Benzyl Isothiocyanate Induces Apoptosis via Reactive Oxygen Species-Initiated Mitochondrial Dysfunction and DR4 and DR5 Death Receptor Activation in Gastric Adenocarcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Cell Culture and Morphological Observation
2.3. Cell Viability Measurement (MTT Assay)
2.4. Measurement of Intracellular ROS
2.5. Cell Viability Inhibition Assay
2.6. Preparation of Cellular Extracts and Western Blot Analysis
2.7. Statistical Analysis
3. Results
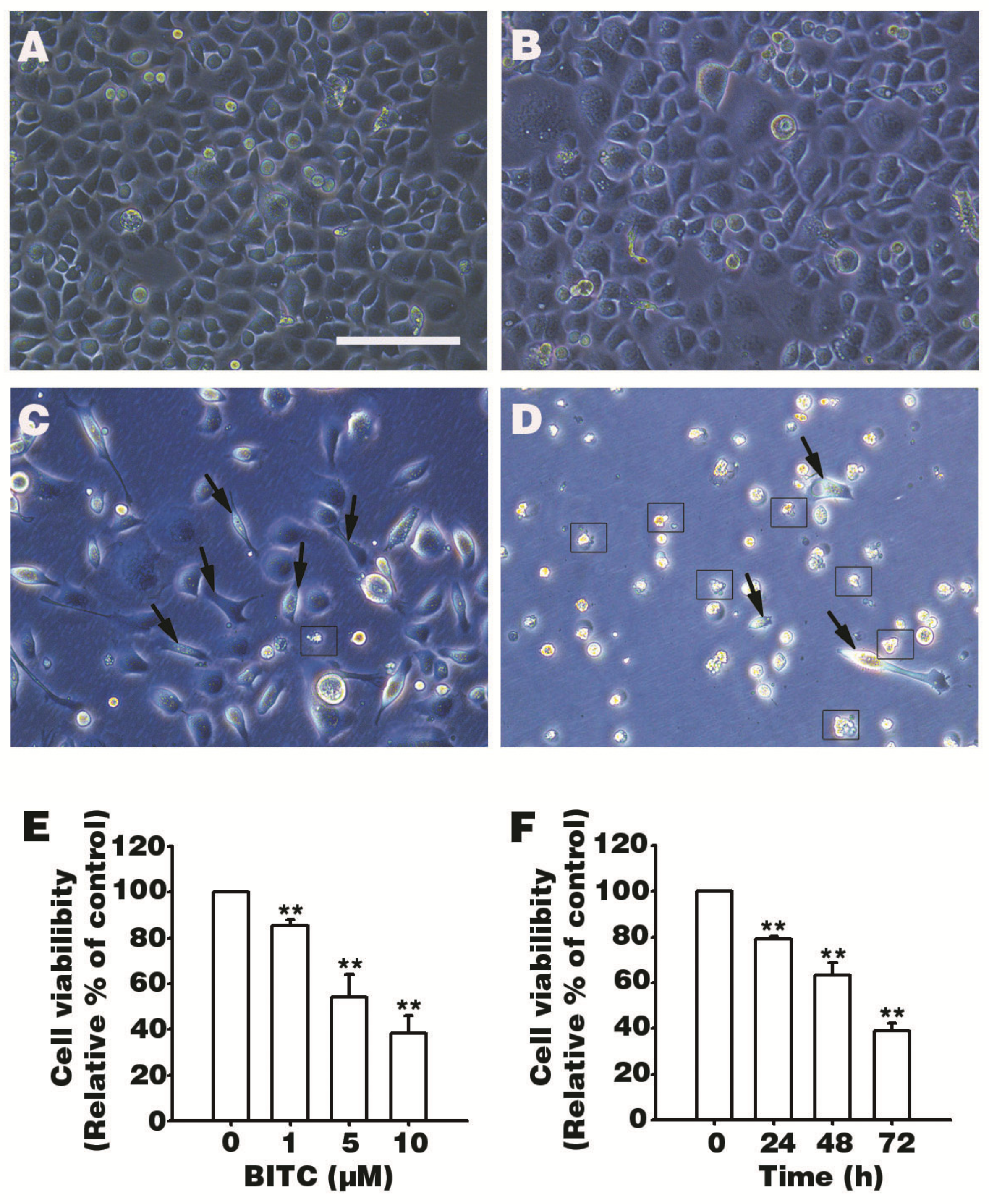
3.1. BITC Inhibits AGS Cells Survival
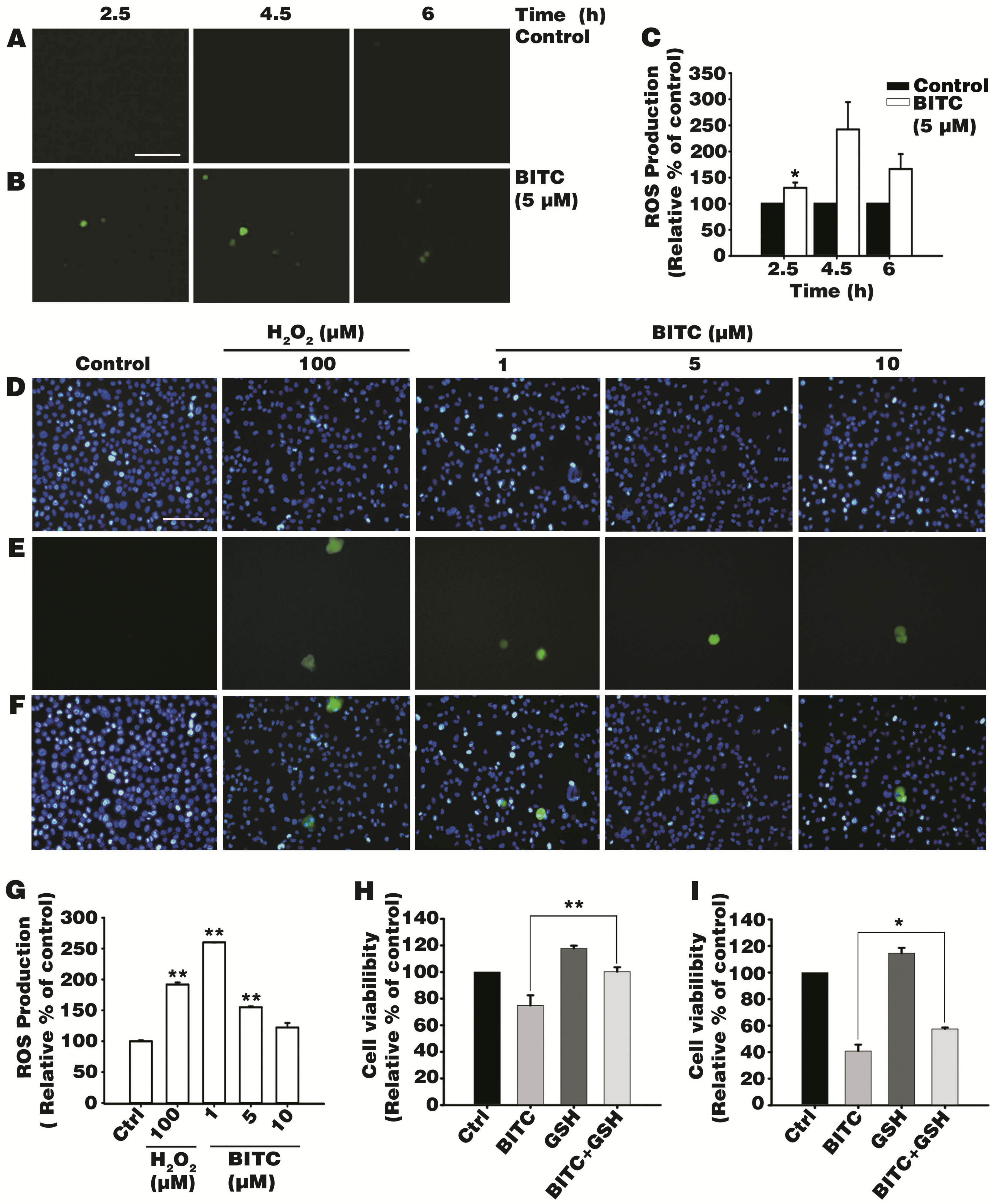
3.2. BITC Induces Intracellular ROS Production
3.3. Antioxidant Glutathione Ameliorated BITC-Induced AGS Cell Death
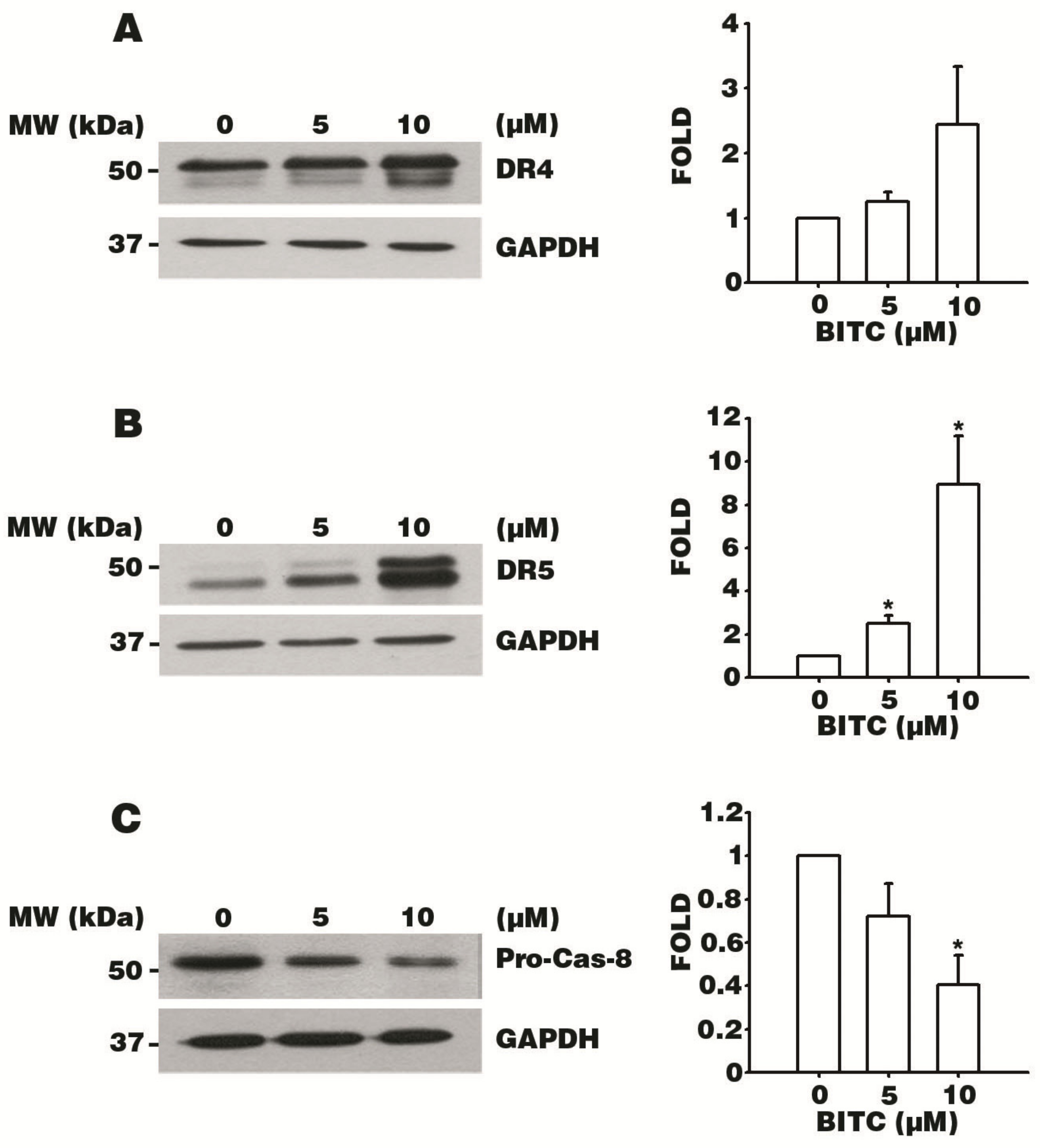
3.4. BITC Increases the Expression of DR4 and DR5 TRAIL Death Receptors
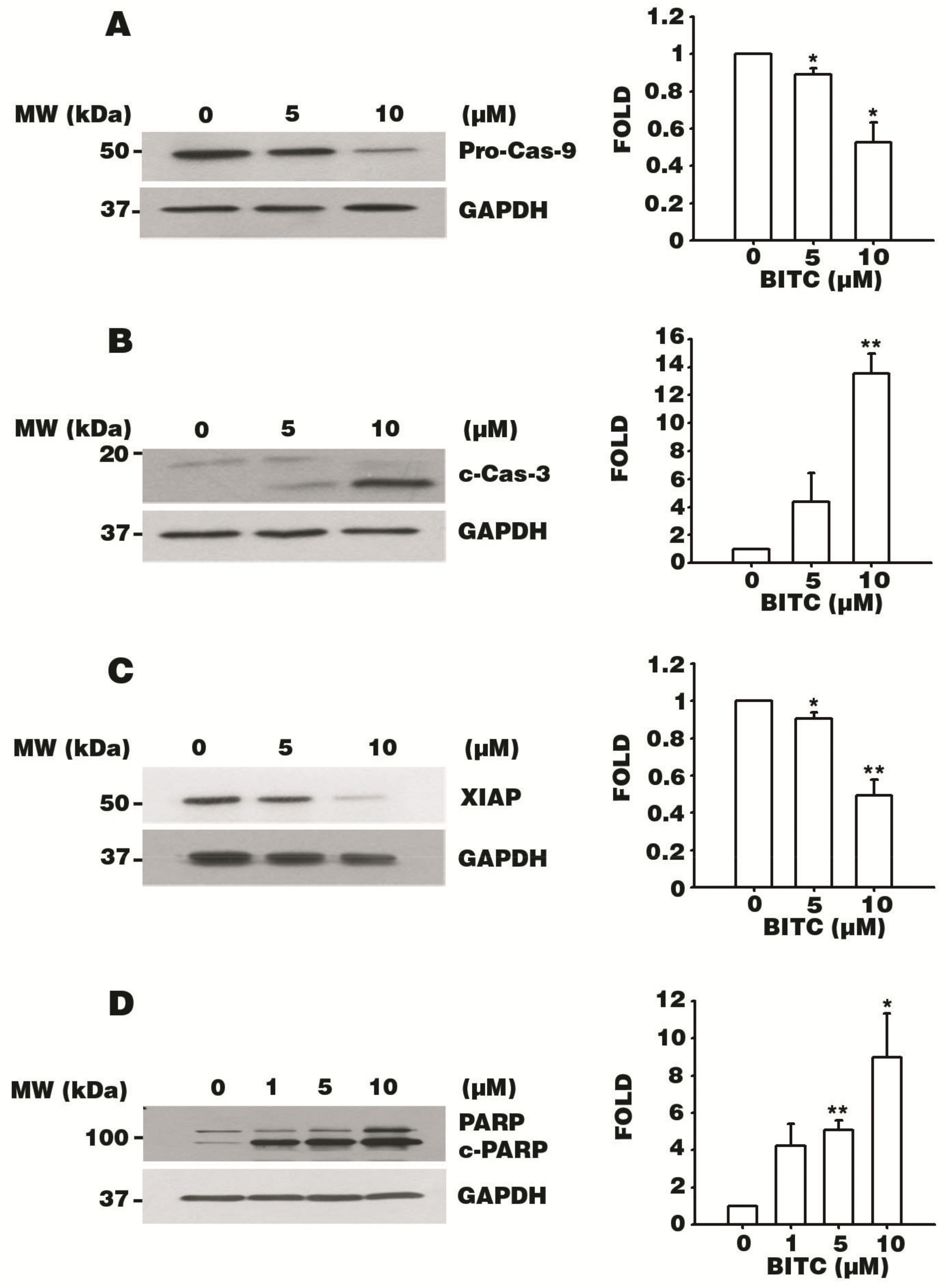
3.5. BITC Activates the Caspase Signaling Cascade
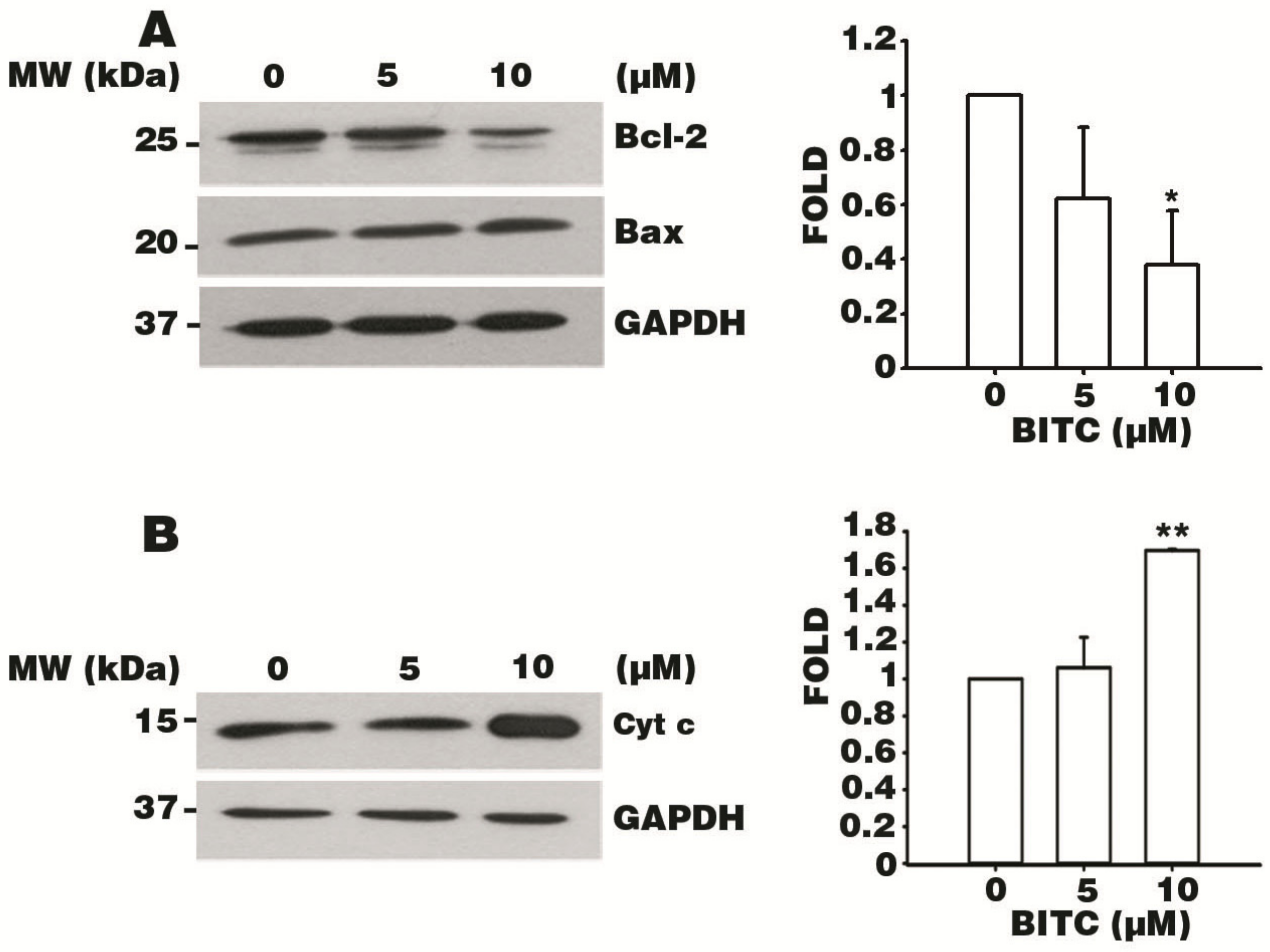
3.6. BITC Induces the Bcl-2-Regulated Apoptotic Pathway
4. Discussion
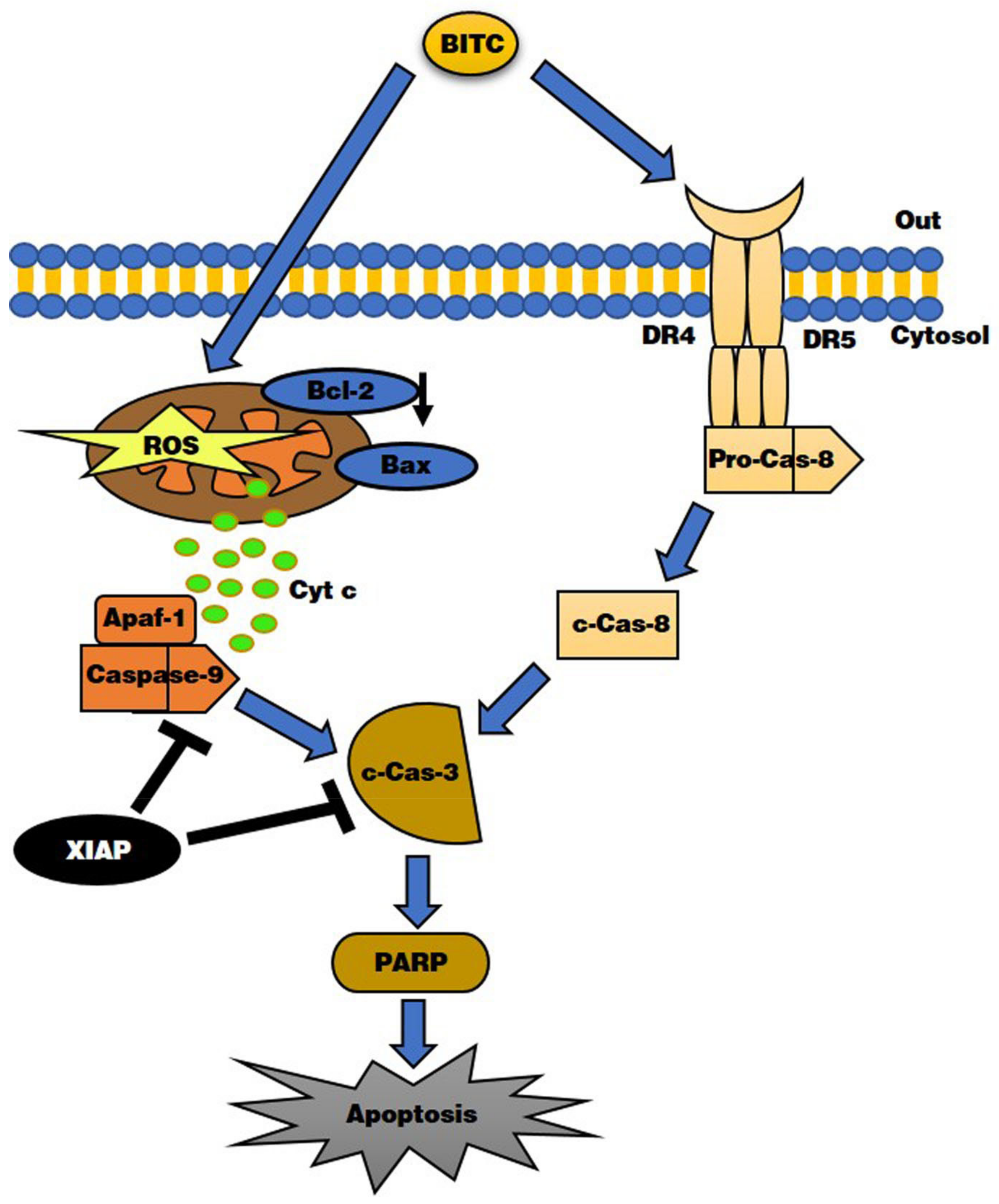
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Heron, M.; Anderson, R.N. Changes in the Leading Cause of Death: Recent Patterns in Heart Disease and Cancer Mortality. NCHS Data Brief 2016, 254, 1–8. [Google Scholar]
- Ma, X.; Yu, H. Global burden of cancer. Yale J. Biol. Med. 2006, 79, 85–94. [Google Scholar] [PubMed]
- Donaldson, M.S. Nutrition and cancer: A review of the evidence for an anti-cancer diet. Nutr. J. 2004, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stan, S.D.; Kar, S.; Stoner, G.D.; Singh, S.V. Bioactive food components and cancer risk reduction. J. Cell. Biochem. 2008, 104, 339–356. [Google Scholar] [CrossRef]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef]
- Orditura, M.; Galizia, G.; Sforza, V.; Gambardella, V.; Fabozzi, A.; Laterza, M.M.; Andreozzi, F.; Ventriglia, J.; Savastano, B.; Mabilia, A.; et al. Treatment of gastric cancer. World J. Gastroenterol. 2014, 20, 1635–1649. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Z.; Lu, N. Molecular targeted therapy for the treatment of gastric cancer. J. Exp. Clin. Cancer Res. 2016, 35, 1. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.V.; Singh, K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis 2012, 33, 1833–1842. [Google Scholar] [CrossRef] [Green Version]
- Moy, K.A.; Yuan, J.M.; Chung, F.L.; Wang, X.L.; Van Den Berg, D.; Wang, R.; Gao, Y.T.; Yu, M.C. Isothiocyanates, glutathione S-transferase M1 and T1 polymorphisms and gastric cancer risk: A prospective study of men in Shanghai, China. Int. J. Cancer 2009, 125, 2652–2659. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.F.; Chiang, N.N.; Lu, Y.H.; Huang, Y.S.; Yang, J.S.; Tsai, S.C.; Lu, C.C.; Chen, F.A. Benzyl isothiocyanate (BITC) triggers mitochondria-mediated apoptotic machinery in human cisplatin-resistant oral cancer CAR cells. Biomedicine (Taipei) 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Kasiappan, R.; Jutooru, I.; Karki, K.; Hedrick, E.; Safe, S. Benzyl Isothiocyanate (BITC) Induces Reactive Oxygen Species-dependent Repression of STAT3 Protein by Down-regulation of Specificity Proteins in Pancreatic Cancer. J. Biol. Chem. 2016, 291, 27122–27133. [Google Scholar] [CrossRef] [Green Version]
- Shang, H.S.; Shih, Y.L.; Lu, T.J.; Lee, C.H.; Hsueh, S.C.; Chou, Y.C.; Lu, H.F.; Liao, N.C.; Chung, J.G. Benzyl isothiocyanate (BITC) induces apoptosis of GBM 8401 human brain glioblastoma multiforms cells via activation of caspase-8/Bid and the reactive oxygen species-dependent mitochondrial pathway. Environ. Toxicol. 2016, 31, 1751–1760. [Google Scholar] [CrossRef]
- Huang, S.H.; Wu, L.W.; Huang, A.C.; Yu, C.C.; Lien, J.C.; Huang, Y.P.; Yang, J.S.; Yang, J.H.; Hsiao, Y.P.; Wood, W.G.; et al. Benzyl isothiocyanate (BITC) induces G2/M phase arrest and apoptosis in human melanoma A375.S2 cells through reactive oxygen species (ROS) and both mitochondria-dependent and death receptor-mediated multiple signaling pathways. J. Agric. Food Chem. 2012, 60, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.Y.; Hsiao, Y.P.; Hsu, S.C.; Hsueh, S.C.; Chang, C.H.; Ji, B.C.; Yang, J.S.; Lu, H.F.; Chung, J.G. Oral administration of benzyl-isothiocyanate inhibits in vivo growth of subcutaneous xenograft tumors of human malignant melanoma A375.S2 cells. In Vivo 2013, 27, 623–626. [Google Scholar] [PubMed]
- Ho, C.C.; Lai, K.C.; Hsu, S.C.; Kuo, C.L.; Ma, C.Y.; Lin, M.L.; Yang, J.S.; Chung, J.G. Benzyl isothiocyanate (BITC) inhibits migration and invasion of human gastric cancer AGS cells via suppressing ERK signal pathways. Hum. Exp. Toxicol. 2011, 30, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Kermanshai, R.; McCarry, B.E.; Rosenfeld, J.; Summers, P.S.; Weretilnyk, E.A.; Sorger, G.J. Benzyl isothiocyanate is the chief or sole anthelmintic in papaya seed extracts. Phytochemistry 2001, 57, 427–435. [Google Scholar] [CrossRef]
- Lee, Y.M.; Seon, M.R.; Cho, H.J.; Kim, J.S.; Park, J.H. Benzyl isothiocyanate exhibits anti-inflammatory effects in murine macrophages and in mouse skin. J. Mol. Med. (Berl) 2009, 87, 1251–1261. [Google Scholar] [CrossRef]
- Chuang, W.T.; Liu, Y.T.; Huang, C.S.; Lo, C.W.; Yao, H.T.; Chen, H.W.; Lii, C.K. Benzyl Isothiocyanate and Phenethyl Isothiocyanate Inhibit Adipogenesis and Hepatosteatosis in Mice with Obesity Induced by a High-Fat Diet. J. Agric. Food Chem. 2019, 67, 7136–7146. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther. 2005, 4, 139–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.Y.; Yang, X. Proteases for cell suicide: Functions and regulation of caspases. Microbiol. Mol. Biol. Rev. 2000, 64, 821–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; Stuart, K.; Gilley, R.; Sale, M.J. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017, 284, 4177–4195. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.F.; Tsai, T.F.; Yang, S.C.; Lin, Y.C.; Chen, H.E.; Chou, K.Y.; Hwang, T.I.S. Benzyl isothiocyanate induces reactive oxygen species-initiated autophagy and apoptosis in human prostate cancer cells. Oncotarget 2017, 8, 20220–20234. [Google Scholar] [CrossRef] [Green Version]
- Nechushtan, A.; Smith, C.L.; Lamensdorf, I.; Yoon, S.H.; Youle, R.J. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 2001, 153, 1265–1276. [Google Scholar] [CrossRef]
- Hundie, G.B.; Woldemeskel, D.; Gessesse, A. Evaluation of Direct Colorimetric MTT Assay for Rapid Detection of Rifampicin and Isoniazid Resistance in Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0169188. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: Forty years of application and controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef]
- Lee, H.R.; Lee, J.; Jajoo, R.; Kong, S.Y.; Shin, J.Y.; Kim, J.O.; Lee, J.; Lee, J.; Kim, H.J. Discovery of a Small Molecule that Enhances Astrocytogenesis by Activation of STAT3, SMAD1/5/8, and ERK1/2 via Induction of Cytokines in Neural Stem Cells. ACS Chem. Neurosci. 2016, 7, 90–99. [Google Scholar] [CrossRef]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Lavrentiadou, S.N.; Chan, C.; Kawcak, T.; Ravid, T.; Tsaba, A.; van der Vliet, A.; Rasooly, R.; Goldkorn, T. Ceramide-mediated apoptosis in lung epithelial cells is regulated by glutathione. Am. J. Respir. Cell Mol. Biol. 2001, 25, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Russi, S.; Verma, H.K.; Laurino, S.; Mazzone, P.; Storto, G.; Nardelli, A.; Zoppoli, P.; Calice, G.; La Rocca, F.; Sgambato, A.; et al. Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.J.; Gao, J.B. Molecular mechanisms of chemoresistance in gastric cancer. World J. Gastrointest. Oncol. 2016, 8, 673–681. [Google Scholar] [CrossRef]
- Bui, H.T.T.; Le, N.H.; Le, Q.A.; Kim, S.E.; Lee, S.; Kang, D. Synergistic apoptosis of human gastric cancer cells by bortezomib and TRAIL. Int. J. Med. Sci. 2019, 16, 1412–1423. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.Y.; Jin, C.Y.; Han, M.H.; Kim, G.Y.; Kim, N.D.; Lee, W.H.; Kim, S.K.; Choi, Y.H. Sanguinarine sensitizes human gastric adenocarcinoma AGS cells to TRAIL-mediated apoptosis via down-regulation of AKT and activation of caspase-3. Anticancer Res. 2009, 29, 4457–4465. [Google Scholar]
- Hylander, B.L.; Sen, A.; Beachy, S.H.; Pitoniak, R.; Ullas, S.; Gibbs, J.F.; Qiu, J.; Prey, J.D.; Fetterly, G.J.; Repasky, E.A. Tumor priming by Apo2L/TRAIL reduces interstitial fluid pressure and enhances efficacy of liposomal gemcitabine in a patient derived xenograft tumor model. J. Control. Release 2015, 217, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Kim, B.; Kim, S.H.; Srivastava, S.K. Molecular targets of isothiocyanates in cancer: Recent advances. Mol. Nutr. Food Res. 2014, 58, 1685–1707. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Kawakami, M.; Yoshihiro, A.; Miyoshi, N.; Ohigashi, H.; Kawai, K.; Osawa, T.; Uchida, K. Involvement of the mitochondrial death pathway in chemopreventive benzyl isothiocyanate-induced apoptosis. J. Biol. Chem. 2002, 277, 8492–8499. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; Calissano, P.; Bobba, A.; Azzariti, A.; Marra, E.; Passarella, S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J. Biol. Chem. 2000, 275, 37159–37166. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.H. ROS-mediated activation of AMPK plays a critical role in sulforaphane-induced apoptosis and mitotic arrest in AGS human gastric cancer cells. Gen. Physiol. Biophys. 2018, 37, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, C.; He, J.; Hu, Z.; Guan, W.C.; Liu, S.H. Protective effect of reduced glutathione C60 derivative against hydrogen peroxide-induced apoptosis in HEK 293T cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2016, 36, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Zunino, F.; Pratesi, G.; Micheloni, A.; Cavalletti, E.; Sala, F.; Tofanetti, O. Protective effect of reduced glutathione against cisplatin-induced renal and systemic toxicity and its influence on the therapeutic activity of the antitumor drug. Chem. Biol. Interact. 1989, 70, 89–101. [Google Scholar] [CrossRef]
- Lee, C.S.; Kim, Y.J.; Jang, E.R.; Kim, W.; Myung, S.C. Fluoxetine induces apoptosis in ovarian carcinoma cell line OVCAR-3 through reactive oxygen species-dependent activation of nuclear factor-kappaB. Basic Clin. Pharmacol. Toxicol. 2010, 106, 446–453. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y. Mitochondria are the primary target in isothiocyanate-induced apoptosis in human bladder cancer cells. Mol. Cancer Ther. 2005, 4, 1250–1259. [Google Scholar] [CrossRef] [Green Version]
- Checinska, A.; Hoogeland, B.S.; Rodriguez, J.A.; Giaccone, G.; Kruyt, F.A. Role of XIAP in inhibiting cisplatin-induced caspase activation in non-small cell lung cancer cells: A small molecule Smac mimic sensitizes for chemotherapy-induced apoptosis by enhancing caspase-3 activation. Exp. Cell Res. 2007, 313, 1215–1224. [Google Scholar] [CrossRef]
- Saraei, R.; Soleimani, M.; Movassaghpour Akbari, A.A.; Farshdousti Hagh, M.; Hassanzadeh, A.; Solali, S. The role of XIAP in resistance to TNF-related apoptosis-inducing ligand (TRAIL) in Leukemia. Biomed. Pharm. 2018, 107, 1010–1019. [Google Scholar] [CrossRef]
- Schulze-Osthoff, K.; Ferrari, D.; Los, M.; Wesselborg, S.; Peter, M.E. Apoptosis signaling by death receptors. Eur. J. Biochem. 1998, 254, 439–459. [Google Scholar] [CrossRef]
- Golstein, P. Cell death: TRAIL and its receptors. Curr. Biol. 1997, 7, R750–R753. [Google Scholar] [CrossRef]
- Okinaga, T.; Kasai, H.; Tsujisawa, T.; Nishihara, T. Role of caspases in cleavage of lamin A/C and PARP during apoptosis in macrophages infected with a periodontopathic bacterium. J. Med. Microbiol. 2007, 56, 1399–1404. [Google Scholar] [CrossRef]
- Saralamma, V.V.; Nagappan, A.; Hong, G.E.; Lee, H.J.; Yumnam, S.; Raha, S.; Heo, J.D.; Lee, S.J.; Lee, W.S.; Kim, E.H.; et al. Poncirin Induces Apoptosis in AGS Human Gastric Cancer Cells through Extrinsic Apoptotic Pathway by up-Regulation of Fas Ligand. Int. J. Mol. Sci. 2015, 16, 22676–22691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Cao, A.; Shi, J.; Yin, P.; Wang, L.; Ji, G.; Xie, J.; Wu, D. Tangeretin, a citrus polymethoxyflavonoid, induces apoptosis of human gastric cancer AGS cells through extrinsic and intrinsic signaling pathways. Oncol. Rep. 2014, 31, 1788–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.S.; Kim, E.J.; Kim, S.H. Chestnut extract induces apoptosis in AGS human gastric cancer cells. Nutr. Res. Pract. 2011, 5, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.C.; Jeon, H.J.; Kee, K.H.; Lee, M.J.; Hong, R.; Han, S.I. Andrographolide induces apoptotic and non-apoptotic death and enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in gastric cancer cells. Oncol. Lett. 2017, 13, 3837–3844. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Kim, D.W.; Lee, H.C.; Lee, J.H.; Lee, T.H. Phenethyl isothiocyanate sensitizes glioma cells to TRAIL-induced apoptosis. Biochem. Biophys. Res. Commun. 2014, 446, 815–821. [Google Scholar] [CrossRef]
- Wang, D.; Upadhyaya, B.; Liu, Y.; Knudsen, D.; Dey, M. Phenethyl isothiocyanate upregulates death receptors 4 and 5 and inhibits proliferation in human cancer stem-like cells. BMC Cancer 2014, 14, 591. [Google Scholar] [CrossRef] [Green Version]
- Akazawa, Y.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Kahraman, A.; Guicciardi, M.E.; Meng, X.W.; Kohno, S.; Shah, V.H.; Kaufmann, S.H.; et al. Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology 2009, 136, 2365–2376.e7. [Google Scholar] [CrossRef] [Green Version]
- Langsenlehner, T.; Langsenlehner, U.; Renner, W.; Kapp, K.S.; Krippl, P.; Hofmann, G.; Clar, H.; Pummer, K.; Mayer, R. The Glu228Ala polymorphism in the ligand binding domain of death receptor 4 is associated with increased risk for prostate cancer metastases. Prostate 2008, 68, 264–268. [Google Scholar] [CrossRef]
- Fimognari, C.; Turrini, E.; Ferruzzi, L.; Lenzi, M.; Hrelia, P. Natural isothiocyanates: Genotoxic potential versus chemoprevention. Mutat. Res. 2012, 750, 107–131. [Google Scholar] [CrossRef]
- Yeh, Y.T.; Hsu, Y.N.; Huang, S.Y.; Lin, J.S.; Chen, Z.F.; Chow, N.H.; Su, S.H.; Shyu, H.W.; Lin, C.C.; Huang, W.T.; et al. Benzyl isothiocyanate promotes apoptosis of oral cancer cells via an acute redox stress-mediated DNA damage response. Food Chem. Toxicol. 2016, 97, 336–345. [Google Scholar] [CrossRef]
- Sehrawat, A.; Croix, C.S.; Baty, C.J.; Watkins, S.; Tailor, D.; Singh, R.P.; Singh, S.V. Inhibition of mitochondrial fusion is an early and critical event in breast cancer cell apoptosis by dietary chemopreventative benzyl isothiocyanate. Mitochondrion 2016, 30, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, D.; Vogel, V.; Singh, S.V. Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol. Cancer Ther. 2006, 5, 2931–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, S.; Sahu, R.P.; Kandala, P.K.; Srivastava, S.K. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Mol. Cancer Ther. 2010, 9, 1596–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, K.W.W.; Po, W.W.; Sohn, U.D.; Kim, H.-J. Benzyl Isothiocyanate Induces Apoptosis via Reactive Oxygen Species-Initiated Mitochondrial Dysfunction and DR4 and DR5 Death Receptor Activation in Gastric Adenocarcinoma Cells. Biomolecules 2019, 9, 839. https://doi.org/10.3390/biom9120839
Han KWW, Po WW, Sohn UD, Kim H-J. Benzyl Isothiocyanate Induces Apoptosis via Reactive Oxygen Species-Initiated Mitochondrial Dysfunction and DR4 and DR5 Death Receptor Activation in Gastric Adenocarcinoma Cells. Biomolecules. 2019; 9(12):839. https://doi.org/10.3390/biom9120839
Chicago/Turabian StyleHan, Khin Wah Wah, Wah Wah Po, Uy Dong Sohn, and Hyun-Jung Kim. 2019. "Benzyl Isothiocyanate Induces Apoptosis via Reactive Oxygen Species-Initiated Mitochondrial Dysfunction and DR4 and DR5 Death Receptor Activation in Gastric Adenocarcinoma Cells" Biomolecules 9, no. 12: 839. https://doi.org/10.3390/biom9120839
APA StyleHan, K. W. W., Po, W. W., Sohn, U. D., & Kim, H. -J. (2019). Benzyl Isothiocyanate Induces Apoptosis via Reactive Oxygen Species-Initiated Mitochondrial Dysfunction and DR4 and DR5 Death Receptor Activation in Gastric Adenocarcinoma Cells. Biomolecules, 9(12), 839. https://doi.org/10.3390/biom9120839