New Insights into the Control of Cell Fate Choices and Differentiation by Retinoic Acid in Cranial, Axial and Caudal Structures

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
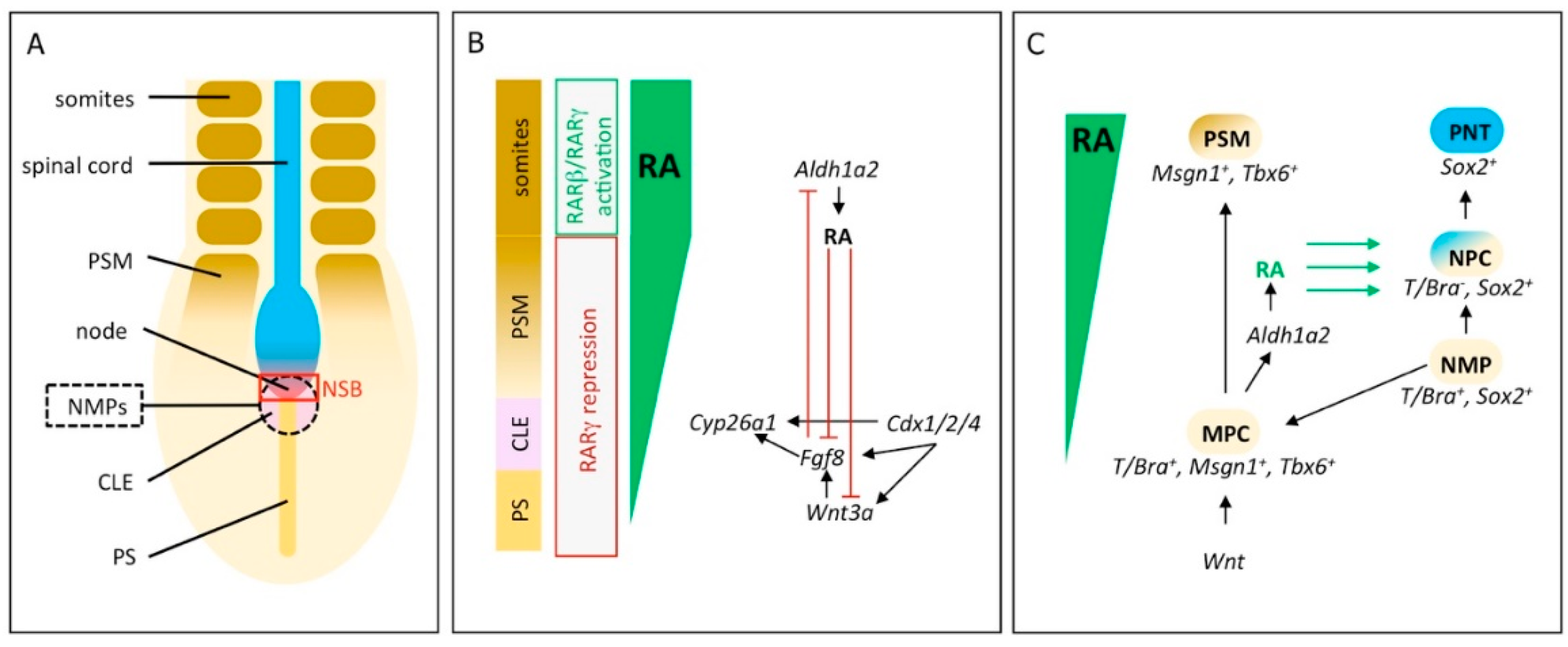
2. RA Signaling Controls Induction and Differentiation of Neuromesodermal Progenitors
3. Initiation of Vertebrae Formation in Zebrafish Relies on Precisely Regulated RA-Signaling
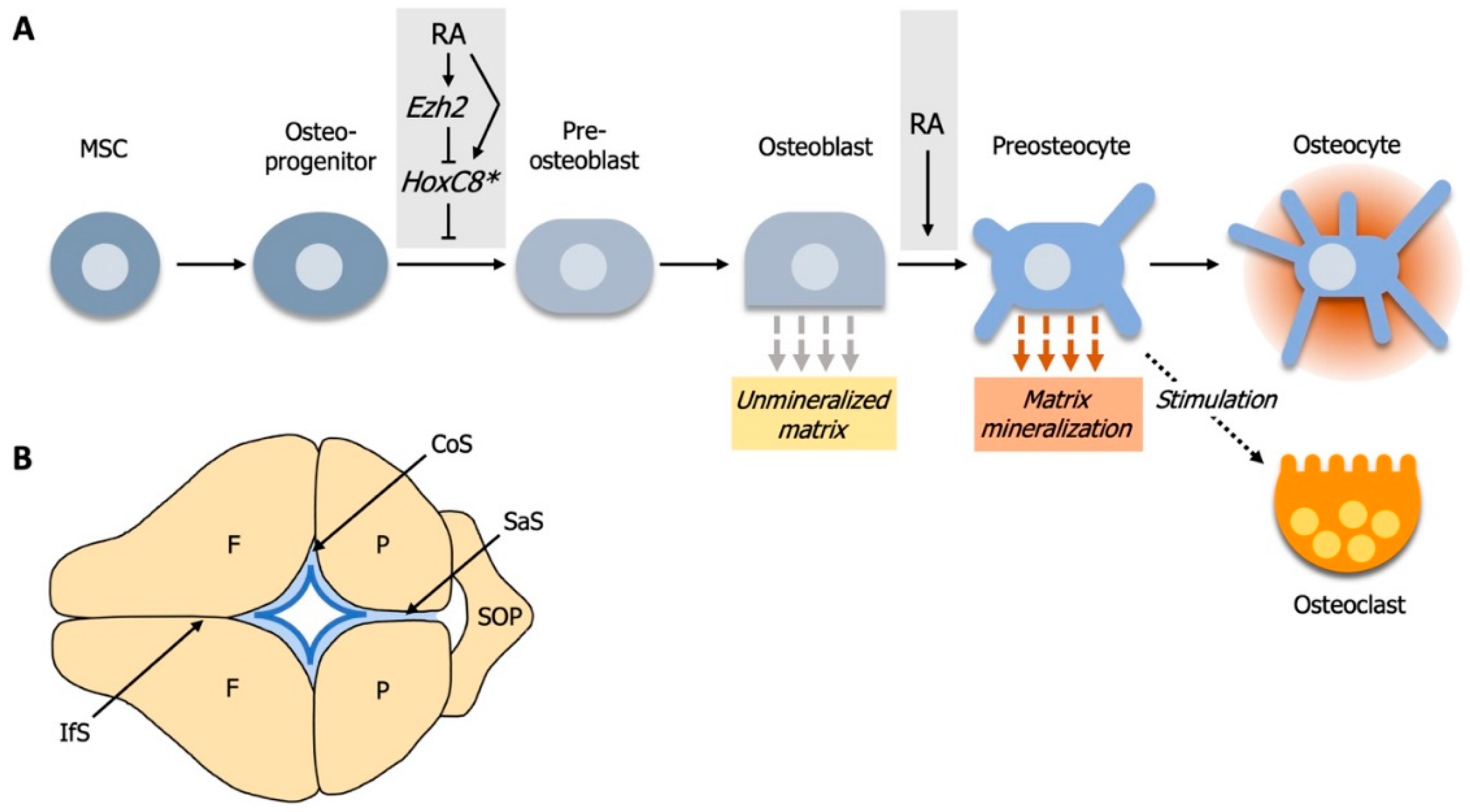
4. RA Controls Cell Fate Determination during Calvarial Bone Development
4.1. Elevated RA-Signaling Leads to Premature Osteoblast to Preosteocyte Transition
4.2. RA-Signaling and Ezh2 Act in Opposition for Calvarial Bone Lineage Commitment
5. RA Controls the Development and Number of Pharyngeal Teeth in Zebrafish
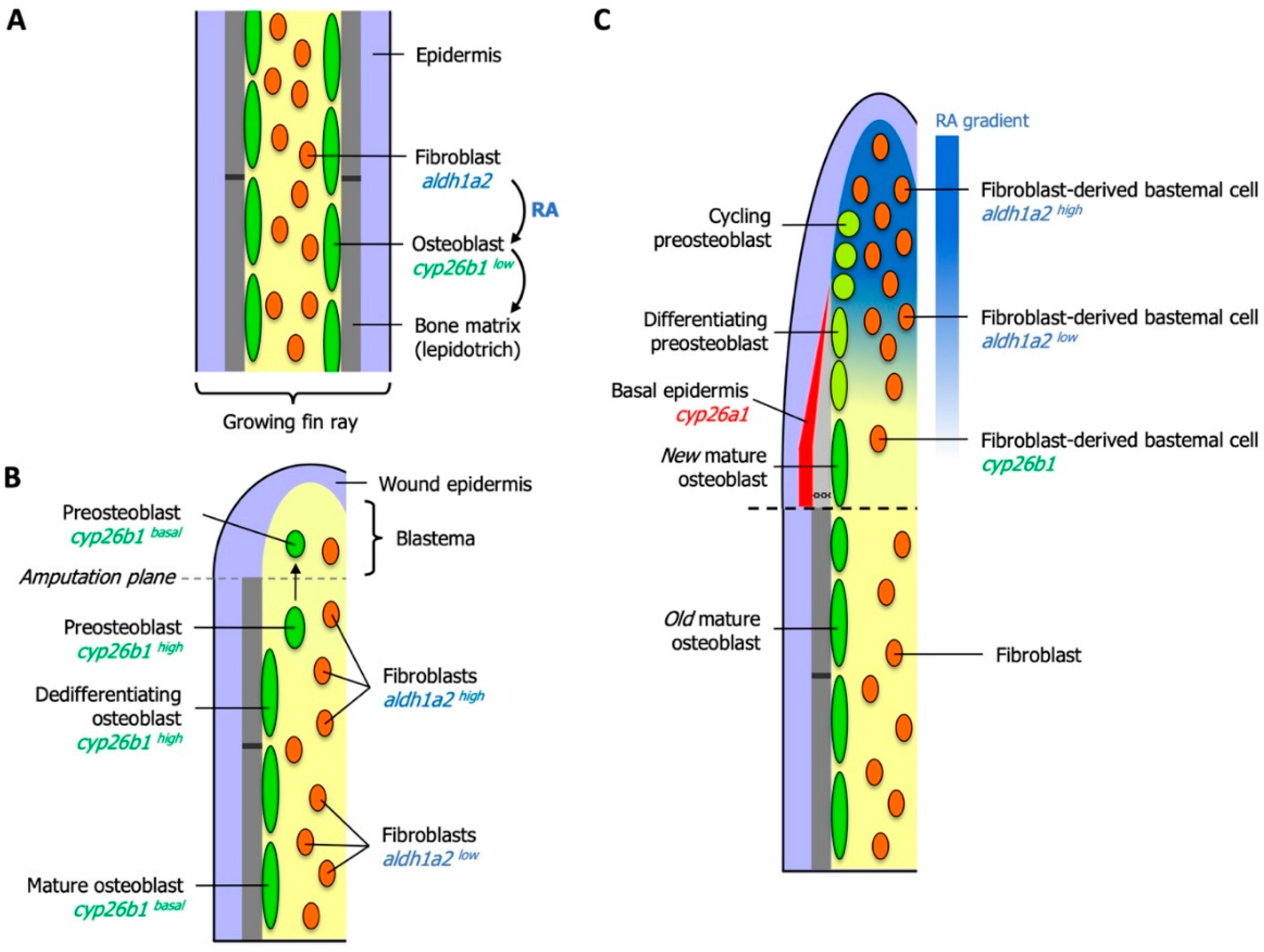
6. Essential Roles for RA in Zebrafish Fin Regeneration
6.1. RA Controls Blastema Formation and Maintenance
6.2. Local Degradation of RA Controls Morphogenetic Processes of Osteoblasts and Osteoclasts
6.3. RA Controls Cell Fate in the Preosteoblast Lineage
6.4. Growth Control Upstream of RA in Zebrafish Fins
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tang, X.-H.; Gudas, L.J. Retinoids, Retinoic Acid Receptors, and Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaller, C.; Eichele, G. Identification and spatial distribution of retinoids in the developing chick limb bud. Nature 1987, 327, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Dollé, P. Retinoic acid in development: towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Kam, R.K.T.; Deng, Y.; Chen, Y.; Zhao, H. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci. 2012, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Reboul, E. Absorption of Vitamin A and Carotenoids by the Enterocyte: Focus on Transport Proteins. Nutrients 2013, 5, 3563–3581. [Google Scholar] [CrossRef] [Green Version]
- Widjaja-Adhi, M.A.K.; Lobo, G.P.; Golczak, M.; Von Lintig, J. A genetic dissection of intestinal fat-soluble vitamin and carotenoid absorption. Hum. Mol. Genet. 2015, 24, 3206–3219. [Google Scholar] [CrossRef] [Green Version]
- Napoli, J.L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 2012, 1821, 152–167. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A Metabolism: An Update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [Green Version]
- Chelstowska, S.; Widjaja-Adhi, M.; Silvaroli, J.; Golczak, M. Molecular Basis for Vitamin A Uptake and Storage in Vertebrates. Nutrients 2016, 8, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellovino, D.; Apreda, M.; Gragnoli, S.; Massimi, M.; Gaetani, S. Vitamin A transport: in vitro models for the study of RBP secretion. Mol. Asp. Med. 2003, 24, 411–420. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Zhong, M.; Kassai, M.; Ter-Stepanian, M.; Sun, H. Vitamin A Transport Mechanism of the Multitransmembrane Cell-Surface Receptor STRA6. Membranes 2015, 5, 425–453. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.N.; Davis, G.J.; Hurley, T.D.; Stone, C.L.; Li, T.K.; Bosron, W.F. Catalytic efficiency of human alcohol dehydrogenases for retinol oxidation and retinal reduction. Alcohol. Clin. Exp. Res. 1994, 18, 587–591. [Google Scholar] [CrossRef]
- Kim, C.I.; Leo, M.A.; Lieber, C.S. Retinol forms retinoic acid via retinal. Arch. Biochem. Biophys. 1992, 294, 388–393. [Google Scholar] [CrossRef]
- Boleda, M.D.; Saubi, N.; Farrés, J.; Parés, X. Physiological substrates for rat alcohol dehydrogenase classes: aldehydes of lipid peroxidation, omega-hydroxyfatty acids, and retinoids. Arch. Biochem. Biophys. 1993, 307, 85–90. [Google Scholar] [CrossRef]
- Kedishvili, N.Y. Retinoic acid synthesis and degradation. In The Biochemistry of Retinoid Signaling II; Springer: Berlin, Germany, 2016; pp. 127–161. [Google Scholar]
- Niederreither, K.; Subbarayan, V.; Dollé, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Dubey, A.; Rose, R.E.; Jones, D.R.; Saint-Jeannet, J.-P. Generating retinoic acid gradients by local degradation during craniofacial development: One cell’s cue is another cell’s poison. Genesis 2018, 56, e23091. [Google Scholar] [CrossRef]
- Pennimpede, T.; Cameron, D.A.; MacLean, G.A.; Li, H.; Abu-Abed, S.; Petkovich, M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Reijntjes, S.; Blentic, A.; Gale, E.; Maden, M. The control of morphogen signalling: Regulation of the synthesis and catabolism of retinoic acid in the developing embryo. Dev. Biol. 2005, 285, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbs-McAuliffe, B.; Zhao, Q.; Linney, E. Feedback mechanisms regulate retinoic acid production and degradation in the zebrafish embryo. Mech. Dev. 2004, 121, 339–350. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, E.; Rydeen, A.B.; Anderson, J.L.; Mandal, A.; Waxman, J.S. Depletion of retinoic acid receptors initiates a novel positive feedback mechanism that promotes teratogenic increases in retinoic acid. PLoS Genet. 2013, 9, e1003689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydeen, A.; Voisin, N.; D’Aniello, E.; Ravisankar, P.; Devignes, C.-S.; Waxman, J.S. Excessive feedback of Cyp26a1 promotes cell non-autonomous loss of retinoic acid signaling. Dev. Biol. 2015, 405, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Cai, A.Q.; Radtke, K.; Linville, A.; Lander, A.D.; Nie, Q.; Schilling, T.F. Cellular retinoic acid-binding proteins are essential for hindbrain patterning and signal robustness in zebrafish. Development 2012, 139, 2150–2155. [Google Scholar] [CrossRef] [Green Version]
- Rochette-Egly, C.; Germain, P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, nrs.07005. [Google Scholar] [CrossRef]
- Escriva, H.; Bertrand, S.; Germain, P.; Robinson-Rechavi, M.; Umbhauer, M.; Cartry, J.; Duffraisse, M.; Holland, L.; Gronemeyer, H.; Laudet, V. Neofunctionalization in Vertebrates: The Example of Retinoic Acid Receptors. PLoS Genet 2006, 2, e102. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.K.; Saxena, V.; Liu, R.-Z.; Thisse, C.; Thisse, B.; Denovan-Wright, E.M.; Wright, J.M. Differential expression of the duplicated cellular retinoic acid-binding protein 2 genes (crabp2a and crabp2b) during zebrafish embryonic development. Gene Expr. Patterns 2005, 5, 371–379. [Google Scholar] [CrossRef]
- Taylor, J.S.; Braasch, I.; Frickey, T.; Meyer, A.; Van de Peer, Y. Genome Duplication, a Trait Shared by 22,000 Species of Ray-Finned Fish. Genome Res. 2003, 13, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarut, E.; Gaudin, C.; Hughes, S.; Gillet, B.; De Bernard, S.; Jouve, P.-E.; Buffat, L.; Allot, A.; Lecompte, O.; Berekelya, L.; et al. Retinoic acid receptor subtype-specific transcriptotypes in the early zebrafish embryo. Mol. Endocrinol. 2014, 28, 260–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, nrs.07002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 940–954. [Google Scholar] [CrossRef]
- Kane, M.A.; Folias, A.E.; Pingitore, A.; Perri, M.; Obrochta, K.M.; Krois, C.R.; Cione, E.; Ryu, J.Y.; Napoli, J.L. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 21884–21889. [Google Scholar] [CrossRef] [Green Version]
- Blaner, W.S.; Olson, J.A. Retinol and retinoic acid metabolism. In The Retinoids: Biology, Chemistry and Medicine; Sporn, M.B., Roberts, A.B., Goodmann, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 229–255. [Google Scholar]
- Duester, G. Retinoic Acid Synthesis and Signaling during Early Organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Al Tanoury, Z.; Piskunov, A.; Rochette-Egly, C. Vitamin A and retinoid signaling: genomic and nongenomic effects: Thematic Review Series: Fat-Soluble Vitamins: Vitamin A. J. Lipid Res. 2013, 54, 1761–1775. [Google Scholar] [CrossRef] [Green Version]
- Conaway, H.H.; Henning, P.; Lerner, U.H. Vitamin A Metabolism, Action, and Role in Skeletal Homeostasis. Endocr. Rev. 2013, 34, 766–797. [Google Scholar] [CrossRef] [Green Version]
- Shannon, S.R.; Moise, A.R.; Trainor, P.A. New insights and changing paradigms in the regulation of vitamin A metabolism in development: Regulation of vitamin A metabolism. Wires Dev. Biol. 2017, 6, e264. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Duester, G. Retinoic acid controls body axis extension by directly repressing Fgf8 transcription. Development 2014, 141, 2972–2977. [Google Scholar] [CrossRef] [Green Version]
- Rochette-Egly, C. Retinoic acid signaling and mouse embryonic stem cell differentiation: Cross talk between genomic and non-genomic effects of RA. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2015, 1851, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: an overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar] [CrossRef] [PubMed]
- Mezquita, B.; Mezquita, C. Two Opposing Faces of Retinoic Acid: Induction of Stemness or Induction of Differentiation Depending on Cell-Type. Biomolecules 2019, 9, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janesick, A.; Wu, S.C.; Blumberg, B. Retinoic acid signaling and neuronal differentiation. Cell. Mol. Life Sci. 2015, 72, 1559–1576. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Mazariegos, J.; Schubert, M.; Laudet, V. Evolution of Retinoic Acid Receptors and Retinoic Acid Signaling. In The Biochemistry of Retinoic Acid Receptors I: Structure, Activation, and Function at the Molecular Level; Asson-Batres, M.A., Rochette-Egly, C., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2014; pp. 55–73. [Google Scholar] [CrossRef]
- Metzler, M.; Sandell, L. Enzymatic Metabolism of Vitamin A in Developing Vertebrate Embryos. Nutrients 2016, 8, 812. [Google Scholar] [CrossRef]
- Napoli, J.L. Functions of Intracellular Retinoid Binding-Proteins. In The Biochemistry of Retinoid Signaling II; Asson-Batres, M.A., Rochette-Egly, C., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2016; Volume 81, pp. 21–76. [Google Scholar] [CrossRef] [Green Version]
- Ghyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Long, X.; Xie, Y.; Zeng, X.; Chen, X.; Mo, Z. The roles of retinoic acid in the differentiation of spermatogonia and spermatogenic disorders. Clin. Chim. Acta 2019, 497, 54–60. [Google Scholar] [CrossRef]
- Stefanovic, S.; Zaffran, S. Mechanisms of retinoic acid signaling during cardiogenesis. Mech. Dev. 2017, 143, 9–19. [Google Scholar] [CrossRef]
- Williams, A.L.; Bohnsack, B.L. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 2019, e23308. [Google Scholar] [CrossRef]
- Cañete, A.; Cano, E.; Muñoz-Chápuli, R.; Carmona, R. Role of Vitamin A/Retinoic Acid in Regulation of Embryonic and Adult Hematopoiesis. Nutrients 2017, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Zieger, E.; Schubert, M. Chapter One - New Insights into the Roles of Retinoic Acid Signaling in Nervous System Development and the Establishment of Neurotransmitter Systems. In International Review of Cell and Molecular Biology; Galluzzi, L., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 330, pp. 1–84. [Google Scholar] [CrossRef]
- Green, A.C.; Martin, T.J.; Purton, L.E. The role of vitamin A and retinoic acid receptor signaling in post-natal maintenance of bone. J. Steroid Biochem. Mol. Biol. 2016, 155, 135–146. [Google Scholar] [CrossRef] [PubMed]
- von Boehmer, H. Oral tolerance: is it all retinoic acid? J. Exp. Med. 2007, 204, 1737–1739. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Grainger, J.R.; Spencer, S.P.; Belkaid, Y. The Role of Retinoic Acid in Tolerance and Immunity. Immunity 2011, 35, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Mendonça Oliveira, L.; Teixeira, F.M.E.; Sato, M.N. Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases. Mediat. Inflamm. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bono, M.; Tejon, G.; Flores-Santibañez, F.; Fernandez, D.; Rosemblatt, M.; Sauma, D. Retinoic Acid as a Modulator of T Cell Immunity. Nutrients 2016, 8, 349. [Google Scholar] [CrossRef] [Green Version]
- Raverdeau, M.; Mills, K.H.G. Modulation of T Cell and Innate Immune Responses by Retinoic Acid. J. Immunol. 2014, 192, 2953–2958. [Google Scholar] [CrossRef]
- Harris, M.P.; Arratia, G. Notochord: Patterning the spine. eLife 2018, 7, e37288. [Google Scholar] [CrossRef]
- Willems, B.; Büttner, A.; Huysseune, A.; Renn, J.; Witten, P.E.; Winkler, C. Conditional ablation of osteoblasts in medaka. Dev. Biol. 2012, 364, 128–137. [Google Scholar] [CrossRef]
- Fleming, A.; Kishida, M.G.; Kimmel, C.B.; Keynes, R.J. Building the backbone: the development and evolution of vertebral patterning. Development 2015, 142, 1733–1744. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Keynes, R.; Tannahill, D. A central role for the notochord in vertebral patterning. Development 2004, 131, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Laue, K.; Jänicke, M.; Plaster, N.; Sonntag, C.; Hammerschmidt, M. Restriction of retinoic acid activity by Cyp26b1 is required for proper timing and patterning of osteogenesis during zebrafish development. Development 2008, 135, 3775–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spoorendonk, K.M.; Peterson-Maduro, J.; Renn, J.; Trowe, T.; Kranenbarg, S.; Winkler, C.; Schulte-Merker, S. Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton. Development 2008, 135, 3765–3774. [Google Scholar] [CrossRef] [Green Version]
- Steventon, B.; Martinez Arias, A. Evo-engineering and the cellular and molecular origins of the vertebrate spinal cord. Dev. Biol. 2017, 432, 3–13. [Google Scholar] [CrossRef]
- Solnica-Krezel, L. Conserved Patterns of Cell Movements during Vertebrate Gastrulation. Curr. Biol. 2005, 15, R213–R228. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, T.J.; Colas, A.; Duester, G. Early molecular events during retinoic acid induced differentiation of neuromesodermal progenitors. Biol. Open 2016, 5, 1821–1833. [Google Scholar] [CrossRef] [Green Version]
- Gouti, M.; Delile, J.; Stamataki, D.; Wymeersch, F.J.; Huang, Y.; Kleinjung, J.; Wilson, V.; Briscoe, J. A Gene Regulatory Network Balances Neural and Mesoderm Specification during Vertebrate Trunk Development. Dev. Cell 2017, 41, 243–261.e7. [Google Scholar] [CrossRef]
- Janesick, A.; Nguyen, T.T.L.; Aisaki, K.-I.; Igarashi, K.; Kitajima, S.; Chandraratna, R.A.S.; Kanno, J.; Blumberg, B. Active repression by RAR signaling is required for vertebrate axial elongation. Development 2014, 141, 2260–2270. [Google Scholar] [CrossRef] [Green Version]
- Janesick, A.; Tang, W.; Nguyen, T.T.L.; Blumberg, B. RARβ2 is required for vertebrate somitogenesis. Development 2017, 144, 1997–2008. [Google Scholar] [CrossRef] [Green Version]
- Wilson, V.; Olivera-Martinez, I.; Storey, K.G. Stem cells, signals and vertebrate body axis extension. Development 2009, 136, 1591–1604. [Google Scholar] [CrossRef] [Green Version]
- Cambray, N.; Wilson, V. Axial progenitors with extensive potency are localised to the mouse chordoneural hinge. Development 2002, 129, 4855–4866. [Google Scholar]
- Cambray, N.; Wilson, V. Two distinct sources for a population of maturing axial progenitors. Development 2007, 134, 2829–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivera-Martinez, I.; Harada, H.; Halley, P.A.; Storey, K.G. Loss of FGF-Dependent Mesoderm Identity and Rise of Endogenous Retinoid Signalling Determine Cessation of Body Axis Elongation. PLoS Biol. 2012, 10, e1001415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wymeersch, F.J.; Huang, Y.; Blin, G.; Cambray, N.; Wilkie, R.; Wong, F.C.; Wilson, V. Position-dependent plasticity of distinct progenitor types in the primitive streak. eLife 2016, 5, e10042. [Google Scholar] [CrossRef] [PubMed]
- Ribes, V.; Le Roux, I.; Rhinn, M.; Schuhbaur, B.; Dolle, P. Early mouse caudal development relies on crosstalk between retinoic acid, Shh and Fgf signalling pathways. Development 2009, 136, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Sirbu, I.O.; Duester, G. Retinoic-acid signalling in node ectoderm and posterior neural plate directs left–right patterning of somitic mesoderm. Nat. Cell Biol. 2006, 8, 271–277. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Brade, T.; Sandell, L.L.; Lewandoski, M.; Trainor, P.A.; Colas, A.; Mercola, M.; Duester, G. Retinoic Acid Activity in Undifferentiated Neural Progenitors Is Sufficient to Fulfill Its Role in Restricting Fgf8 Expression for Somitogenesis. PLoS ONE 2015, 10, e0137894. [Google Scholar] [CrossRef]
- del Corral, R.D.; Olivera-Martinez, I.; Goriely, A.; Gale, E.; Maden, M.; Storey, K. Opposing FGF and Retinoid Pathways Control Ventral Neural Pattern, Neuronal Differentiation, and Segmentation during Body Axis Extension. Neuron 2003, 40, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Olivera-Martinez, I.; Storey, K.G. Wnt signals provide a timing mechanism for the FGF-retinoid differentiation switch during vertebrate body axis extension. Development 2007, 134, 2125–2135. [Google Scholar] [CrossRef] [Green Version]
- Wilson, V.; Olivera-Martinez, I.; Storey, K.G. Erratum: Stem cells signals and vertebrate body axis extension (Development vol. 136 (1591-1604)). Development 2009, 136, 2133. [Google Scholar] [CrossRef] [Green Version]
- Garriock, R.J.; Chalamalasetty, R.B.; Kennedy, M.W.; Canizales, L.C.; Lewandoski, M.; Yamaguchi, T.P. Lineage tracing of neuromesodermal progenitors reveals novel wnt-dependent roles in trunk progenitor cell maintenance and differentiation. Development 2015, 142, 1628–1638. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.L.; Kimelman, D. Canonical Wnt Signaling Dynamically Controls Multiple Stem Cell Fate Decisions during Vertebrate Body Formation. Dev. Cell 2012, 22, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouti, M.; Tsakiridis, A.; Wymeersch, F.J.; Huang, Y.; Kleinjung, J.; Wilson, V.; Briscoe, J. In Vitro Generation of Neuromesodermal Progenitors Reveals Distinct Roles for Wnt Signalling in the Specification of Spinal Cord and Paraxial Mesoderm Identity. PLoS Biol. 2014, 12, e1001937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrier, L.; Davidson, L.; Gierliński, M.; Dady, A.; Storey, K.G. Neural differentiation, selection and transcriptomic profiling of human neuromesodermal progenitor-like cells in vitro. Development 2018, 145, dev166215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalamalasetty, R.B.; Dunty, W.C., Jr.; Biris, K.K.; Ajima, R.; Iacovino, M.; Beisaw, A.; Lionel, F.; Chapman, D.L.; Yoon, J.K.; Kyba, M.; et al. The Wnt3a/β-catenin target gene Mesogenin1 controls the segmentation clock by activating a Notch signalling program. Nat. Commun. 2011, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouti, M.; Metzis, V.; Briscoe, J. The route to spinal cord cell types: a tale of signals and switches. Trends Genet. 2015, 31, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Tsakiridis, A.; Wilson, V. Assessing the bipotency of in vitro-derived neuromesodermal progenitors. F1000Research 2015, 4. [Google Scholar] [CrossRef]
- Iulianella, A.; Beckett, B.; Petkovich, M.; Lohnes, D. A Molecular Basis for Retinoic Acid-Induced Axial Truncation. Dev. Biol. 1999, 205, 33–48. [Google Scholar] [CrossRef]
- Martin, B.L.; Kimelman, D. Brachyury establishes the embryonic mesodermal progenitor niche. Genes Dev. 2010, 24, 2778–2783. [Google Scholar] [CrossRef] [Green Version]
- Sakai, Y.; Meno, C.; Fujii, H.; Nishino, J.; Shiratori, H.; Saijoh, Y.; Rossant, J.; Hamada, H. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001, 15, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Chawengsaksophak, K.; de Graaff, W.; Rossant, J.; Deschamps, J.; Beck, F. Cdx2 is essential for axial elongation in mouse development. Proc. Natl. Acad. Sci. USA 2004, 101, 7641–7645. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, V.; Meyer, B.I.; Gruss, P. Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell 1995, 83, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Moreno, T.A.; Kintner, C. Regulation of Segmental Patterning by Retinoic Acid Signaling during Xenopus Somitogenesis. Dev. Cell 2004, 6, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Dahmann, C.; Oates, A.C.; Brand, M. Boundary formation and maintenance in tissue development. Nat. Rev. Genet. 2011, 12, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Hitachi, K.; Kondow, A.; Danno, H.; Inui, M.; Uchiyama, H.; Asashima, M. Tbx6, Thylacine1, and E47 synergistically activate bowline expression in Xenopus somitogenesis. Dev. Biol. 2008, 313, 816–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.P.; Fu, Y.; Liu, Y.; Maye, P. Inverse agonism of retinoic acid receptors directs epiblast cells into the paraxial mesoderm lineage. Stem Cell Res. 2018, 30, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Inohaya, K.; Takano, Y.; Kudo, A. The teleost intervertebral region acts as a growth center of the centrum: In vivo visualization of osteoblasts and their progenitors in transgenic fish. Dev. Dyn. 2007, 236, 3031–3046. [Google Scholar] [CrossRef]
- Wopat, S.; Bagwell, J.; Sumigray, K.D.; Dickson, A.L.; Huitema, L.F.A.; Poss, K.D.; Schulte-Merker, S.; Bagnat, M. Spine Patterning Is Guided by Segmentation of the Notochord Sheath. Cell Rep. 2018, 22, 2026–2038. [Google Scholar] [CrossRef] [Green Version]
- Forero, L.L.; Narayanan, R.; Huitema, L.F.; VanBergen, M.; Apschner, A.; Peterson-Maduro, J.; Logister, I.; Valentin, G.; Morelli, L.G.; Oates, A.C. Segmentation of the zebrafish axial skeleton relies on notochord sheath cells and not on the segmentation clock. eLife 2018, 7, e33843. [Google Scholar] [CrossRef]
- Pogoda, H.-M.; Riedl-Quinkertz, I.; Löhr, H.; Waxman, J.S.; Dale, R.M.; Topczewski, J.; Schulte-Merker, S.; Hammerschmidt, M. Direct activation of chordoblasts by retinoic acid is required for segmented centra mineralization during zebrafish spine development. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; Bagwell, J.; Njaine, B.; Norman, J.; Levic, D.S.; Wopat, S.; Miller, S.E.; Liu, X.; Locasale, J.W.; Stainier, D.Y. Sheath cell invasion and trans-differentiation repair mechanical damage caused by loss of caveolae in the zebrafish notochord. Curr. Biol. 2017, 27, 1982–1989.e3. [Google Scholar] [CrossRef] [Green Version]
- Dale, R.M.; Topczewski, J. Identification of an evolutionarily conserved regulatory element of the zebrafish col2a1a gene. Dev. Biol. 2011, 357, 518–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huitema, L.F.A.; Apschner, A.; Logister, I.; Spoorendonk, K.M.; Bussmann, J.; Hammond, C.L.; Schulte-Merker, S. Entpd5 is essential for skeletal mineralization and regulates phosphate homeostasis in zebrafish. Proc. Natl. Acad. Sci. USA 2012, 109, 21372–21377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Chen, Q.; Washio, Y.; Yokoi, H.; Suzuki, T. Excess Retinoic Acid Induces Fusion of Centra by Degenerating Intervertebral Ligament Cells in Japanese flounder, Paralichthys olivaceus. J. Exp. Zool. (Mol. Dev. Evol.) 2016, 326B, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: how osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. N. Y. Acad. Sci. 2010, 1192, 437. [Google Scholar] [CrossRef] [Green Version]
- Laue, K.; Pogoda, H.-M.; Daniel, P.B.; van Haeringen, A.; Alanay, Y.; von Ameln, S.; Rachwalski, M.; Morgan, T.; Gray, M.J.; Breuning, M.H.; et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am. J. Hum. Genet. 2011, 89, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Jeradi, S.; Hammerschmidt, M. Retinoic acid-induced premature osteoblast-to-preosteocyte transitioning has multiple effects on calvarial development. Development 2016, 143, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Yip, J.E.; Kokich, V.G.; Shepard, T.H. The effect of high doses of retinoic acid on prenatal craniofacial development in Macaca nemestrina. Teratology 1980, 21, 29–38. [Google Scholar] [CrossRef]
- Maclean, G.; Dollé, P.; Petkovich, M. Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2009, 238, 732–745. [Google Scholar] [CrossRef]
- James, A.W.; Levi, B.; Xu, Y.; Carre, A.L.; Longaker, M.T. Retinoic acid enhances osteogenesis in cranial suture-derived mesenchymal cells: potential mechanisms of retinoid-induced craniosynostosis. Plast. Reconstr. Surg. 2010, 125, 1352–1361. [Google Scholar] [CrossRef] [Green Version]
- Lind, T.; Öhman, C.; Calounova, G.; Rasmusson, A.; Andersson, G.; Pejler, G.; Melhus, H. Excessive dietary intake of vitamin A reduces skull bone thickness in mice. PLoS ONE 2017, 12, e0176217. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of Osteoblast and Odontoblast Differentiation by RUNX2. J. Oral Biosci. 2010, 52, 22–25. [Google Scholar] [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 2010, 339, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T. Signaling networks in RUNX2-dependent bone development. J. Cell. Biochem. 2011, 112, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.R.; Bland, R.; Sheppard, M.C. Characterization of thyroid hormone (T3) receptors in three osteosarcoma cell lines of distinct osteoblast phenotype: interactions among T3, vitamin D3, and retinoid signaling. Endocrinology 1994, 135, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Sheppard, C. Retinoids Modify Regulation of Endogenous Gene Expression by Vitamin D, and Thyroid Hormone in Three Osteosarcoma Cell Lines. Endocrinology 1995, 136, 4304–4314. [Google Scholar] [CrossRef]
- Williams, G.; Robson, H.; Shalet, S. Thyroid hormone actions on cartilage and bone: interactions with other hormones at the epiphyseal plate and effects on linear growth. J. Endocrinol. 1998, 157, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.L.; Cohen, A.J.; Lassová, L. Integration of signaling pathways regulating chondrocyte differentiation during endochondral bone formation. J. Cell. Physiol. 2007, 213, 635–641. [Google Scholar] [CrossRef]
- Lim, J.; Park, E.K. Effect of fibroblast growth factor-2 and retinoic acid on lineage commitment of bone marrow mesenchymal stem cells. Tissue Eng. Regen. Med. 2016, 13, 47–56. [Google Scholar] [CrossRef]
- Fernández, I.; Ortiz-Delgado, J.B.; Darias, M.J.; Hontoria, F.; Andree, K.B.; Manchado, M.; Sarasquete, C.; Gisbert, E. Vitamin A Affects Flatfish Development in a Thyroid Hormone Signaling and Metamorphic Stage Dependent Manner. Front. Physiol. 2017, 8, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, A.C.C.; de Souza Cardozo, F.T.G.; de Souza Magini, R.; Simões, C.M.O. Retinoic acid increases the effect of bone morphogenetic protein type 2 on osteogenic differentiation of human adipose-derived stem cells. J. Appl. Oral Sci. 2019, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roa, L.A.; Bloemen, M.; Carels, C.E.L.; Wagener, F.A.D.T.G.; Von den Hoff, J.W. Retinoic acid disrupts osteogenesis in pre-osteoblasts by down-regulating WNT signaling. Int. J. Biochem. Cell Biol. 2019, 116, 105597. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, Z. The osteoclast: A multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J. Cell. Biochem. 2007, 102, 1130–1139. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.-L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin Ligand Is a Cytokine that Regulates Osteoclast Differentiation and Activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Mochizuki, S.-I.; Yano, K.; Fujise, N.; Sato, Y.; Goto, M.; Yamaguchi, K.; Kuriyama, M.; et al. Identity of Osteoclastogenesis Inhibitory Factor (OCIF) and Osteoprotegerin (OPG): A Mechanism by which OPG/OCIF Inhibits Osteoclastogenesis in Vitro. Endocrinology 1998, 139, 1329–1337. [Google Scholar] [CrossRef]
- Burger, E.H.; Klein-Nulend, J.; Smit, T.H. Strain-derived canalicular fluid flow regulates osteoclast activity in a remodelling osteon—A proposal. J. Biomech. 2003, 36, 1453–1459. [Google Scholar] [CrossRef]
- Mackay, E.W.; Apschner, A.; Schulte-Merker, S. A bone to pick with zebrafish. Bonekey Rep. 2013, 2. [Google Scholar] [CrossRef] [Green Version]
- Siegenthaler, J.A.; Ashique, A.M.; Zarbalis, K.; Patterson, K.P.; Hecht, J.H.; Kane, M.A.; Folias, A.E.; Choe, Y.; May, S.R.; Kume, T.; et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell 2009, 139, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Pittlik, S.; Begemann, G. New sources of retinoic acid synthesis revealed by live imaging of an Aldh1a2-GFP reporter fusion protein throughout zebrafish development. Dev. Dyn. 2012, 241, 1205–1216. [Google Scholar] [CrossRef]
- Kindle, L.; Rothe, L.; Kriss, M.; Osdoby, P.; Collin-Osdoby, P. Human Microvascular Endothelial Cell Activation by IL-1 and TNF-α Stimulates the Adhesion and Transendothelial Migration of Circulating Human CD14+ Monocytes That Develop with RANKL Into Functional Osteoclasts. J. Bone Min. Res. 2005, 21, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast–osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Maderspacher, F. Formation of the adult pigment pattern in zebrafish requires leopard and obelix dependent cell interactions. Development 2003, 130, 3447–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatani, M.; Takano, Y.; Kudo, A. Osteoclasts in bone modeling, as revealed by in vivo imaging, are essential for organogenesis in fish. Dev. Biol. 2011, 360, 96–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Holdway, J.E.; Poss, K.D. Regeneration of amputated zebrafish fin rays from de novo osteoblasts. Dev. Cell 2012, 22, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Loudig, O.; Maclean, G.A.; Dore, N.L.; Luu, L.; Petkovich, M. Transcriptional co-operativity between distant retinoic acid response elements in regulation of Cyp26A1 inducibility. Biochem. J. 2005, 392, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, J.W.; Devarajan, M.; Atit, R.P. Stage-specific roles of Ezh2 and Retinoic acid signaling ensure calvarial bone lineage commitment. Dev. Biol. 2018, 443, 173–187. [Google Scholar] [CrossRef]
- Ferguson, J.; Devarajan, M.; DiNuoscio, G.; Saiakhova, A.; Liu, C.-F.; Lefebvre, V.; Scacheri, P.C.; Atit, R.P. PRC2 Is Dispensable in Vivo for β-Catenin-Mediated Repression of Chondrogenesis in the Mouse Embryonic Cranial Mesenchyme. G3 2018, 8, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.; Varum, S.; Zemke, M.; Scholer, A.; Baggiolini, A.; Draganova, K.; Koseki, H.; Schubeler, D.; Sommer, L. Ezh2 is required for neural crest-derived cartilage and bone formation. Development 2014, 141, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Dudakovic, A.; Camilleri, E.T.; Xu, F.; Riester, S.M.; McGee-Lawrence, M.E.; Bradley, E.W.; Paradise, C.R.; Lewallen, E.A.; Thaler, R.; Deyle, D.R.; et al. Epigenetic Control of Skeletal Development by the Histone Methyltransferase Ezh2. J. Biol. Chem. 2015, 290, 27604–27617. [Google Scholar] [CrossRef] [Green Version]
- Weaver, D.D.; Graham, C.B.; Thomas, I.T.; Smith, D.W. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J. Pediatrics 1974, 84, 547–552. [Google Scholar] [CrossRef]
- Cole, T.R.; Dennis, N.R.; Hughes, H.E. Weaver syndrome. J. Med. Genet. 1992, 29, 332–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatton-Brown, K.; Hanks, S.; Ruark, E.; Zachariou, A.; Duarte, S.D.V.; Ramsay, E.; Snape, K.; Murray, A.; Perdeaux, E.R.; Seal, S.; et al. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, W.T.; Hood, R.L.; Zhan, S.H.; Bulman, D.E.; Fejes, A.P.; Moore, R.; Mungall, A.J.; Eydoux, P.; Babul-Hirji, R.; An, J.; et al. Mutations in EZH2 Cause Weaver Syndrome. Am. J. Hum. Genet. 2012, 90, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alon, U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007, 8, 450–461. [Google Scholar] [CrossRef]
- Morkmued, S.; Laugel-Haushalter, V.; Mathieu, E.; Schuhbaur, B.; Hemmerlé, J.; Dollé, P.; Bloch-Zupan, A.; Niederreither, K. Retinoic Acid Excess Impairs Amelogenesis Inducing Enamel Defects. Front. Physiol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Pasco-Viel, E.; Charles, C.; Chevret, P.; Semon, M.; Tafforeau, P.; Viriot, L.; Laudet, V. Evolutionary Trends of the Pharyngeal Dentition in Cypriniformes (Actinopterygii: Ostariophysi). PLoS ONE 2010, 5, e11293. [Google Scholar] [CrossRef] [Green Version]
- Pasco-Viel, E.; Yang, L.; Veran, M.; Balter, V.; Mayden, R.L.; Laudet, V.; Viriot, L. Stability versus diversity of the dentition during evolutionary radiation in cyprinine fish. Proc. R. Soc. B 2014, 281, 20132688. [Google Scholar] [CrossRef]
- Gibert, Y.; Samarut, E.; Pasco-Viel, E.; Bernard, L.; Borday-Birraux, V.; Sadier, A.; Labbé, C.; Viriot, L.; Laudet, V. Altered retinoic acid signalling underpins dentition evolution. Proc. R. Soc. B Biol. Sci. 2015, 282, 20142764. [Google Scholar] [CrossRef] [Green Version]
- Gibert, Y.; Bernard, L.; Debiais-Thibaud, M.; Bourrat, F.; Joly, J.-S.; Pottin, K.; Meyer, A.; Retaux, S.; Stock, D.W.; Jackman, W.R.; et al. Formation of oral and pharyngeal dentition in teleosts depends on differential recruitment of retinoic acid signaling. FASEB J. 2010, 24, 3298–3309. [Google Scholar] [CrossRef] [Green Version]
- Yelick, P.C.; Schilling, T.F. Molecular dissection of craniofacial development using zebrafish. Crit. Rev. Oral Biol. Med. 2002, 13, 308–322. [Google Scholar] [CrossRef]
- Jackman, W.R.; Draper, B.W.; Stock, D.W. Fgf signaling is required for zebrafish tooth development. Dev. Biol. 2004, 274, 139–157. [Google Scholar] [CrossRef] [Green Version]
- Gibert, Y.; Samarut, E.; Ellis, M.K.; Jackman, W.R.; Laudet, V. The first formed tooth serves as a signalling centre to induce the formation of the dental row in zebrafish. Proc. R. Soc. B 2019, 286, 20190401. [Google Scholar] [CrossRef] [Green Version]
- Seritrakul, P.; Samarut, E.; Lama, T.T.S.; Gibert, Y.; Laudet, V.; Jackman, W.R. Retinoic acid expands the evolutionarily reduced dentition of zebrafish. FASEB J. 2012, 26, 5014–5024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pispa, J.; Thesleff, I. Mechanisms of ectodermal organogenesis. Dev. Biol. 2003, 262, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Woltmann, I.; Shkil, F.; De Clercq, A.; Huysseune, A.; Witten, P.E. Supernumerary teeth in the pharyngeal dentition of slow-developing zebrafish (Danio rerio, Hamilton, 1822). J. Appl. Ichthyol. 2018, 34, 455–464. [Google Scholar] [CrossRef]
- Lee, L.R.; Mortensen, R.M.; Larson, C.A.; Brent, G.A. Thyroid hormone receptor-alpha inhibits retinoic acid-responsive gene expression and modulates retinoic acid-stimulated neural differentiation in mouse embryonic stem cells. Mol. Endocrinol. 1994, 8, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, B.L.; Kahana, A. Thyroid hormone and retinoic acid interact to regulate zebrafish craniofacial neural crest development. Dev. Biol. 2013, 373, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Bohnsack, B.L.; Gallina, D.; Kahana, A. Phenothiourea Sensitizes Zebrafish Cranial Neural Crest and Extraocular Muscle Development to Changes in Retinoic Acid and IGF Signaling. PLoS ONE 2011, 6, e22991. [Google Scholar] [CrossRef] [Green Version]
- Kogai, T.; Liu, Y.-Y.; Richter, L.L.; Mody, K.; Kagechika, H.; Brent, G.A. Retinoic Acid Induces Expression of the Thyroid Hormone Transporter, Monocarboxylate Transporter 8 (Mct8). J. Biol. Chem. 2010, 285, 27279–27288. [Google Scholar] [CrossRef] [Green Version]
- Pfefferli, C.; Jaźwińska, A. The art of fin regeneration in zebrafish. Regeneration (Oxford, England) 2015, 2, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Wehner, D.; Weidinger, G. Signaling networks organizing regenerative growth of the zebrafish fin. Trends Genet. 2015, 31, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Blum, N.; Begemann, G. Osteoblast de- and redifferentiation are controlled by a dynamic response to retinoic acid during zebrafish fin regeneration. Development 2015, 142, 2894–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, N.; Begemann, G. Retinoic acid signaling controls the formation, proliferation and survival of the blastema during adult zebrafish fin regeneration. Development 2012, 139, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Spencer, J.A.; Koh, B.I.; Kobayashi, T.; Fujisaki, J.; Clemens, T.L.; Lin, C.P.; Kronenberg, H.M.; Scadden, D.T. Endogenous Bone Marrow MSCs Are Dynamic, Fate-Restricted Participants in Bone Maintenance and Regeneration. Cell Stem Cell 2012, 10, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Shibata, E.; Hans, S.; Brand, M.; Kawakami, A. Osteoblast Production by Reserved Progenitor Cells in Zebrafish Bone Regeneration and Maintenance. Dev. Cell 2017, 43, 643–650.e3. [Google Scholar] [CrossRef] [Green Version]
- Knopf, F.; Hammond, C.; Chekuru, A.; Kurth, T.; Hans, S.; Weber, C.W.; Mahatma, G.; Fisher, S.; Brand, M.; Schulte-Merker, S.; et al. Bone regenerates via dedifferentiation of osteoblasts in the zebrafish fin. Dev. Cell 2011, 20, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Stewart, S.; Stankunas, K. Limited dedifferentiation provides replacement tissue during zebrafish fin regeneration. Dev. Biol. 2012, 365, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.; Afonso, N.; Bensimon-Brito, A.; Fonseca, M.; Simões, M.; Leon, J.; Roehl, H.; Cancela, M.L.; Jacinto, A. Differentiated skeletal cells contribute to blastema formation during zebrafish fin regeneration. Development 2011, 138, 3897–3905. [Google Scholar] [CrossRef] [Green Version]
- Addison, M.; Xu, Q.; Cayuso, J.; Wilkinson, D.G. Cell Identity Switching Regulated by Retinoic Acid Signaling Maintains Homogeneous Segments in the Hindbrain. Dev. Cell 2018. [Google Scholar] [CrossRef] [Green Version]
- Blum, N.; Begemann, G. Retinoic acid signaling spatially restricts osteoblasts and controls ray-interray organization during zebrafish fin regeneration. Development 2015, 142, 2888–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardeira, J.; Gavaia, P.J.; Fernández, I.; Cengiz, I.F.; Moreira-Silva, J.; Oliveira, J.M.; Reis, R.L.; Cancela, M.L.; Laizé, V. Quantitative assessment of the regenerative and mineralogenic performances of the zebrafish caudal fin. Sci. Rep. 2016, 6, 39191. [Google Scholar] [CrossRef] [PubMed]
- Conaway, H.H.; Persson, E.; Halén, M.; Granholm, S.; Svensson, O.; Pettersson, U.; Lie, A.; Lerner, U.H. Retinoids inhibit differentiation of hematopoetic osteoclast progenitors. FASEB J. 2009, 23, 3526–3538. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lind, T.; Sundqvist, A.; Jacobson, A.; Melhus, H. Retinoic Acid Increases Proliferation of Human Osteoclast Progenitors and Inhibits RANKL-Stimulated Osteoclast Differentiation by Suppressing RANK. PLoS ONE 2010, 5, e13305. [Google Scholar] [CrossRef] [Green Version]
- McMillan, S.C.; Zhang, J.; Phan, H.-E.; Jeradi, S.; Probst, L.; Hammerschmidt, M.; Akimenko, M.-A. A regulatory pathway involving retinoic acid and calcineurin demarcates and maintains joint cells and osteoblasts in regenerating fin. Development 2018, 145, dev161158. [Google Scholar] [CrossRef] [Green Version]
- Sims, K.; Eble, D.M.; Iovine, M.K. Connexin43 regulates joint location in zebrafish fins. Dev. Biol. 2009, 327, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Kujawski, S.; Lin, W.; Kitte, F.; Börmel, M.; Fuchs, S.; Arulmozhivarman, G.; Vogt, S.; Theil, D.; Zhang, Y.; Antos, C.L. Calcineurin regulates coordinated outgrowth of zebrafish regenerating fins. Dev. Cell 2014, 28, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Budhu, A.S.; Noy, N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol. Cell. Biol. 2002, 22, 2632–2641. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Perathoner, S.; Daane, J.M.; Henrion, U.; Seebohm, G.; Higdon, C.W.; Johnson, S.L.; Nüsslein-Volhard, C.; Harris, M.P. Bioelectric signaling regulates size in zebrafish fins. PLoS Genet. 2014, 10, e1004080. [Google Scholar] [CrossRef] [Green Version]
- Maden, M. The effect of vitamin A on the regenerating axolotl limb. J. Embryol. Exp. Morphol. 1983, 77, 273–295. [Google Scholar] [PubMed]
- Maden, M. Retinoids as endogenous components of the regenerating limb and tail. Wound Rep. Reg. 1998, 6, 358–365. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Boffa, M.B.; Jones, B.; Petkovich, M. A zebrafish retinoic acid receptor expressed in the regenerating caudal fin. Development 1994, 120, 1861–1872. [Google Scholar] [PubMed]
- Geraudie, J.; Monnot, M.J.; Brulfert, A.; Ferretti, P. Caudal fin regeneration in wild type and long-fin mutant zebrafish is affected by retinoic acid. Int. J. Dev. Biol. 1995, 39, 373–381. [Google Scholar] [PubMed]
- Daane, J.M.; Lanni, J.; Rothenberg, I.; Seebohm, G.; Higdon, C.W.; Johnson, S.L.; Harris, M.P. Bioelectric-calcineurin signaling module regulates allometric growth and size of the zebrafish fin. Sci. Rep. 2018, 8, 10391. [Google Scholar] [CrossRef] [Green Version]
- Schilling, T.F.; Nie, Q.; Lander, A.D. Dynamics and precision in retinoic acid morphogen gradients. Curr. Opin. Genet. Dev. 2012, 22, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Aulehla, A.; Pourquié, O. Signaling gradients during paraxial mesoderm development. Cold Spring Harb. Perspect. Biol. 2010, 2, a000869. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Draut, H.; Liebenstein, T.; Begemann, G. New Insights into the Control of Cell Fate Choices and Differentiation by Retinoic Acid in Cranial, Axial and Caudal Structures. Biomolecules 2019, 9, 860. https://doi.org/10.3390/biom9120860
Draut H, Liebenstein T, Begemann G. New Insights into the Control of Cell Fate Choices and Differentiation by Retinoic Acid in Cranial, Axial and Caudal Structures. Biomolecules. 2019; 9(12):860. https://doi.org/10.3390/biom9120860
Chicago/Turabian StyleDraut, Heidrun, Thomas Liebenstein, and Gerrit Begemann. 2019. "New Insights into the Control of Cell Fate Choices and Differentiation by Retinoic Acid in Cranial, Axial and Caudal Structures" Biomolecules 9, no. 12: 860. https://doi.org/10.3390/biom9120860
APA StyleDraut, H., Liebenstein, T., & Begemann, G. (2019). New Insights into the Control of Cell Fate Choices and Differentiation by Retinoic Acid in Cranial, Axial and Caudal Structures. Biomolecules, 9(12), 860. https://doi.org/10.3390/biom9120860