The TGF-β1/p53/PAI-1 Signaling Axis in Vascular Senescence: Role of Caveolin-1


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Vascular smooth muscle cell (VSMC) Senescence and Vascular Disease
2. Caveolin-1 and the Senescent Phenotype: Involvement of p53
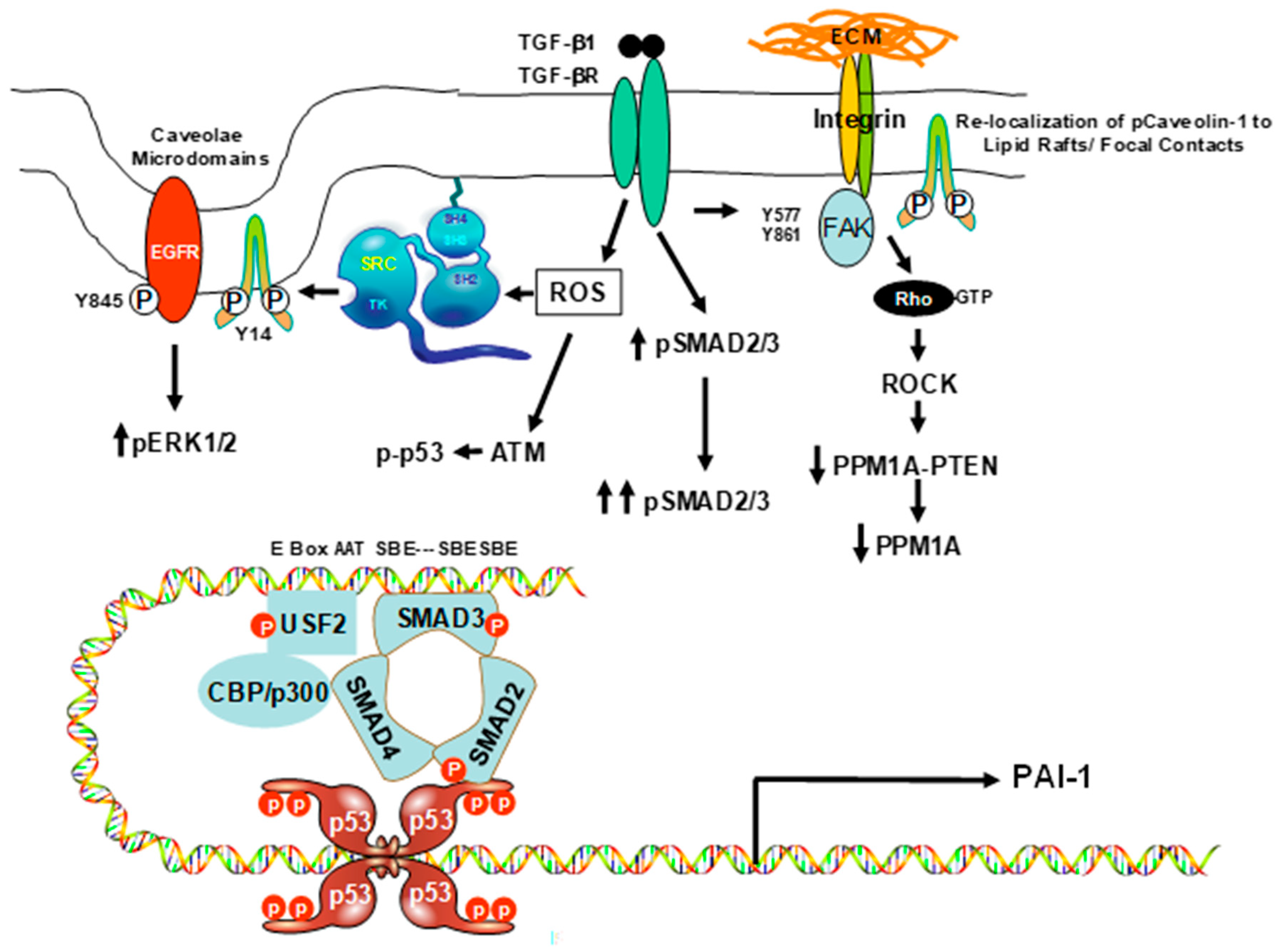
3. Caveolin-1 Signaling Is Required for Expression of the Senescence-Inducing p53-Target PAI-1 Gene in VSMCs
4. Mechanisms of Caveolin-1 Signaling
5. Controversies and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Naylor, R.M.; Baker, D.J.; van Deursen, J.M. Senescent cells: A novel therapeutic target for aging and age-related diseases. Clin. Pharmacol. Ther. 2013, 93, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Li, H.; Van Deursen, J.M. Senescent cells: A therapeutic target for cardiovascular disease. J. Clin. Investig. 2018, 128, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Li, D.-J.; Jiang, Y.-J.; Tong, J.; Fu, H.; Wu, Y.-H.; Shen, F.-M. Vascular smooth muscle cell senescence and age-related diseases: State of the art. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1810–1821. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Niño, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.J.; Roks, A.J.M. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Halazonetis, T.D. Oncogene-induced senescence: The bright and dark side of the response. Curr. Opin. Cell Boil. 2010, 22, 816–827. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Ermolaeva, M.; Neri, F.; Ori, A.; Rudolph, K.L. Cellular and epigenetic drivers of stem cell ageing. Nat. Rev. Mol. Cell Boil. 2018, 19, 594–610. [Google Scholar] [CrossRef]
- Adams, P.D. Healing and Hurting: Molecular Mechanisms, Functions, and Pathologies of Cellular Senescence. Mol. Cell 2009, 36, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Lan, Y.; Li, C.Y.; Sidoli, S.; Donahue, G.; Dou, Z.; Frederick, B.; Chen, Q.; Luense, L.J.; Garcia, B.A.; et al. Histone Acetyltransferase p300 Induces De Novo Super-Enhancers to Drive Cellular Senescence. Mol. Cell 2019, 73, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Gorenne, I.; Kavurma, M.; Scott, S.; Bennett, M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc. Res. 2006, 72, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uryga, A.K.; Bennett, M.R. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J. Physiol. 2016, 594, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.C.T.; Cho, K.A. Versatile Functions of Caveolin-1 in Aging-related Diseases. Chonnam Med. J. 2017, 53, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Samarakoon, R.; Higgins, C.E.; Higgins, S.P.; Kutz, S.M.; Higgins, P.J. Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-β1-stimulated smooth muscle cells is pp60c-src/MEK-dependent. J. Cell. Physiol. 2005, 204, 236–246. [Google Scholar] [CrossRef]
- Matsui, T.; Higashimoto, Y.; Taira, J.; Yamagishi, S.-I. Pigment epithelium-derived factor (PEDF) binds to caveolin-1 and inhibits the pro-inflammatory effects of caveolin-1 in endothelial cells. Biochem. Biophys. Res. Commun. 2013, 441, 405–410. [Google Scholar] [CrossRef]
- Otsuka, G.; Agah, R.; Frutkin, A.D.; Wight, T.A.; Dichek, D.A. TGF-β1 induces neointima formation through PAI-1 dependent pathways. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar]
- Burton, D.G.; Giles, P.J.; Sheerin, A.N.; Smith, S.K.; Lawton, J.J.; Ostler, E.L.; Rhys-Williams, W.; Kipling, D.; Faragher, R.G. Microarray analysis of senescent vascular smooth muscle cells: A link to atherosclerosis and vascular calcification. Exp. Gerontol. 2009, 44, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.; Matsubara, H.; Ikeda, K.; Burton, D. Pathophysiology of vascular calcification: Pivotal role of cellular senescence in vascular smooth muscle cells. Exp. Gerontol. 2010, 45, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Stoppani, E.; Volonte, D.; Galbiati, F. Caveolin-1, cellular senescence and age-related diseases. Mech. Ageing Dev. 2011, 132, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Volonte, D.; Zhang, K.; Lisanti, M.P.; Galbiati, F. Expression of Caveolin-1 Induces Premature Cellular Senescence in Primary Cultures of Murine Fibroblasts. Mol. Boil. Cell 2002, 13, 2502–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.-P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological Adjustment of Senescent Cells by Modulating Caveolin-1 Status. J. Boil. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, F.; Volonte’, D.; Liu, J.; Capozza, F.; Frank, P.G.; Zhu, L.; Pestell, R.G.; Lisanti, M.P. Caveolin-1 Expression Negatively Regulates Cell Cycle Progression by Inducing G0/G1 Arrest via a p53/p21WAF1/Cip1-dependent Mechanism. Mol. Boil. Cell 2001, 12, 2229–2244. [Google Scholar] [CrossRef]
- Volonte, D.; Galbiati, F. Polymerase I and Transcript Release Factor (PTRF)/Cavin-1 Is a Novel Regulator of Stress-induced Premature Senescence. J. Boil. Chem. 2011, 286, 28657–28661. [Google Scholar] [CrossRef] [Green Version]
- Overstreet, J.M.; Samarakoon, R.; Meldrum, K.K.; Higgins, P.J. Redox control pf p53 in the transcriptional regulation of TGF-β1 target genes through SMAD cooperativity. Cell Signal. 2014, 26, 1427–1436. [Google Scholar] [CrossRef]
- Bartholomew, J.N.; Volonte, D.; Galbiati, F. Caveolin-1 regulates the antagonistic pleiotropic properties of cellular senescence through a novel Mdm2/p53-mediated pathway. Cancer Res. 2009, 69, 2878–2886. [Google Scholar] [CrossRef]
- Bitar, M.S.; Abdel-Halim, S.M.; Al-Mulla, F. Caveolin-1/PTRF upregulation constitutes a mechanism for mediating p53-induced cellular senescence: Implications for evidence-based therapy of delayed wound healing in diabetes. Am. J. Physiol. Metab. 2013, 305, E951–E963. [Google Scholar] [CrossRef]
- Ovadya, Y.; Krizhanovsky, V. Strategies targeting cellular senescence. J. Clin. Investig. 2018, 128, 1247–1254. [Google Scholar] [CrossRef]
- Miao, S.-B.; Xie, X.-L.; Yin, Y.-J.; Zhao, L.-L.; Zhang, F.; Shu, Y.-N.; Chen, R.; Chen, P.; Dong, L.-H.; Lin, Y.-L.; et al. Accumulation of Smooth Muscle 22α Protein Accelerates Senescence of Vascular Smooth Muscle Cells via Stabilization of p53 In Vitro and In Vivo. Arter. Thromb. Vasc. Boil. 2017, 37, 1849–1859. [Google Scholar] [CrossRef]
- Vaughan, D.E. PAI-1 and TGF-β: Unmasking the real driver of TGF-β-induced vascular pathology. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 679–680. [Google Scholar] [CrossRef]
- Weisberg, A.D.; Albornoz, F.; Griffin, J.P.; Crandall, D.L.; Elokdah, H.; Fogo, A.B.; Vaughan, D.E.; Brown, N.J. Pharmacological inhibition and genetic deficiency of PAI-1 attenuates angiotensin II/salt-induced aortic remodeling. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 365–371. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen activator inhibitor-1 is a marker and mediator of senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Singh, N.N.; Ramji, D.P. The role of TGF-β in atherosclerosis. Cytokine Growth Factor Rev. 2006, 17, 487–499. [Google Scholar] [CrossRef]
- Otsuka, G.; Stempien-Otero, A.; Frutkin, A.D.; Dichek, D.A. Mechanisms of TGF-β1-induced intimal growth: Plasminogen-independent activities of plasminogen activator inhibitor-1 and heterogenous origin of intimal cells. Circ. Res. 2007, 100, 1300–1307. [Google Scholar] [CrossRef]
- Kwon, I.-S.; Kim, J.; Rhee, D.-K.; Kim, B.-O.; Pyo, S. Pneumolysin induces cellular senescence by increasing ROS production and activation of MAPK/NF-κB signal pathway in glial cells. Toxicon 2017, 129, 100–112. [Google Scholar] [CrossRef]
- You, W.; Hong, Y.; He, H.; Huang, X.; Tao, W.; Liang, X.; Zhang, Y.; Li, X. TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging 2019, 11, 3574–3584. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nature 2006, 8, 877–884. [Google Scholar] [CrossRef]
- Hiebert, P.; Wietecha, M.S.; Cangkrama, M.; Haertel, E.; Mavrogonatou, E.; Stumpe, M.; Steenbock, H.; Grossi, S.; Beer, H.-D.; Angel, P.; et al. Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome. Dev. Cell 2018, 46, 145–161. [Google Scholar] [CrossRef]
- Özcan, S.; Alessio, N.; Acar, M.B.; Mert, E.; Omerli, F.; Peluso, G.; Galderisi, U. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging 2016, 8, 1316–1327. [Google Scholar] [CrossRef]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.-A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362. [Google Scholar] [CrossRef]
- Khan, S.S.; Shah, S.J.; Klyachko, E.; Baldridge, A.S.; Eren, M.; Place, A.T.; Aviv, A.; Puterman, E.; Lloyd-Jones, D.M.; Heiman, M.; et al. A null mutation in SERPINE1 protects against biological aging in humans. Sci. Adv. 2017, 3, eaao1617. [Google Scholar] [CrossRef]
- Seo, J.Y.; Park, J.; Yu, M.R.; Kim, Y.S.; Ha, H.; Lee, H.B. Positive feedback loop between plasminogen activator inhibitor-1 and transforming growth factor-β1 during renal fibrosis in diabetes. Am. J. Nephrol. 2009, 30, 481–490. [Google Scholar] [CrossRef]
- Higgins, P.J.; Mu, X.-C.; Mu, X. Differential growth state-dependent regulation of plasminogen activator inhibitor type-1 expression in senescent IMR-90 human diploid fibroblasts. J. Cell. Physiol. 1995, 165, 647–657. [Google Scholar]
- Mu, X.-C.; Staiano-Coico, L.; Higgins, P.J. Increased transcription and modified growth state-dependent expression of the plasminogen activator inhibitor type-1 gene characterize the senescent phenotype in human diploid fibroblasts. J. Cell. Physiol. 1998, 174, 90–98. [Google Scholar] [CrossRef]
- Samarakoon, R.; Chitnis, S.S.; Higgins, S.P.; Higgins, C.E.; Krepinsky, J.C.; Higgins, P.J. Redox-initiated Src kinase and caveolin-1 signaling in TGF-β1-initiated SMAD2/3 activation and PAI-1 expression. PLoS ONE 2011, 6, e22896. [Google Scholar] [CrossRef]
- Samarakoon, R.; Dobberfuhl, A.D.; Cooley, C.; Overstreet, J.M.; Patel, S.; Goldschmeding, R.; Meldrum, K.K.; Higgins, P.J. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013, 25, 2198–2209. [Google Scholar] [CrossRef]
- Samarakoon, R.; Higgins, S.; Higgins, C.E.; Higgins, P.J. Cooperative Rho/Rock and EGFR signaling in modulating TGF-β1-induced PAI-1 expression in vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2008, 44, 527–538. [Google Scholar] [CrossRef]
- Samarakoon, R.; Higgins, P.J. Integration of non-SMAD and SMAD signaling in TGF-β1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb. Haemost. 2009, 100, 976–983. [Google Scholar] [CrossRef]
- Samarakoon, R.; Goppelt-Struebe, M.; Higgins, P.J. Linking Cell Structure to Gene Regulation: Signaling Events and Expression Controls on the Model Genes PAI-1 and CTGF. Cell Signal. 2010, 22, 1413–1419. [Google Scholar] [CrossRef]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nature 2003, 5, 410–421. [Google Scholar] [CrossRef]
- Gvaramia, D.; Blaauboer, M.E.; Hanemaaijer, R.; Everts, V. Role of caveolin-1 in fibrotic diseases. Matrix Boil. 2013, 32, 307–315. [Google Scholar] [CrossRef]
- Yu, D.; Jung, S.H.; An, H.; Lee, S.; Hong, J.; Park, J.S.; Lee, H.; Lee, H.; Bahn, M.; Lee, H.C.; et al. Caveolin-1 deficiency induces premature senescence with mitochondrial dysfunction. Aging Cell 2017, 16, 773–784. [Google Scholar] [CrossRef]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.-P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrosis. Cell Signal. 2018, 43, 1–10. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chu, C.; Lin, A.; Jo, H.; Ikezu, T.; Okamoto, T.; Kohtz, D.; Lisanti, M.P. Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. FEBS Lett. 1998, 428, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Schlegel, A.; Scherer, P.E.; Lisanti, M.P. Caveolins, a Family of Scaffolding Proteins for Organizing “Preassembled Signaling Complexes” at the Plasma Membrane. J. Boil. Chem. 1998, 273, 5419–5422. [Google Scholar] [CrossRef]
- Mayoral, R.; Valverde, A.M.; Llorente Izquierdo, C.; Gonzalez-Rodriguez, A.; Bosca, L.; Martin-Sanz, P. Impairment of TGF-β signaling in caveolin-1 deficient hepatocytes: Role in liver regeneration. J. Biol. Chem. 2010, 285, 3633–3642. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signaling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Dijke, P.T.; Hill, C.S. New insights into TGF-β–Smad signalling. Trends Biochem. Sci. 2004, 29, 265–273. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Li, S.C.; Kabeer, M.H. Spatiotemporal switching signals for cancer stem cell activation in pediatric origins of adulthood cancer: Towards a watch-and-wait lifetime strategy for cancer treatment. World J. Stem Cells 2018, 10, 15–22. [Google Scholar] [CrossRef]
- Moreno-Caceres, J.; Caballero-Diaz, D.; Nwosu, Z.C.; Meyer, C.; Lopez-Luque, J.; Malfettone, A.; Lastra, R.; Serrano, T.; Ramos, E.; Dooley, S.; et al. The level of caveolin-1 expression determines response to TGF-β as a tumor suppressor in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e3098. [Google Scholar] [CrossRef]
- Gottlieb-Abraham, E.; Shvartsman, D.E.; Donaldson, J.C.; Ehrlich, M.; Gutman, O.; Martin, G.S.; Henis, Y. Src-mediated caveolin-1 phosphorylation affects the targeting if active Src to specific membrane sites. Mol. Biol. Cell 2013, 24, 3881–3895. [Google Scholar]
- Li, S.; Seitz, R.; Lisanti, M.P. Phosphorylation of Caveolin by Src Tyrosine Kinases. J. Boil. Chem. 1996, 271, 3863–3868. [Google Scholar] [CrossRef] [Green Version]
- Couet, J.; Sargiacomo, M.; Lisanti, M.P. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J. Boil. Chem. 1997, 272, 30429–30438. [Google Scholar] [CrossRef]
- Wang, Y.; Roche, O.; Xu, C.; Moriyama, E.H.; Heir, P.; Chung, J.; Roos, F.C.; Chen, Y.; Finak, G.; Milosevic, M.; et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor–mediated upregulation of caveolin-1. Proc. Natl. Acad. Sci. USA 2012, 109, 4892–4897. [Google Scholar] [CrossRef]
- Boscher, C.; Nabi, I.R. Galectin-3- and phospho-caveolin-1-dependent outside-in integrin signaling mediates the EGF motogenic response in mammary cancer cells. Mol. Boil. Cell 2013, 24, 2134–2145. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, L.; Chen, W.; Xu, J.; Zhang, R.; Liu, R.; Zhou, L.; Hu, W.; Ju, R.; Lee, C.; et al. Inhibitory effect of caveolin-1 in vascular endothelial cells, pericytes and smooth muscle cells. Oncotarget 2017, 8, 76165–76173. [Google Scholar] [CrossRef] [Green Version]
- Overmiller, A.M.; McGuinn, K.P.; Roberts, B.J.; Cooper, F.; Brennan-Crispi, D.M.; Deguchi, T.; Peltonen, S.; Wahl, J.K.; Machoney, M.G. c-Src/Cav1-dependent activation of the EGFR by Dsg2. Oncotarget 2016, 7, 37536–37555. [Google Scholar] [CrossRef]
- Moreno-Càceres, J.; Caja, L.; Mainez, J.; Mayoral, R.; Martín-Sanz, P.; Moreno-Vicente, R.; Del Pozo, M.A.; Dooley, S.; Egea, G.; Fabregat, I. Caveolin-1 is required for TGF- β -induced transactivation of the EGF receptor pathway in hepatocytes through the activation of the metalloprotease TACE/ADAM17. Cell Death Dis. 2014, 5, e1326. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, T.; Zhao, R.; Gao, D.; Cui, Y.; Wang, K.; Guo, Y. Caveolin-1 Inhibits Proliferation, Migration, and Invasion of Human Colorectal Cancer Cells by Suppressing Phosphorylation of Epidermal Growth Factor Receptor. Med. Sci. Monit. 2018, 24, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Hassan, G.S.; Williams, T.M.; Frank, P.G.; Lisanti, M.P. Caveolin-1-deficient aortic smooth muscle cells show autonomous abnormalities in proliferation, migration, and endothelian-based signal transduction. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2393–H2401. [Google Scholar] [CrossRef]
- Peterson, T.E.; Guicciardi, M.E.; Gulati, R.; Kleppe, L.S.; Mueske, C.S.; Mookadam, M.; Sowa, G.; Gores, G.J.; Sessa, W.C.; Simari, R.D. Caveolin-1 Can Regulate Vascular Smooth Muscle Cell Fate by Switching Platelet-Derived Growth Factor Signaling from a Proliferative to an Apoptotic Pathway. Arter. Thromb. Vasc. Boil. 2003, 23, 1521–1527. [Google Scholar] [CrossRef]
- Kim, S.R.; Jiang, K.; Ogrodnik, M.; Chen, X.; Zhu, X.-Y.; Lohmeier, H.; Ahmed, L.; Tang, H.; Tchkonia, T.; Hickson, L.J.; et al. Increased renal cellular senescence in murine high-fat diet: Effect of the senolytic drug quercetin. Transl. Res. 2019. accepted for publication. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; DeMaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef]
- Kohli, J.; Campisi, J.; Demaria, M. A novel suicide gene therapy for the treatment of p16Ink41-overexpressing tumors. Oncotarget 2018, 9, 7274–7281. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. The TGF-β1/p53/PAI-1 Signaling Axis in Vascular Senescence: Role of Caveolin-1. Biomolecules 2019, 9, 341. https://doi.org/10.3390/biom9080341
Samarakoon R, Higgins SP, Higgins CE, Higgins PJ. The TGF-β1/p53/PAI-1 Signaling Axis in Vascular Senescence: Role of Caveolin-1. Biomolecules. 2019; 9(8):341. https://doi.org/10.3390/biom9080341
Chicago/Turabian StyleSamarakoon, Rohan, Stephen P. Higgins, Craig E. Higgins, and Paul J. Higgins. 2019. "The TGF-β1/p53/PAI-1 Signaling Axis in Vascular Senescence: Role of Caveolin-1" Biomolecules 9, no. 8: 341. https://doi.org/10.3390/biom9080341
APA StyleSamarakoon, R., Higgins, S. P., Higgins, C. E., & Higgins, P. J. (2019). The TGF-β1/p53/PAI-1 Signaling Axis in Vascular Senescence: Role of Caveolin-1. Biomolecules, 9(8), 341. https://doi.org/10.3390/biom9080341