Conjugate of Buthionine Sulfoximine. Anticancer Agents Med. Chem. 2019. [CrossRef]
- Zhao, Y.; Tanaka, S.; Yuan, B.; Sugiyama, K.; Onda, K.; Kiyomi, A.; Takagi, N.; Sugiura, M.; Hirano, T. Arsenic Disulfide Combined with L-Buthionine-(S, R)-Sulfoximine Induces Synergistic Antitumor Effects in Two-Dimensional and Three-Dimensional Models of MCF-7 Breast Carcinoma Cells. Am. J. Chin. Med. 2019, 47, 1149–1170. [Google Scholar] [CrossRef]
- Nazmeen, A.; Maiti, S. Oxidant stress induction and signalling in xenografted (human breast cancer-tissues) plus estradiol treated or N-ethyl-N-nitrosourea treated female rats via altered estrogen sulfotransferase (rSULT1E1) expressions and SOD1/catalase regulations. Mol. Biol. Rep. 2018, 45, 2571–2584. [Google Scholar] [CrossRef]
- Mesbahi, Y.; Zekri, A.; Ghaffari, S.H.; Tabatabaie, P.S.; Ahmadian, S.; Ghavamzadeh, A. Blockade of JAK2/STAT3 intensifies the anti-tumor activity of arsenic trioxide in acute myeloid leukemia cells: Novel synergistic mechanism via the mediation of reactive oxygen species. Eur. J. Pharmacol. 2018, 834, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, J.; Zhao, H.; Wang, Y.; Liu, J.; Shao, Y.; Xue, Y.; Xing, M. Synergistic effect of copper and arsenic upon oxidative stress, inflammation and autophagy alterations in brain tissues of Gallus gallus. J. Inorg. Biochem. 2018, 178, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Pearson, H.B.; Clatworthy, S.A.; Smith, Z.M.; Francis, P.S.; Llanos, R.M.; Volitakis, I.; Phillips, W.A.; Meggyesy, P.M.; Masaldan, S.; et al. Copper as a target for prostate cancer therapeutics: Copper-ionophore pharmacology and altering systemic copper distribution. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Cadavid-Vargas, J.F.; Leon, I.E.; Etcheverry, S.B.; Santi, E.; Torre, M.H.; Di Virgilio, A.L. Copper (II) complexes with saccharinate and glutamine as antitumor agents: Cytoand genotoxicity in human osteosarcoma cells. Anticancer Agents Med. Chem. 2017, 17, 424–433. [Google Scholar] [CrossRef]
- Somwar, R.; Erdjument-Bromage, H.; Larsson, E.; Shum, D.; Lockwood, W.W.; Yang, G.; Sander, C.; Ouerfelli, O.; Tempst, P.J.; Djaballah, H.; et al. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 16375–16380. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mandal, A.K.; Son, Y.-O.; Pratheeshkumar, P.; Wise, J.T.; Wang, L.; Zhang, Z.; Shi, X.; Chen, Z. Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol. Appl. Pharmacol. 2018, 353, 23–30. [Google Scholar] [CrossRef]
- Lee, J.H.; Khor, T.O.; Shu, L.; Su, Z.Y.; Fuentes, F.; Kong, A.-N.T. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacol. Ther. 2013, 137, 153–171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Mohammadi, F.; Soltani, A.; Ghahremanloo, A.; Javid, H.; Hashemy, S.I. The thioredoxin system and cancer therapy: A review. Cancer. Chemother. Pharmacol. 2019, 1–11. [Google Scholar] [CrossRef]
- Benhar, M.; Shytaj, I.L.; Stamler, J.S.; Savarino, A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J. Clin. Invest. 2016, 126, 1630–1639. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.R.; Khuntia, D. Motexafin gadolinium injection for the treatment of brain metastases in patients with non-small cell lung cancer. Int. J. Nanomedicine 2007, 2, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.M. Motexafin gadolinium: A redox-active tumor selective agent for the treatment of cancer. Curr. Opin. Oncol. 2004, 16, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.; Kim, Y.; Lee, H.; Lee, D.S.; Lee, H.S. Interplay Between Mitochondrial Peroxiredoxins and ROS in Cancer Development and Progression. Int. J. Mol. Sci. 2019, 20, 4407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.F.; Liu, H.; Luo, X.J.; Zhao, Z.; Zou, Z.Y.; Li, J.; Lin, X.J.; Liang, Y. The roles of reactive oxygen species (ROS) and autophagy in the survival and death of leukemia cells. Crit. Rev. Oncol. Hematol. 2017, 112, 21–30. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Ramaekers, B.L.; Riemsma, R.; Grimm, S.; Fayter, D.; Deshpande, S.; Armstrong, N.; Witlox, W.; Pouwels, X.; Duffy, S.; Worthy, G.; et al. Arsenic Trioxide for Treating Acute Promyelocytic Leukaemia: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. Pharmacoeconomics 2019, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and mitochondrial ROS in cancer: Novel targets for anticancer therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Yokoyama, C.; Sueyoshi, Y.; Ema, M.; Mori, Y.; Takaishi, K.; Hisatomi, H. Induction of oxidative stress by anticancer drugs in the presence and absence of cells. Oncol. Lett. 2017, 14, 6066–6070. [Google Scholar] [CrossRef] [Green Version]
- Horstman, M.G.; Meadows, G.G.; Yost, G.S. Separate mechanisms for procarbazine spermatotoxicity and anticancer activity. Cancer Res. 1987, 47, 1547–1550. [Google Scholar]
- Skoetz, N.; Will, A.; Monsef, I.; Brillant, C.; Engert, A.; von Tresckow, B. Comparison of first--line chemotherapy including escalated BEACOPP versus chemotherapy including ABVD for people with early unfavourable or advanced stage Hodgkin lymphoma. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef]
- Donovan, L.E.; Lassman, A.B. Chemotherapy Treatment and Trials in Low-Grade Gliomas. Neurosurg. Clin. N. Am. 2019, 30, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Parasramka, S.; Talari, G.; Rosenfeld, M.; Guo, J.; Villano, J.L. Procarbazine, lomustine and vincristine for recurrent high--grade glioma. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef]
- Panchuk, R.R.; Skorokhyd, N.R.; Kozak, Y.S.; Lehka, L.V.; Moiseenok, A.G.; Stoika, R.S. Tissue-protective activity of selenomethionine and D-panthetine in B16 melanoma-bearing mice under doxorubicin treatment is not connected with their ROS scavenging potential. Croat. Med. J. 2017, 58, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Megias-Vericat, J.; Montesinos, P.; Herrero, M.; Moscardo, F.; Boso, V.; Rojas, L.; Martínez-Cuadrón, D.; Rodríguez-Veiga, R.; Sendra, L.; Cervera, J.; et al. Impact of NADPH oxidase functional polymorphisms in acute myeloid leukemia induction chemotherapy. Pharmacogenomics J. 2018, 18. [Google Scholar] [CrossRef]
- Platzbecker, U.; Avvisati, G.; Cicconi, L.; Thiede, C.; Paoloni, F.; Vignetti, M.; Ferrara, F.; Divona, M.; Albano, F.; Efficace, F.; et al. In Improved outcomes with retinoic acid and arsenic trioxide compared with retinoic acid and chemotherapy in non-high-risk acute promyelocytic leukemia: Final results of the randomized Italian-German APL0406 trial. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Graczyk-Jarzynka, A.; Zagozdzon, R.; Muchowicz, A.; Siernicka, M.; Juszczynski, P.; Firczuk, M. New insights into redox homeostasis as a therapeutic target in B-cell malignancies. Curr. Opin. Hematol. 2017, 24. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Mazieres, J.; Barlesi, F.; Filleron, T.; Besse, B.; Monnet, I.; Beau-Faller, M.; Peters, S.; Dansin, E.; Früh, M.; Pless, M.; et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: Results from the European EUHER2 cohort. Ann. Oncol. 2015, 27, 281–286. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Wang, J.; Chen, Y.; Yin, X.; Shi, G.; Li, H.; Hu, Z.; Liang, X. Emodin enhances cisplatin-induced cytotoxicity in human bladder cancer cells through ROS elevation and MRP1 downregulation. BMC Cancer 2016, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, N.; Zhang, H.; Su, S.; Ding, Y.; Yu, X.; Tang, Y.; Wang, Q.; Liu, P. Emodin enhances the chemosensitivity of endometrial cancer by inhibiting ROS-mediated Cisplatin-resistance. Anticancer Agents Med. Chem. 2018, 18, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ishak Gabra, M.B.; Hanse, E.A.; Lowman, X.H.; Tran, T.Q.; Li, H.; Milman, N.; Liu, J.; Reid, M.A.; Locasale, J.W.; et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, M.A.; Wang, W.I.; Rosales, K.R.; Welliver, M.X.; Pan, M.; Kong, M. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol. Cell 2013, 50, 200–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Liu, X.; Yin, Y.; Zheng, J.T.; Jiang, C.F.; Wang, J.; Shen, H.; Li, C.Y.; Wang, M.; Liu, L.Z.; et al. Insulin regulates glucose consumption and lactate production through reactive oxygen species and pyruvate kinase M2. Oxid. Med. Cell Longev. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qiang, W.; Xu, X.; Dong, R.; Karst, A.M.; Liu, Z.; Kong, B.; Drapkin, R.I.; Wei, J.J. Role of miR-182 in response to oxidative stress in the cell fate of human fallopian tube epithelial cells. Oncotarget 2015, 6, 38983–38998. [Google Scholar] [CrossRef]
- He, J.; Xu, Q.; Jing, Y.; Agani, F.; Qian, X.; Carpenter, R.; Li, Q.; Wang, X.R.; Peiper, S.S.; Lu, Z.; et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012, 13, 1116–1122. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.F.; Ou, C.C.; Chien, P.J.; Chang, H.Y.; Ko, J.L.; Wang, B.Y. Chidamide-induced ROS accumulation and miR-129-3p-dependent cell cycle arrest in non-small lung cancer cells. Phytomedicine 2019, 56, 94–102. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, L.; Zhang, P.; Luo, M.; Du, J.; Gao, T.; O’Connell, D.; Wang, G.; Wang, H.; Yang, Y. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol. Carcinog. 2018, 57, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Z.; Hu, Y.Y.; Zhao, J.; Zhao, Y.B.; Sun, J.D.; Yang, Y.F.; Ji, C.C.; Liu, Z.B.; Cao, W.D.; Qu, Y.; et al. MicroRNA-34a induces apoptosis in the human glioma cell line, A172, through enhanced ROS production and NOX2 expression. Biochem. Biophys. Res. Commun. 2014, 444, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Dworakowski, R.; Anilkumar, N.; Zhang, M.; Shah, A.M. Redox signalling involving NADPH oxidase-derived reactive oxygen species. Biochem. Soc. Trans. 2006, 34, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Redon, C.E.; Parekh, P.R.; Dupuy, C.; Bonner, W.M. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anticancer Agents Med. Chem. 2013, 13, 502–514. [Google Scholar] [PubMed]
- Liu, W.; Zabirnyk, O.; Wang, H.; Shiao, Y.H.; Nickerson, M.L.; Khalil, S.; Anderson, L.M.; Perantoni, A.O.; Phang, J.M. miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 2010, 29, 4914–4924. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Borchert, G.L.; Surazynski, A.; Hu, C.A.; Phang, J.M. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: The role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 2006, 25, 5640–5647. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Li, Z.; Zhang, X.; Yin, K.; Wang, W.; Xu, Z.; Li, B.; Zhang, L.; Xu, J.; Sun, G.; et al. MiR-422a regulates cellular metabolism and malignancy by targeting pyruvate dehydrogenase kinase 2 in gastric cancer. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Zhang, L.; Li, L.; Su, Y. miR-148b Functions as a Tumor Suppressor by Targeting Endoplasmic Reticulum Metallo Protease 1 in Human Endometrial Cancer Cells. Oncol. Res. 2018, 27, 81–88. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef]
- Assi, M. The differential role of reactive oxygen species in early and late stages of cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R646–R653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramani, T.; Yeap, S.K.; Ho, W.Y.; Ho, C.L.; Omar, A.R.; Aziz, S.A.; Rahman, N.M.A.N.A.; Alitheen, N.B. Vitamin C suppresses cell death in MCF--7 human breast cancer cells induced by tamoxifen. J. Cell. Mol. Med. 2014, 18, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Batteux, F.; Nicco, C.; Chéreau, C.; Laurent, A.; Guillevin, L.; Weill, B.; Goldwasser, F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel--induced cancer cell death both in vitro and in vivo. Int. J. Cancer 2006, 119, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Yamabe, N.; Zhu, B.T. Resveratrol attenuates the anticancer efficacy of paclitaxel in human breast cancer cells invitro and in vivo. Eur. J. Cancer 2010, 46, 1882–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, Q.; Zhang, J.; Zhao, T.; Xue, F.; Gao, F.; Ma, S.; Wang, Y. Vitamin E promotes breast cancer cell proliferation by reducing ROS production and p53 expression. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2710–2717. [Google Scholar] [PubMed]
- Podmore, I.D.; Griffiths, H.R.; Herbert, K.E.; Mistry, N.; Mistry, P.; Lunec, J. Vitamin C exhibits pro-oxidant properties. Nature 1998, 392, 559. [Google Scholar] [CrossRef]
- Uetaki, M.; Tabata, S.; Nakasuka, F.; Soga, T.; Tomita, M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Syed, F.A.; Singh, S.; Hadi, S. Prooxidant activity of resveratrol in the presence of copper ions: Mutagenicity in plasmid DNA. Toxicol. lett. 2005, 159, 1–12. [Google Scholar] [CrossRef]
- Khan, H.Y.; Zubair, H.; Faisal, M.; Ullah, M.F.; Farhan, M.; Sarkar, F.H.; Ahmad, A.; Hadi, S.M. Plant polyphenol induced cell death in human cancer cells involves mobilization of intracellular copper ions and reactive oxygen species generation: A mechanism for cancer chemopreventive action. Mol. Nutr. Food Res. 2014, 58, 437–446. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect | Mechanism | Cell Line | References |
|---|---|---|---|
| Oxidative Stress | Aquaporin AQP5-mediated H2O2 influx rate indicates the presence of a highly efficient peroxiporin activity and consequently activates signaling networks related to cell survival and cancer progression | Pancreatic Carcinoma line-3 (BxPC3) | [7] |
| PCB118 promotes hepatocellular carcinoma cell (HCC) proliferation via Pyruvate kinase M2 (PKM2)-dependent up-regulation of glycolysis, which is mediated by Aryl hydrocarbon receptor/Nicotinamide adenine dinucleotide phosphate oxidase (AhR/NADPH oxidase)-induced ROS prouction | SMMC-7721 | [8] | |
| Enhanced ROS of exposed cells alters the mitochondrial metabolic activities in terms of increased mitochondrial mass and DNA content and initiates cancer progression through modifying cellar biomarkers | MOE1A | [9] | |
| Inflammatory markers | Serum ROS and damaged mtDNA may be markers of mitochondrial metabolism through oxygenation of the primary tumor and results in systemic inflammation and adverse outcomes of locally advanced rectal cancer (LARC) | HCT-116, HT-29, and LoVo | [10] |
| Inflammation in the stroma induces TNF-α signaling and the NOX1/ROS signaling pathway is activated downstream with expression of TLR2 which is an important tumor-promoting mechanism stimulated by inflammation | Mouse Model | [11] | |
| Alkylating agents may evoke inflammatory responses that seem to contribute to malignant progression in specific breast cancer cells | MDA-MB231, Hs578T, SKBR3 and MCF7 | [12] | |
| Metastasis | ROS induce epithelial-mesenchymal transition (EMT), the glycolytic switch, and mitochondrial repression by activating the Distal-less homeobox-2 (Dlx-2)/Snail axis, thereby playing crucial roles in metastasis | MCF-7 | [13] |
| Elevated mitochondrial ROS via fatty acid β-oxidation, activates the MAPK cascades, results in EMT process of ROS high tumor spheres (RH-TS) cells, and enhances metastasis | 4 T1, SW480, HCT116 and HT29 | [14] | |
| Loss of TMEM126A induces ROS production with mitochondrial dysfunction and subsequently metastasis by activating extracellular matrix (ECM) remodeling and EMT | MDA-MB-231HM | [15] | |
| PM2.5 exposure induces ROS, which activates loc146880 expression and promotes the malignant behavior | A549 | [16] | |
| Angiogenesis | ROS-ERK1/2-HIF-1α-VEGF-induces angiogenesis by increased level of RRM2 | C33A and MCF-7 | [17] |
| High glucose increases angiogenesis and decreases apoptosis due to activation of the NF-κB pathway by increasing ROS | MCF-7 | [18] | |
| 27-Hydroxycholesterol (27HC) enhanced the generation of ROS and activates the STAT-3/VEGF signaling in an ER independent manner which results in induced angiogenesis | Breast Cancer Cells | [19] |
| Effect | Mechanism | Cell Line | References |
|---|---|---|---|
| Apoptosis | Increase in cell oxidation by c-Met-Nrf2-HO-1 pathway and promotes apoptotic cell death | 786-O and ACHN | [116] |
| Apoptosis enhanced by ROS by affecting MAPK & AKT signaling and DNA damage mediated p53 phosphorylation | HePG-2 Cells | [117] | |
| ↓ ROS by expression of GPx3 and leads to G2/M arrest | H157, H460, A549, H1299, H1650, and H1975 lung cancer cells | [118] | |
| ↑ ROS by knockdown of nicotinamide nucleotide transhydrogenase and significant cell apoptosis under oxidative Stress | GES-1, SGC7901, SNU216, MKN45, MKN74, BGC823, HGC27 and MGC803 | [119] | |
| Short mRNA | Salviamiltiorrhiza treatment induces apoptosis through regulation of miR-216b and ROS/ER stress pathways | U266 and U937 Cells | [120] |
| miR-21 silencing effect the ROS-induced activation, invasion, migration, and glycolysis of Pancreatic stellate cells (PSCs) | Human PSCs, Panc-1 | [121] | |
| Down-regulation of NOX2 using siRNA technology in decreased cell viability and ROS content | SNU719 cells | [122] | |
| Melanoma differentiation-associated gene-7/interleukin-24 (mda-7/IL-24) regulates miRNA biogenesis through alteration of ROS-dependent MITF-DICER pathway | Animal cancer model | [123] | |
| Autophagy | Silencing of YAP enhanced autophagic flux by increasing RAC1-driven ROS, through inactivation of mTOR | BEL/FU, BEL-7402 | [124] |
| Zinc Oxide Nanoparticle (ZON) evoked autophagy by accelerating the intracellular dissolution of ZONs and ROS generation. | MCF-7/ADR | [125] | |
| Cell killing was due to the summative effect of caspase-dependent intrinsic apoptosis and caspase-independent autophagy by activation of MAPK family members (ERK1/2 and JNK) with generation of ROS | SNU-719 | [126] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. https://doi.org/10.3390/biom9110735
Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, Varol M, Jain A, Khan MA, Sethi G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules. 2019; 9(11):735. https://doi.org/10.3390/biom9110735
Chicago/Turabian StyleAggarwal, Vaishali, Hardeep Singh Tuli, Ayşegül Varol, Falak Thakral, Mukerrem Betul Yerer, Katrin Sak, Mehmet Varol, Aklank Jain, Md. Asaduzzaman Khan, and Gautam Sethi. 2019. "Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements" Biomolecules 9, no. 11: 735. https://doi.org/10.3390/biom9110735
APA StyleAggarwal, V., Tuli, H. S., Varol, A., Thakral, F., Yerer, M. B., Sak, K., Varol, M., Jain, A., Khan, M. A., & Sethi, G. (2019). Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules, 9(11), 735. https://doi.org/10.3390/biom9110735