1. Introduction
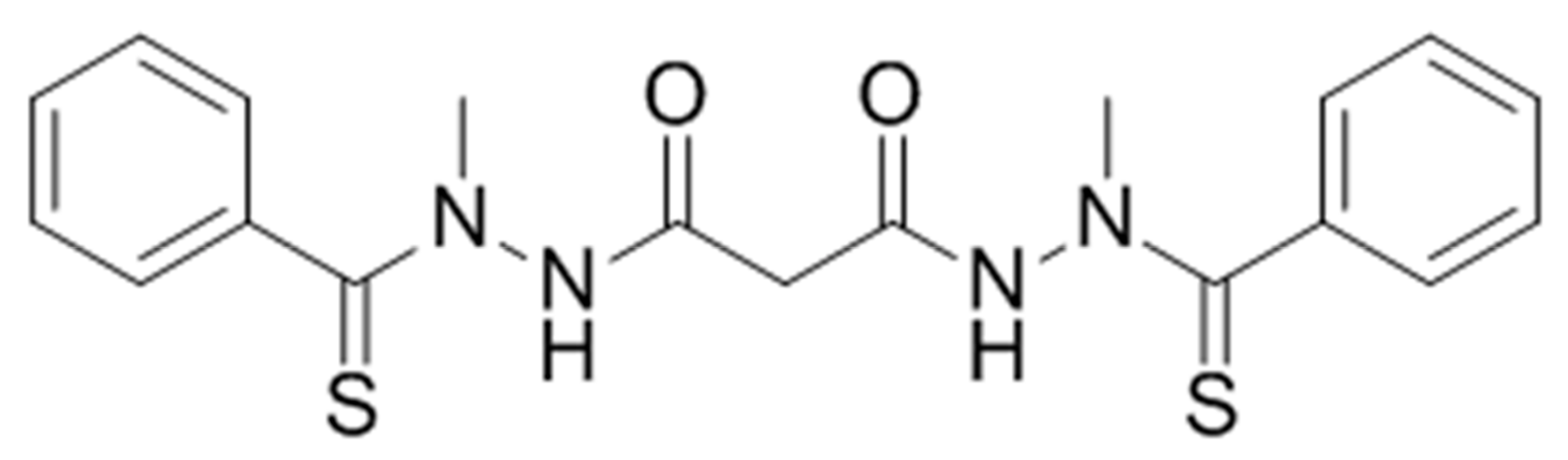
Elesclomol (
N-malonyl-bis(
N′-methyl-
N′-thiobenzoylhydrazide)); formerly STA-4783;
Figure 1) is a mitochondria-targeted chemotherapeutic agent that has exhibited antitumor activity against a broad range of cancer cell types in vitro, enhanced the potency of known chemotherapeutic agents in human tumor models in vivo, and displayed encouraging but limited therapeutic benefit as a single agent and in combination with other anti-cancer compounds in Phase I through Phase III clinical trials [
1,
2,
3,
4,
5,
6].
Elesclomol has been shown to induce a rapid accumulation of intracellular reactive oxygen species (ROS) and a gene transcription profile characteristic of an oxidative stress response in vitro [
7]. Interestingly, the antioxidant
N-acetylcysteine blocks elesclomol induced gene expression and cell death, suggesting that ROS generation may be the primary means of cancer cell killing by the drug [
7].
The biologically active form of elesclomol is a deprotonated copper chelate [
8]. Upon therapeutic administration, this chelate forms when elesclomol acquires Cu
2+ in the bloodstream. However, in vitro studies investigating the mechanism of action of elesclomol have demonstrated that the addition of a pre-formed elesclomol:copper (E:C) complex is necessary to induce cytotoxicity [
8]. In one such in vitro study, comparative growth assays using deletion mutants of a yeast model yield evidence that E:C works through a biologically coherent set of processes occurring in the mitochondrion [
9]. The results suggest that E:C interacts with the electron transport chain (ETC), a major component of the process of oxidative phosphorylation, to generate high levels of ROS within the organelle and ultimately induce cell death. The authors concluded that E:C mediated cytotoxicity occurs via general disruption of the process of electron flow down the ETC, rather than by targeting any particular protein or enzyme function within the ETC.
The purpose of this study was to further explore the mechanism of cellular and mitochondrial toxicity of E:C by examining its direct effect on mitochondrial bioenergetic function in whole cells of non-tumorigenic origin and in isolated mammalian mitochondria. The results obtained indicate that E:C treatment in whole cells induces a rapid and substantial increase in mitochondrial superoxide levels and dissipation of mitochondrial membrane potential. Furthermore, E:C acts as an uncoupler of oxidative phosphorylation and generalized inhibitor of electron transport activity in isolated, intact mitochondria, and induces a dose-dependent inhibition of mitochondrial NADH-ubiquinone oxidoreductase activity in freeze-thawed mitochondrial preparations. The results of this study are important in that they are the first to demonstrate a direct effect of the E:C chelate on bioenergetic function in isolated mammalian mitochondria, and suggest the possibility that the increase in ROS production and cytotoxicity induced by E:C may in part be due to uncoupling of mitochondrial oxidative phosphorylation or inhibition of electron transport activity, or both.
2. Materials and Methods
2.1. Materials
Elesclomol was obtained from MedChemExpress (
https://www.medchemexpress.com/) and made fresh daily at a concentration ranging from 1–10 mM in dimethyl sulfoxide (DMSO). The E:C complex was formed by mixing equimolar and equivolume additions of elesclomol and copper chloride (dissolved in H
2O). MitoSOX Red mitochondrial superoxide indicator for live-cell imaging was obtained from Molecular Probes (Eugene, OR, USA); the mitochondrial superoxide scavenger MitoTEMPO was obtained from Sigma Aldrich (St. Louis, MO, USA); the mitochondrial membrane potential probe TMRE (tetramethylrhodamine, ethyl ester) was obtained from Biotium (Fremont, CA, USA); and the live cell nuclear stain, NucBlue Live, and the cell viability stain, NucGreen Dead, were obtained from ThermoFisher Scientific (Waltham, MA, USA). All solutions were used according to the manufacturer’s specifications.
2.2. Cell Cultures
The CV-1 African green monkey kidney epithelial cells (ATCC® CCL-70™) were grown in Eagle′s minimal essential medium (EMEM) supplemented with 10% fetal bovine serum. All cells were maintained at 37 °C in a 5% CO2 atmosphere.
2.3. Mitochondrial Superoxide Production in Whole Cells
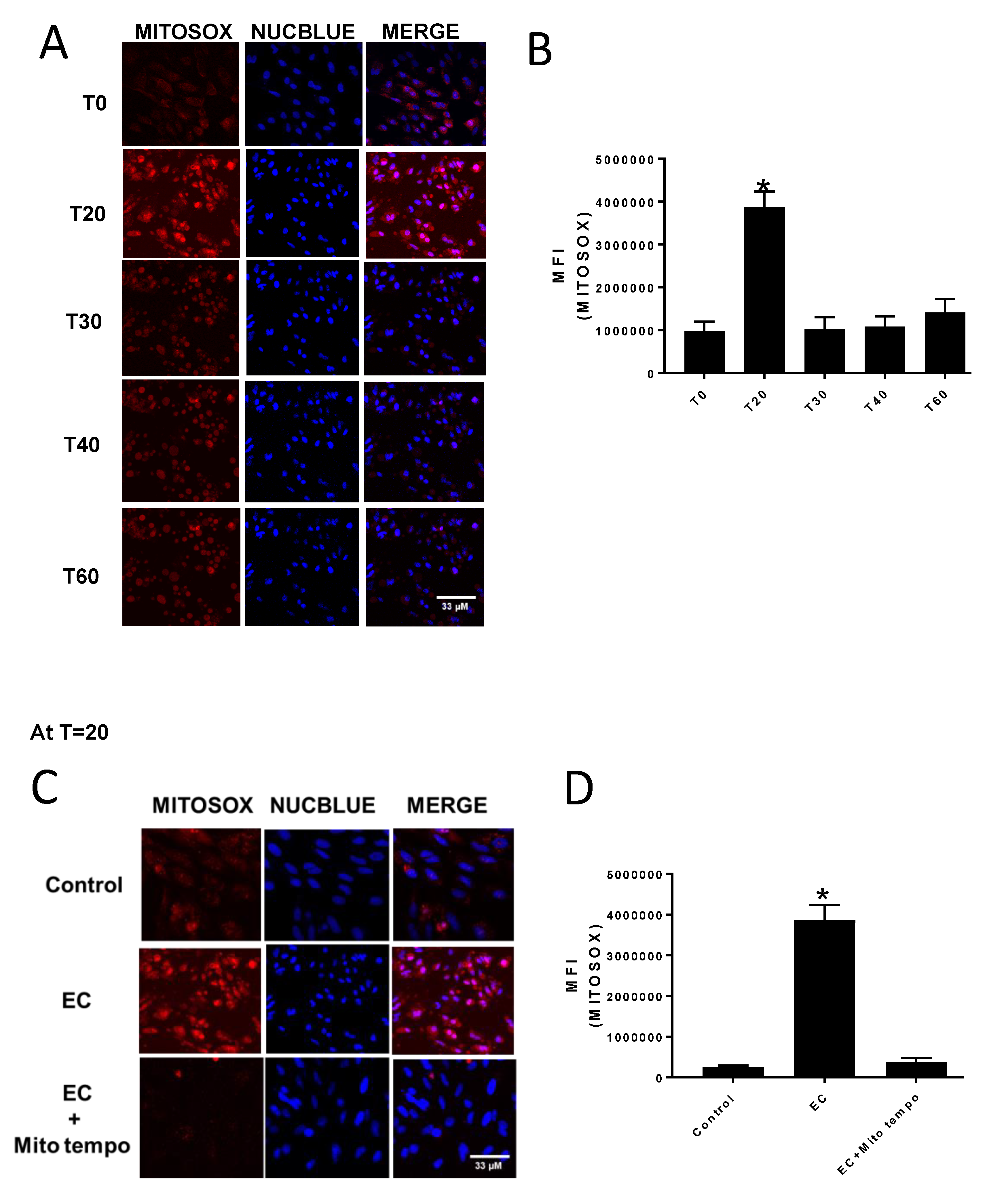
Mitochondrial superoxide production was assessed by measuring MitoSOX fluorescence using a Zeiss 800 confocal microscope (Carl Zeiss Microscopy GmbH, Jena, Germany). The CV-1 cells were seeded in glass bottom 35 mm plates (MatTek) and grown to 90% confluency. To initiate an experiment, cells were incubated for 10 min with 5 μM MitoSOX in phenol red free EMEM, washed twice with sterile phosphate buffered saline, and replenished with phenol red free EMEM and 2 drops/mL media of NucBlue or NucGreen. Cells were then treated with either DMSO (control) or 70 μM E:C, and MitoSOX, NucBlue, and/or NucGreen fluorescence was monitored over a period of 1 h. To block mitochondrial superoxide accumulation, some cells underwent a 1-h pre-treatment with 5 μM of the free radical scavenger MitoTEMPO prior to addition of DMSO or E:C.
2.4. Mitochondrial Membrane Potential in Whole Cells
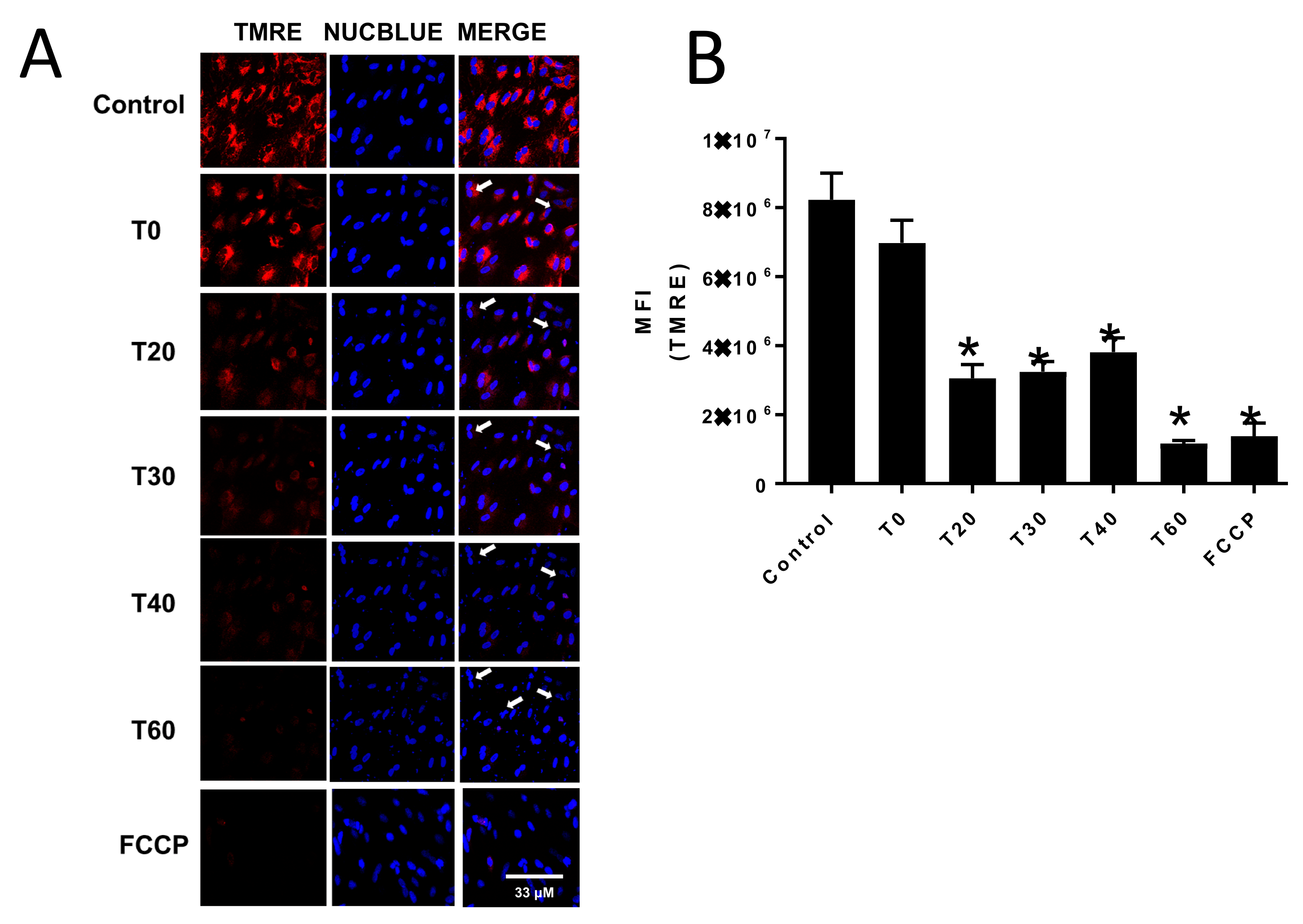
Mitochondrial membrane potential was assessed by measuring TMRE fluorescence using a Zeiss 800 confocal microscope. The CV-1 cells were seeded in glass bottom 35 mm plates (MatTek) and grown to 90% confluency. To initiate an experiment, cells were washed twice with sterile phosphate buffered saline and incubated for 10 min with 100 nM TMRE in phenol red free EMEM and two drops/ml media of NucBlue or NucGreen. Cells were then treated with either DMSO (control) or 70 μM E:C, and TMRE and NucBlue and/or NucGreen fluorescence was monitored over a period of 1 h. As a positive control for uncoupling of oxidative phosphorylation and dissipation of mitochondrial membrane potential, cells were incubated for 10 min in the presence of the ionophore FCCP (carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; 100 μM).
2.5. Isolation of Mitochondria
Mitochondria were isolated from bovine or rat liver by a process of differential centrifugation at 4 °C [
10]. Briefly, 5–6 g of tissue were minced and homogenized in STE (250 mM sucrose, 1 mM Tris-HCI, and 1 mM EDTA (ethylenediaminetetraacetic acid, pH 7.4)), 20%
w/v, and centrifuged at 600×
g for 10 min. The supernatant was collected and then centrifuged for 10 min at 8000×
g. The resulting mitochondrial pellet was re-suspended and washed twice by centrifugation for 10 min at 8000×
g in STE, followed by an additional wash in ST (250 mM sucrose and 1 mM Tris-HC1 (pH 7.4)). The final pellet was re-suspended in ST and the protein concentration of the mitochondrial suspension was determined by the method of Lowry [
11].
2.6. Mitochondrial Respiration
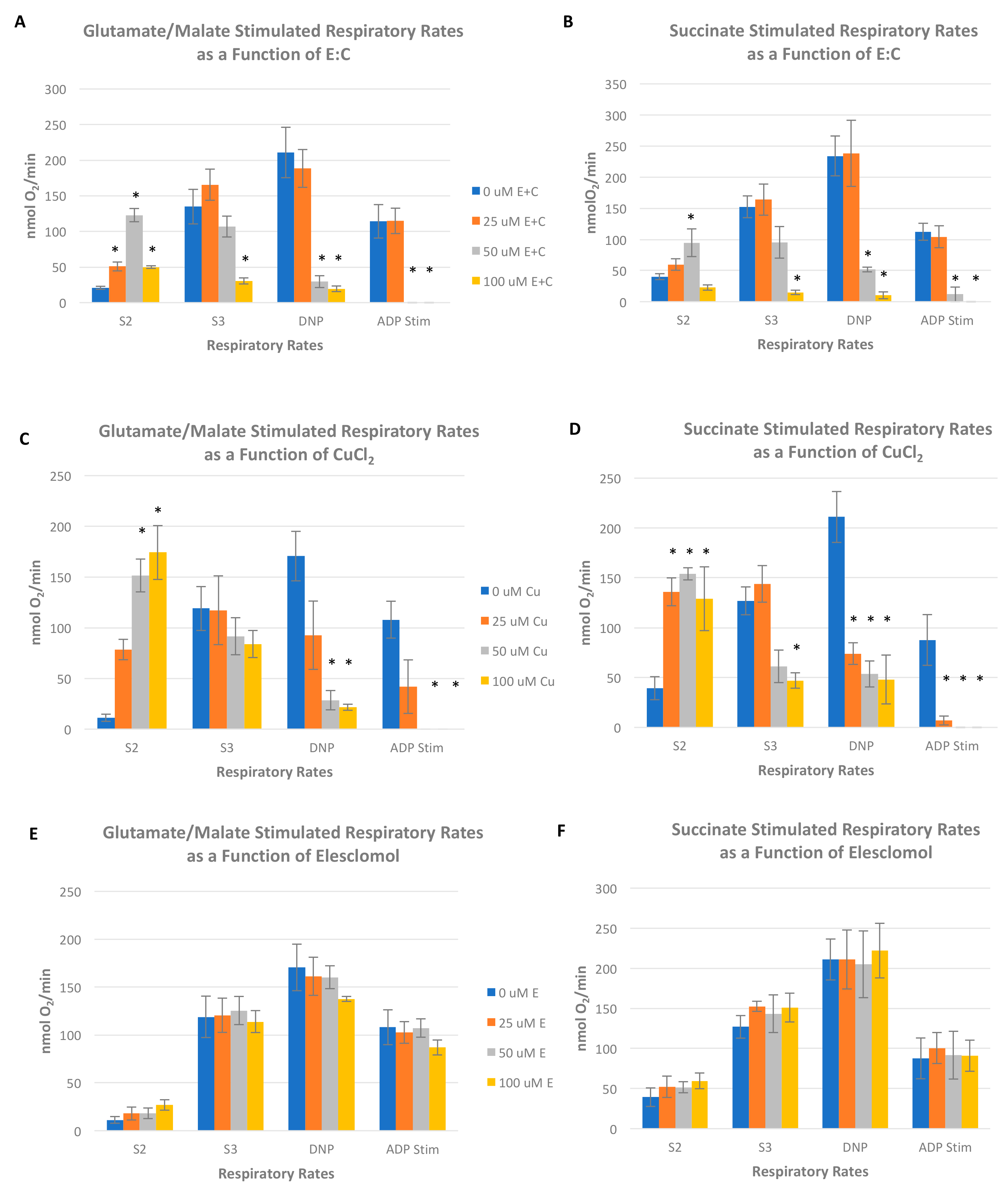
Oxygen consumption was measured in freshly isolated rat liver mitochondria using a Clark-type polarographic oxygen electrode (Oxytherm, Hansatech Instruments, PP Systems Inc., Amesbury, MA, USA) in a solid-state Peltier temperature controlled chamber maintained at 30 °C. The basic respiratory assay medium consisted of 225 mM sucrose, 10 mM KCl, 1 mg/mL 10 mM K2HPO4-KH2PO4, 5 mM MgCl2, and 10 mM Tris-HCl (pH 7.4). An initial rate of oxygen consumption (state 2 rate) was recorded after the addition of mitochondria (approximately 1.5 mg) and respiratory substrate (5 mM each glutamate plus malate or 10 mM succinate). The ADP (120 nmol) was then added to obtain a state 3 rate, which is a measure of oxygen consumption that is coupled to the process of ADP phosphorylation to ATP. Following a measurable state 4 rate (i.e., the respiratory rate after all added ADP has been phosphorylated), 80 µM of the chemical uncoupler 2,4-dinitrophenol was added to obtain a respiratory rate in the absence of coupled oxidative phosphorylation. Respiratory rates were measured in the absence or presence of varying concentrations of E:C (25–100 μM).
2.7. Enzyme Assays
Spectrophotometric measurements of mitochondrial electron transport enzyme activities were obtained using a single-wavelength, temperature-controlled spectrophotometer at 37 °C essentially as described previously [
10,
12]. All enzyme activities were assayed in a final reaction volume of 1.0 mL. As elesclomol, which is dissolved in DMSO, was varied in the assays, additional DMSO was added as necessary to maintain a constant final volume of the solvent in each reaction mixture. For each assay, triplicate values of enzyme activity were obtained for control runs and at every concentration of E:C tested.
The NADH-ubiquinone oxidoreductase activity (a measure of electron transport through respiratory complex I) was determined by measuring the rate of decrease in absorbance at 340 nm due to the oxidation of NADH. Mitochondria (300 µg) that had been subjected to three cycles of freeze-thawing in liquid nitrogen were added to a cuvette containing 50 mM potassium phosphate, 3 mg/mL fatty acid free BSA, 300 μM KCN, 100 μM NADH in the absence or presence of varying concentrations of E:C (25–100 μM). The reaction was initiated by adding 10 μM ubiquinone1 to the cuvette, and the change in the absorbance was recorded over time in the presence and absence of 10 μM rotenone. The rotenone sensitive activity of complex I of the mitochondrial ETC was determined by subtracting the rate measured in the presence of rotenone from the rate obtained in the absence of rotenone.
Succinate-cytochrome c reductase activity (a measure of electron transport through respiratory complexes II and III) was determined by measuring the rate of increase in absorbance at 550 nm due to the reduction of oxidized cytochrome c. Freeze-thawed (−20 °C) preparations of isolated mitochondria (300 μg) were added to a cuvette containing dH2O, 50 mM potassium phosphate (KH2P04/K2HP04; pH 7.4), 30 μM KCN, in the absence or presence of varying concentrations of E:C (25–100 μM). The sample was pre-incubated with the 20 mM succinate for 10 min at 37 °C in order to fully activate the enzyme. The reaction was initiated by adding 50 μM oxidized cytochrome c to the cuvette, and a change in the absorbance was recorded over time.
Cytochrome c oxidase activity (a measure of electron transport through complex IV) was determined by measuring the decrease in absorbance at 550 nm due to the oxidation of reduced cytochrome c. Freeze-thawed (−20 °C) preparations of isolated mitochondria (300 μg) were added to a cuvette containing dH2O, 40 mM potassium phosphate (KH2PO4/K2HPO4; pH 7.0), in the absence or presence of varying concentrations of E:C (25–100 μM). The reaction was initiated by adding 50 μM reduced cytochrome c, and a linear rate was recorded.
4. Discussion
The results of this study indicate that E:C treatment in non-tumorigenic cells induces a rapid and substantial increase in mitochondrial superoxide levels (
Figure 2). This is consistent with data previously shown for a variety of cancer cells types [
1,
2] Mitochondria are the main intracellular source of ROS in most tissues. It has been estimated that under physiological conditions, 1–2% of the molecular oxygen consumed by respiring cells is converted to ROS molecules as a byproduct of oxidative phosphorylation [
13]. The ROS production occurs when a small fraction of reducing equivalents from complex I, II, or III of the mitochondrial ETC leak electrons directly to molecular oxygen, generating the superoxide anion, O
2−•. The mitochondrial enzyme superoxide dismutase converts O
2−• to H
2O
2, which can then acquire an additional electron from a reduced transition metal to generate the highly reactive hydroxyl radical,
.OH. Under controlled conditions, ROS play an important role as signaling molecules that mediate changes in cell proliferation, differentiation, and gene transcription [
14,
15]. However, uncontrolled ROS activity leads to oxidative stress, which can damage intracellular protein and lipid components, affect the integrity of biological membranes, damage both nuclear and mitochondrial DNA (mtDNA), and ultimately lead to irreversible damage and cell death [
16]. Interestingly, higher baseline levels of ROS in tumor versus normal cells are known to contribute to the development or maintenance of the malignant phenotype, or both, and render cancer cells more vulnerable to irreversible oxidative damage and consequent cell death [
17,
18]. It has been suggested that compounds, such as elesclomol, that induce oxidative stress can exploit this unique characteristic of cancer cells by increasing ROS levels beyond the threshold of lethality in cancer cells, while leaving normal cells viable [
7]). In fact, elesclomol is one among a number of mitochondria-targeted ROS inducers that have recently shown efficacy as potential anticancer agents [
19,
20].
Additionally, the data presented here show that treatment of non-tumorigenic cells with the same concentration of E:C that induces a rapid and substantial increase in mitochondrial superoxide levels, also dissipates the mitochondrial membrane potential to an extent sufficient to cause changes in cell morphology suggestive of a loss of membrane integrity or dying/dead cells (
Figure 3 and
Figure 4). Furthermore, results show that E:C acts directly as an uncoupler of oxidative phosphorylation in intact mammalian mitochondria (
Figure 6).
Interestingly, data obtained in this study show that CuCl
2 as a single agent also acts as an uncoupler mitochondrial oxidative phosphorylation irrespective of whether a complex I or complex II electron donor is used as a respiratory substrate (
Figure 6C,D). In addition, CuCl
2 was shown to inhibit succinate stimulated S3 respiration in isolated mitochondria. These results are in line with an earlier published report, which showed inhibition of mitochondrial respiration, a decrease in mitochondrial membrane potential, and a marked increase in ROS levels in mitochondria exposed to CuCl
2 [
21]. The authors of that study concluded that copper at high doses can induce mitochondrial dysfunction through non-specific effects on the mitochondrial ETC, and that antioxidants can protect mitochondrial function by reducing the formation of free radicals. In another study investigating the effects of heavy metals on mitochondrial permeability transition (MPT) in isolated rat liver mitochondria, micromolar concentrations of Cu
2+ as a single agent produced a dose-dependent inhibition of uncoupler-stimulated respiration, membrane potential dissipation, and enhanced membrane permeabilization manifested in mitochondrial swelling and activation of basal respiration [
22]. It is important to note that the data presented in
Figure 6A–D show that copper as a single agent appears to have a more pronounced uncoupling effect on mitochondria than copper that has been chelated to elesclomol. These results indicate that elesclomol binding mitigates the uncoupling effect of copper.
Finally, the results of this study show that E:C acts as a generalized inhibitor of electron transport activity in isolated, intact mammalian mitochondria, and induces a dose-dependent inhibition of mitochondrial NADH-ubiquinone oxidoreductase activity (complex I) in freeze-thawed mitochondrial preparations (
Figure 7A). Complex I is the largest respiratory enzyme complex of the mammalian mitochondrial ETC. It serves as the entry point for electrons donated by NADH, a high-energy electron carrier produced via the citric acid cycle and fatty acid oxidation, and transfers those electrons to the mobile carrier, ubiquinone, thus regenerating the NAD
+ levels in the mitochondrial matrix. This electron transfer at complex I is coupled to the translocation of protons from the mitochondrial matrix to the intermembrane space. Therefore, complex I plays an integral role in generating the electrochemical proton gradient required for the synthesis of ATP from ADP and P
i. A recent review describes the important role that complex I also plays in redox control and the biosynthesis of macromolecules and nucleic acids necessary for cell proliferation [
20]. It is suggested that these complex I-dependent events contribute to tumor formation, resistance to cell death, and metastasis of cancer cells in part by causing an increase in ROS levels [
20]. Interestingly, a number of inhibitors of mitochondrial ETC complex I have been shown to increase mitochondrial ROS production and, consequently, to induce cell death via apoptosis [
19,
20,
23,
24]. The data presented in this study suggest that the E:C chelate may act similarly. However, additional experiments will be necessary to determine definitively whether E:C inhibits complex I specifically, thus causing an increase in ROS, or whether complex I, which is the most labile of the respiratory complexes, is simply more susceptible to generalized E:C-induced membrane disruption or ROS produced elsewhere in the mitochondria.
It is of interest to consider the relationship between the uncoupling and ROS generating effects shown to be induced by E:C in this study. It is generally accepted that ROS production is dependent upon the mitochondrial transmembrane electric potential difference (ΔΨ
m), and that uncoupling agents, which lower ΔΨ
m, will decrease mitochondrial ROS production [
25,
26]. However, in this study, dissipation of the mitochondrial membrane potential by E:C in whole cells is shown to coincide with an increase in mitochondrial specific superoxide production (
Figure 4). Interestingly, another uncoupler of oxidative phosphorylation, the proton ionophore FCCP, has also been shown to increase mitochondrial ROS production under certain conditions. In one study, exposure of isolated cardiomyocytes to FCCP caused a significant increase in the rate of ROS production compared to control cells, and this increase was shown to be completely blocked by pre-treatment with antioxidants [
27]. The authors proposed that the increase in ROS occurred independently of ΔΨ
m, and raised the possibility that FCCP induced ROS production may be from a source other than the mitochondria. In another study using rat liver mitochondria, the results provided evidence that FCCP caused a burst of H
2O
2 production that preceded membrane permeabilization [
28]. The results presented in this report showing that E:C both dissipates the mitochondrial membrane potential and causes an increase in mitochondrial specific ROS more likely suggest an uncoupling mechanism involving MPT, which is known to be induced by elevated mitochondrial ROS [
29]. Indeed, it has also been shown that low triggering amounts of ROS (possibly generated from mitochondrial ETC inhibition) can cause membrane potential depolarization and a subsequent burst in ROS production, a phenomenon referred to as “ROS-induced ROS release” (RIRR) [
30]. Thus, in one possible scenario, inhibition of mitochondrial respiration by E:C (at complex I or non-specifically) may be sufficient to induce triggering amounts of ROS followed by MPT and a consequent burst of ROS. It is also well known that, if not reversed, MPT can lead to structural breakdown of the mitochondrial matrix accompanied by outer mitochondrial membrane rupture and cell death [
29]. Of course, the possibility also exists that the different biological effects demonstrated by E:C in this study (i.e., ETC inhibition, uncoupling, and ROS induction) may be independent of one another.
Earlier reports have clearly established the ability of elesclomol to generate high levels of mitochondrial ROS and ultimately induce cell death in a variety of cancer cell lines. Several recent studies show that elesclomol can produce a number of other biological effects in different models, suggesting the possibility of multiple mechanisms of action and cellular targets for the compound. For example, it has been shown that low nanomolar concentrations of elesclomol can be used to shuttle sub-lethal concentrations of copper into the mitochondria as a therapeutic strategy to restore mitochondrial respiratory activity in copper deficient yeast mutants [
31]. Using murine, human and zebrafish models, the authors also established broad applicability of elesclomol as a potential therapeutic agent to treat disorders of mitochondrial and cellular copper metabolism. In another study, elesclomol was shown to be a relatively potent bactericidal against a laboratory strain and multidrug resistant clinical isolates of
Mycobacterium tuberculosis (
Mtb), and to display additive interactions with known tuberculosis drugs, such as isoniazid and ethambutol, and a synergistic interaction with rifampicin [
32]. Interestingly, elesclomol exhibited a >65-fold increase in activity against
Mtb in the presence of copper compared to non-copper supplemented growth medium. The authors suggest that the ability of elesclomol to enhance ROS generation and accelerate uptake and intracellular accumulation of copper, which is toxic to
Mtb at high concentrations, as possible mechanisms for the compound’s bactericidal efficacy. Additionally, a very recent report elucidated a unique mechanism of action of cytotoxicity by elesclomol in cancer cells that exhibit proteasome inhibitor resistance or a forced shift away from glycolysis and toward a state of enhanced mitochondrial metabolism, or both [
33]. In these cells, there is an upregulation of the activity of ferredoxin 1 protein (FDX1), which is a critical enzyme component in the Fe–S cluster assembly pathway. Results of the study show that E:C binding to FDX1 interferes with the enzyme’s ability to reduce its natural substrate, thereby inhibiting Fe–S cluster formation and resulting in a unique copper-dependent cell death. The authors suggest that other copper ionophores, such as disulfiram, may promote cancer cell death through a similar mechanism.
The results of the study presented herein provide important information about the mechanisms of mitochondrial and cellular toxicity induced by E:C. First, they reveal at least two additional E:C targets directly affecting mitochondrial bioenergetic function. The data indicate that E:C treatment in non-tumorigenic cells can induce a rapid and substantial increase in mitochondrial superoxide levels and dissipate the mitochondrial membrane potential. Furthermore, the data demonstrate that over the range of concentrations tested, E:C acts as both an uncoupler of oxidative phosphorylation and a generalized inhibitor of electron transport activity in isolated, intact mammalian mitochondria, and induces a dose-dependent inhibition of mitochondrial NADH-ubiquinone oxidoreductase activity in freeze-thawed mitochondrial preparations. These results suggest the possibility that the increase in ROS production and cytotoxicity induced by E:C may at least in part be a result of the uncoupling of mitochondrial oxidative phosphorylation or inhibition of electron transport activity, or both. The results also suggest the possibility that differences between the mitochondria of normal versus cancer cells (e.g., in membrane permeability, response to uncoupling, and complex I structure and function) might somehow contribute to the selective cytotoxicity exhibited by elesclomol for certain cancer cell types. Indeed, nearly a century of scientific research has revealed a number of notable differences in the structure and function of mitochondria between normal and cancer cells, including differences in mtDNA sequence, molecular composition, and metabolic activity (for review see [
34]). Additional studies exploring the possibility that differences at the bioenergetic level may increase the vulnerability of cancer cells to elesclomol is warranted and will ultimately contribute to a better understanding of the therapeutic potential of elesclomol as an anticancer compound.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






