

The Role of Primary Cilia-Associated Phosphoinositide Signaling in Development


Abstract
:1. Introduction
2. Primary Cilia
2.1. Structure of Primary Cilia
2.2. Ciliopathy
3. Cilium-Associated Phosphoinositides and PI-Binding Ciliary Proteins
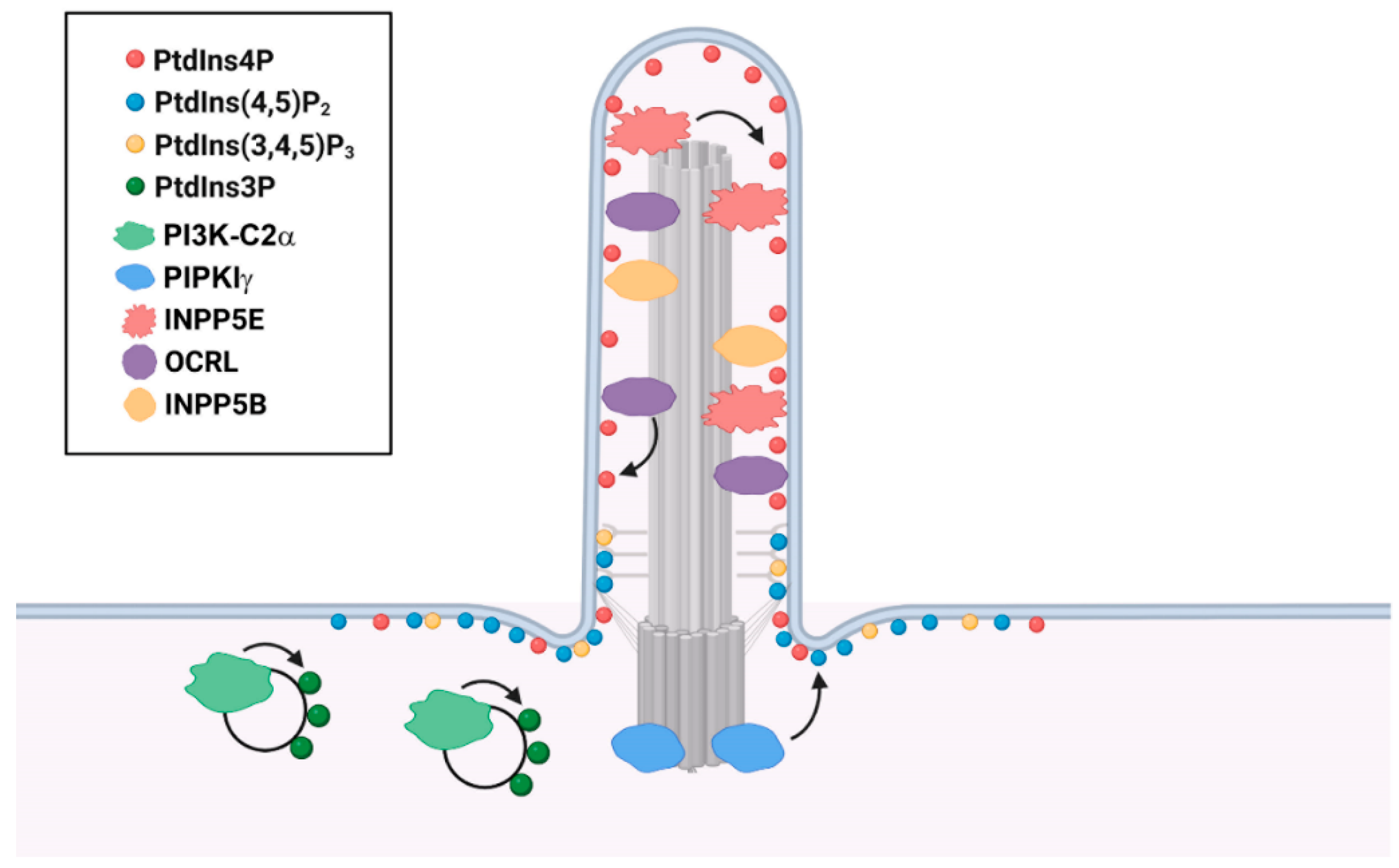
3.1. Cilium-Associated Phosphoinositides
3.2. PI-Binding Ciliary Proteins
4. Primary Cilium-Associated PI Phosphatases
4.1. INPP5E
4.2. OCRL and INPP5B
5. Primary Cilium-Associated PI Kinases
5.1. PI3K-C2α
5.2. PIPKIγ
6. Unanswered Questions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Schink, K.O.; Tan, K.W.; Stenmark, H. Phosphoinositides in Control of Membrane Dynamics. Annu. Rev. Cell Dev. Biol. 2016, 32, 143–171. [Google Scholar] [CrossRef] [PubMed]
- Dickson, E.J.; Hille, B. Understanding phosphoinositides: Rare, dynamic, and essential membrane phospholipids. Biochem. J. 2019, 476, 1–23. [Google Scholar] [CrossRef] [PubMed]
- De Craene, J.O.; Bertazzi, D.L.; Bar, S.; Friant, S. Phosphoinositides, Major Actors in Membrane Trafficking and Lipid Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 634. [Google Scholar] [CrossRef] [Green Version]
- Posor, Y.; Jang, W.; Haucke, V. Phosphoinositides as membrane organizers. Nat. Rev. Mol. Cell Biol. 2022, 23, 797–816. [Google Scholar] [CrossRef]
- Conduit, S.E.; Vanhaesebroeck, B. Phosphoinositide lipids in primary cilia biology. Biochem. J. 2020, 477, 3541–3565. [Google Scholar] [CrossRef]
- Hammond, G.R.V.; Burke, J.E. Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr. Opin. Cell Biol. 2020, 63, 57–67. [Google Scholar] [CrossRef]
- Kutateladze, T.G. Translation of the phosphoinositide code by PI effectors. Nat. Chem. Biol. 2010, 6, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, G.R.; Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta 2015, 1851, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Cullen, P.J.; Cozier, G.E.; Banting, G.; Mellor, H. Modular phosphoinositide-binding domains—Their role in signalling and membrane trafficking. Curr. Biol. 2001, 11, R882–R893. [Google Scholar] [CrossRef]
- Lemmon, M.A. Phosphoinositide recognition domains. Traffic 2003, 4, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Stenmark, H. Analyzing phosphoinositides and their interacting proteins. Nat. Methods 2006, 3, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, R.G.; Mazloumi Gavgani, F.; Edson, A.J.; Goris, M.; Altankhuyag, A.; Lewis, A.E. Polyphosphoinositides in the nucleus: Roadmap of their effectors and mechanisms of interaction. Adv. Biol. Regul. 2019, 72, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Staiano, L.; De Leo, M.G.; Persico, M.; De Matteis, M.A. Mendelian disorders of PI metabolizing enzymes. Biochim. Biophys. Acta 2015, 1851, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bankaitis, V.A. Phosphoinositide phosphatases in cell biology and disease. Prog. Lipid Res. 2010, 49, 201–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Dickson, E.J. Recent advances in understanding phosphoinositide signaling in the nervous system. F1000Res 2019, 8, F1000 Faculty Rev-278. [Google Scholar] [CrossRef] [Green Version]
- Krajnik, A.; Brazzo, J.A., 3rd; Vaidyanathan, K.; Das, T.; Redondo-Munoz, J.; Bae, Y. Phosphoinositide Signaling and Mechanotransduction in Cardiovascular Biology and Disease. Front. Cell Dev. Biol. 2020, 8, 595849. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [Green Version]
- Plotnikova, O.V.; Pugacheva, E.N.; Golemis, E.A. Primary cilia and the cell cycle. Methods Cell Biol. 2009, 94, 137–160. [Google Scholar] [CrossRef]
- Dutta, P.; Ray, K. Ciliary membrane, localised lipid modification and cilia function. J. Cell Physiol. 2022, 237, 2613–2631. [Google Scholar] [CrossRef] [PubMed]
- Bielas, S.L.; Silhavy, J.L.; Brancati, F.; Kisseleva, M.V.; Al-Gazali, L.; Sztriha, L.; Bayoumi, R.A.; Zaki, M.S.; Abdel-Aleem, A.; Rosti, R.O.; et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009, 41, 1032–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacoby, M.; Cox, J.J.; Gayral, S.; Hampshire, D.J.; Ayub, M.; Blockmans, M.; Pernot, E.; Kisseleva, M.V.; Compere, P.; Schiffmann, S.N.; et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009, 41, 1027–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, J.M.; Conduit, S.E.; Feeney, S.J.; Hakim, S.; DiTommaso, T.; Fulcher, A.J.; Sriratana, A.; Ramm, G.; Horan, K.A.; Gurung, R.; et al. INPP5E regulates phosphoinositide-dependent cilia transition zone function. J. Cell Biol. 2017, 216, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Conduit, S.E.; Ramaswamy, V.; Remke, M.; Watkins, D.N.; Wainwright, B.J.; Taylor, M.D.; Mitchell, C.A.; Dyson, J.M. A compartmentalized phosphoinositide signaling axis at cilia is regulated by INPP5E to maintain cilia and promote Sonic Hedgehog medulloblastoma. Oncogene 2017, 36, 5969–5984. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Y.; Wei, Q.; Huang, Y.; Hu, J.; Ling, K. Phosphatidylinositol phosphate kinase PIPKIgamma and phosphatase INPP5E coordinate initiation of ciliogenesis. Nat. Commun. 2016, 7, 10777. [Google Scholar] [CrossRef] [Green Version]
- Chavez, M.; Ena, S.; Van Sande, J.; de Kerchove d’Exaerde, A.; Schurmans, S.; Schiffmann, S.N. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Dev. Cell 2015, 34, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gonzalo, F.R.; Phua, S.C.; Roberson, E.C.; Garcia, G., 3rd; Abedin, M.; Schurmans, S.; Inoue, T.; Reiter, J.F. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev. Cell 2015, 34, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Tang, J.; Li, T.; Zhou, J.; Pan, W. INPP5E and Coordination of Signaling Networks in Cilia. Front. Mol. BioSci. 2022, 9, 885592. [Google Scholar] [CrossRef]
- Nakatsu, F. A Phosphoinositide Code for Primary Cilia. Dev. Cell 2015, 34, 379–380. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linck, R.W.; Chemes, H.; Albertini, D.F. The axoneme: The propulsive engine of spermatozoa and cilia and associated ciliopathies leading to infertility. J. Assist. Reprod. Genet. 2016, 33, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Satir, P. CILIA: Before and after. Cilia 2017, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, J.; Pelletier, L. The Ciliary Transition Zone: Finding the Pieces and Assembling the Gate. Mol. Cells 2017, 40, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Ling, K.; Hu, J. The essential roles of transition fibers in the context of cilia. Curr. Opin. Cell Biol. 2015, 35, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gonzalo, F.R.; Reiter, J.F. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Csh Perspect. Biol. 2017, 9, a028134. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gonzalo, F.R.; Reiter, J.F. Scoring a backstage pass: Mechanisms of ciliogenesis and ciliary access. J. Cell Biol. 2012, 197, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Nachury, M.V. How do cilia organize signalling cascades? Philos. Trans. R. Soc. Lond B Biol. Sci. 2014, 369, 20130465. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H. Involvement of Wnt signaling in primary cilia assembly and disassembly. Febs. J. 2020, 287, 5027–5038. [Google Scholar] [CrossRef]
- Wallingford, J.B. Planar cell polarity signaling, cilia and polarized ciliary beating. Curr. Opin. Cell Biol. 2010, 22, 597–604. [Google Scholar] [CrossRef]
- Schou, K.B.; Pedersen, L.B.; Christensen, S.T. Ins and outs of GPCR signaling in primary cilia. Embo. Rep. 2015, 16, 1099–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, S.T.; Clement, C.A.; Satir, P.; Pedersen, L.B. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J. Pathol. 2012, 226, 172–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, V.; Norris, D.P. Cilia and development. Curr. Opin. Genet. Dev. 2019, 56, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Ferkol, T.W.; Leigh, M.W. Ciliopathies: The central role of cilia in a spectrum of pediatric disorders. J. Pediatr. 2012, 160, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, R.J.; Tobin, J.L.; Beales, P.L. Modeling ciliopathies: Primary cilia in development and disease. Curr. Top Dev. Biol. 2008, 84, 249–310. [Google Scholar] [CrossRef]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef]
- Focsa, I.O.; Budisteanu, M.; Balgradean, M. Clinical and genetic heterogeneity of primary ciliopathies (Review). Int. J. Mol. Med. 2021, 48, 176. [Google Scholar] [CrossRef]
- Powles-Glover, N. Cilia and ciliopathies: Classic examples linking phenotype and genotype-an overview. Reprod. Toxicol. 2014, 48, 98–105. [Google Scholar] [CrossRef]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Csh Perspect. Biol. 2017, 9, a028191. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Wheway, G.; Mitchison, H.M.; Genomics England Research, C. Opportunities and Challenges for Molecular Understanding of Ciliopathies-The 100,000 Genomes Project. Front. Genet. 2019, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conduit, S.E.; Davies, E.M.; Fulcher, A.J.; Oorschot, V.; Mitchell, C.A. Superresolution Microscopy Reveals Distinct Phosphoinositide Subdomains Within the Cilia Transition Zone. Front. Cell Dev. Biol. 2021, 9, 634649. [Google Scholar] [CrossRef] [PubMed]
- Plotnikova, O.V.; Seo, S.; Cottle, D.L.; Conduit, S.; Hakim, S.; Dyson, J.M.; Mitchell, C.A.; Smyth, I.M. INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J. Cell Sci. 2015, 128, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Phua, S.C.; Chiba, S.; Suzuki, M.; Su, E.; Roberson, E.C.; Pusapati, G.V.; Schurmans, S.; Setou, M.; Rohatgi, R.; Reiter, J.F.; et al. Dynamic Remodeling of Membrane Composition Drives Cell Cycle through Primary Cilia Excision. Cell 2017, 168, 264–279.e15. [Google Scholar] [CrossRef] [Green Version]
- Stilling, S.; Kalliakoudas, T.; Benninghoven-Frey, H.; Inoue, T.; Falkenburger, B.H. PIP2 determines length and stability of primary cilia by balancing membrane turnovers. Commun. Biol. 2022, 5, 93. [Google Scholar] [CrossRef]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-Dependent aurora a activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Franco, I.; Gulluni, F.; Campa, C.C.; Costa, C.; Margaria, J.P.; Ciraolo, E.; Martini, M.; Monteyne, D.; De Luca, E.; Germena, G.; et al. PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev. Cell 2014, 28, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Franco, I.; Margaria, J.P.; De Santis, M.C.; Ranghino, A.; Monteyne, D.; Chiaravalli, M.; Pema, M.; Campa, C.C.; Ratto, E.; Gulluni, F.; et al. Phosphoinositide 3-Kinase-C2alpha Regulates Polycystin-2 Ciliary Entry and Protects against Kidney Cyst Formation. J. Am. Soc. Nephrol. 2016, 27, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Otis, J.M.; Suciu, S.K.; Catalano, C.; Xing, L.; Constable, S.; Wachten, D.; Gupton, S.; Lee, J.; Lee, A.; et al. Primary Cilia Signaling Promotes Axonal Tract Development and Is Disrupted in Joubert Syndrome-Related Disorders Models. Dev. Cell 2019, 51, 759–774.e5. [Google Scholar] [CrossRef]
- Suizu, F.; Hirata, N.; Kimura, K.; Edamura, T.; Tanaka, T.; Ishigaki, S.; Donia, T.; Noguchi, H.; Iwanaga, T.; Noguchi, M. Phosphorylation-dependent Akt-Inversin interaction at the basal body of primary cilia. Embo J. 2016, 35, 1346–1363. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowdle, W.E.; Robinson, J.F.; Kneist, A.; Sirerol-Piquer, M.S.; Frints, S.G.; Corbit, K.C.; Zaghloul, N.A.; van Lijnschoten, G.; Mulders, L.; Verver, D.E.; et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am. J. Hum. Genet. 2011, 89, 94–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A. Pleckstrin homology (PH) domains and phosphoinositides. Biochem. Soc. Symp. 2007, 74, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Lenoir, M.; Kufareva, I.; Abagyan, R.; Overduin, M. Membrane and Protein Interactions of the Pleckstrin Homology Domain Superfamily. Membranes 2015, 5, 646–663. [Google Scholar] [CrossRef]
- Higginbotham, H.; Guo, J.M.; Yokota, Y.; Umberger, N.L.; Su, C.Y.; Li, J.J.; Verma, N.; Hirt, J.; Ghukasyan, V.; Caspary, T.; et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat. Neurosci. 2013, 16, 1000–1007. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Wen, X.; Chih, B.; Nelson, C.D.; Lane, W.S.; Scales, S.J.; Jackson, P.K. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010, 24, 2180–2193. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Wen, X.; Ratti, N.; Loktev, A.; Rangell, L.; Scales, S.J.; Jackson, P.K. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 2013, 152, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Miyoshi, K.; Shikada, S.; Amano, G.; Wang, Y.S.Z.M.; Yoshimura, T.; Katayama, T. TULP3 is required for localization of membrane-associated proteins ARL13B and INPP5E to primary cilia. Biochem. Biophys. Res. Commun. 2019, 509, 227–234. [Google Scholar] [CrossRef]
- Badgandi, H.B.; Hwang, S.H.; Shimada, I.S.; Loriot, E.; Mukhopadhyay, S. Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J. Cell Biol. 2017, 216, 743–760. [Google Scholar] [CrossRef]
- Tschaikner, P.; Enzler, F.; Torres-Quesada, O.; Aanstad, P.; Stefan, E. Hedgehog and Gpr161: Regulating cAMP Signaling in the Primary Cilium. Cells 2020, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remans, K.; Burger, M.; Vetter, I.R.; Wittinghofer, A. C2 Domains as Protein-Protein Interaction Modules in the Ciliary Transition Zone. Cell Rep. 2014, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, V.; Li, C.M.; Bowie, R.V.; Clarke, L.; Mohan, S.; Blacque, O.E.; Leroux, M.R. Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance. Embo J. 2015, 34, 2537–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Jensen, V.L.; Park, K.; Kennedy, J.; Garcia-Gonzalo, F.R.; Romani, M.; De Mori, R.; Bruel, A.L.; Gaillard, D.; Doray, B.; et al. MKS5 and CEP290 Dependent Assembly Pathway of the Ciliary Transition Zone. PLoS Biol. 2016, 14, e1002416. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Aravind, L. Identification of novel families and classification of the C2 domain superfamily elucidate the origin and evolution of membrane targeting activities in eukaryotes. Gene 2010, 469, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, M.; Kobayashi, T.; Chiba, S.; Takei, R.; Liang, L.X.X.; Nakayama, K.; Katoh, Y. Formation of the B9-domain protein complex MKS1-B9D2-B9D1 is essential as a diffusion barrier for ciliary membrane proteins. Mol. Biol. Cell 2020, 31, 2259–2268. [Google Scholar] [CrossRef]
- Humbert, M.C.; Weihbrecht, K.; Searby, C.C.; Li, Y.; Pope, R.M.; Sheffield, V.C.; Seo, S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. USA 2012, 109, 19691–19696. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.T.; Fujisawa, S.; Nozaki, S.; Katoh, Y.; Nakayama, K. Interaction of INPP5E with ARL13B is essential for its ciliary membrane retention but dispensable for its ciliary entry. Biol. Open 2021, 10, bio057653. [Google Scholar] [CrossRef]
- Cilleros-Rodriguez, D.; Martin-Morales, R.; Barbeito, P.; Deb Roy, A.; Loukil, A.; Sierra-Rodero, B.; Herranz, G.; Pampliega, O.; Redrejo-Rodriguez, M.; Goetz, S.C.; et al. Multiple ciliary localization signals control INPP5E ciliary targeting. eLife 2022, 11, e78383. [Google Scholar] [CrossRef]
- Shetty, M.; Ramdas, N.; Sahni, S.; Mullapudi, N.; Hegde, S. A Homozygous Missense Variant in INPP5E Associated with Joubert Syndrome and Related Disorders. Mol. SyndroMol. 2017, 8, 313–317. [Google Scholar] [CrossRef]
- Travaglini, L.; Brancati, F.; Silhavy, J.; Iannicelli, M.; Nickerson, E.; Elkhartoufi, N.; Scott, E.; Spencer, E.; Gabriel, S.; Thomas, S.; et al. Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders. Eur. J. Hum. Genet. 2013, 21, 1074–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conduit, S.E.; Dyson, J.M.; Mitchell, C.A. Inositol polyphosphate 5-phosphatases; new players in the regulation of cilia and ciliopathies. Febs. Lett. 2012, 586, 2846–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardee, I.; Soldatos, A.; Davids, M.; Vilboux, T.; Toro, C.; David, K.L.; Ferreira, C.R.; Nehrebecky, M.; Snow, J.; Thurm, A.; et al. Defective ciliogenesis in INPP5E-related Joubert syndrome. Am. J. Med. Genet. A 2017, 173, 3231–3237. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, J.G.; Keeler, L.C.; Parisi, M.A.; Marsh, S.E.; Chance, P.F.; Glass, I.A.; Graham, J.M.; Maria, B.L.; Barkovich, A.J.; Dobyns, W.B. Molar tooth sign of the midbrain-hindbrain junction: Occurrence in multiple distinct syndromes. Am. J. Med. Genet. A 2004, 125a, 125–134. [Google Scholar] [CrossRef]
- Brancati, F.; Dallapiccola, B.; Valente, E.M. Joubert Syndrome and related disorders. Orphanet. J. Rare Dis. 2010, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Tsurusaki, Y.; Kobayashi, Y.; Hisano, M.; Ito, S.; Doi, H.; Nakashima, M.; Saitsu, H.; Matsumoto, N.; Miyake, N. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J. Hum. Genet. 2015, 60, 651. [Google Scholar] [CrossRef] [Green Version]
- de Goede, C.; Yue, W.W.; Yan, G.; Ariyaratnam, S.; Chandler, K.E.; Downes, L.; Khan, N.; Mohan, M.; Lowe, M.; Banka, S. Role of reverse phenotyping in interpretation of next generation sequencing data and a review of INPP5E related disorders. Eur. J. Paediatr. Neuro. 2016, 20, 286–295. [Google Scholar] [CrossRef]
- Torkar, A.D.; Stefanija, M.A.; Bertok, S.; Podkrajsek, K.T.; Debeljak, M.; Kranjc, B.S.; Battelino, T.; Kotnik, P. Novel Insights Into Monogenic Obesity Syndrome Due to INPP5E Gene Variant: A Case Report of a Female Patient. Front. Endocrinol. 2021, 12, 581134. [Google Scholar] [CrossRef]
- Hampshire, D.J.; Ayub, M.; Springell, K.; Roberts, E.; Jafri, H.; Rashid, Y.; Bond, J.; Riley, J.H.; Woods, C.G. MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34. Eur. J. Hum. Genet. 2006, 14, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Hakim, S.; Dyson, J.M.; Feeney, S.J.; Davies, E.M.; Sriratana, A.; Koenig, M.N.; Plotnikova, O.V.; Smyth, I.M.; Ricardo, S.D.; Hobbs, R.M.; et al. Inpp5e suppresses polycystic kidney disease via inhibition of PI3K/Akt-dependent mTORC1 signaling. Hum. Mol. Genet. 2016, 25, 2295–2313. [Google Scholar] [CrossRef]
- Luo, N.; Lu, J.P.; Sun, Y. Evidence of a role of inositol polyphosphate 5-phosphatase INPP5E in cilia formation in zebrafish. Vision Res. 2012, 75, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constable, S.; Long, A.B.; Floyd, K.A.; Schurmans, S.; Caspary, T. The ciliary phosphatidylinositol phosphatase Inpp5e plays positive and negative regulatory roles in Shh signaling. Development 2020, 147, dev183301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhivanan, K.; Ramadesikan, S.; Aguilar, R.C. Role of Ocrl1 and Inpp5E in primary cilia assembly and maintenance: A phosphatidylinositol phosphatase relay system? Res. Rep. Biol. 2016, 7, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Coon, B.G.; Hernandez, V.; Madhivanan, K.; Mukherjee, D.; Hanna, C.B.; Ramirez, I.B.R.; Lowe, M.; Beales, P.L.; Aguilar, R.C. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum. Mol. Genet. 2012, 21, 1835–1847. [Google Scholar] [CrossRef] [Green Version]
- Luo, N.; West, C.C.; Murga-Zamalloa, C.A.; Sun, L.; Anderson, R.M.; Wells, C.D.; Weinreb, R.N.; Travers, J.B.; Khanna, H.; Sun, Y. OCRL localizes to the primary cilium: A new role for cilia in Lowe syndrome. Hum. Mol. Genet. 2012, 21, 3333–3344. [Google Scholar] [CrossRef] [Green Version]
- Montjean, R.; Aoidi, R.; Desbois, P.; Rucci, J.; Trichet, M.; Salomon, R.; Rendu, J.; Faure, J.; Lunardi, J.; Gacon, G.; et al. OCRL-mutated fibroblasts from patients with Dent-2 disease exhibit INPP5B-independent phenotypic variability relatively to Lowe syndrome cells. Hum. Mol. Genet. 2015, 24, 994–1006. [Google Scholar] [CrossRef] [Green Version]
- Prosseda, P.P.; Luo, N.; Wang, B.; Alvarado, J.A.; Hu, Y.; Sun, Y. Loss of OCRL increases ciliary PI(4,5)P2 in Lowe oculocerebrorenal syndrome. J. Cell Sci. 2017, 130, 3447–3454. [Google Scholar] [CrossRef] [Green Version]
- Rbaibi, Y.; Cui, S.; Mo, D.; Carattino, M.; Rohatgi, R.; Satlin, L.M.; Szalinski, C.M.; Swanhart, L.M.; Folsch, H.; Hukriede, N.A.; et al. OCRL1 modulates cilia length in renal epithelial cells. Traffic 2012, 13, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Hagemann, N.; Schoebel, S.; Blankenfeldt, W.; Goody, R.S.; Erdmann, K.S.; Itzen, A. A structural basis for Lowe syndrome caused by mutations in the Rab-binding domain of OCRL1. Embo J. 2011, 30, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Luo, N.; Kumar, A.; Conwell, M.; Weinreb, R.N.; Anderson, R.; Sun, Y. Compensatory Role of Inositol 5-Phosphatase INPP5B to OCRL in Primary Cilia Formation in Oculocerebrorenal Syndrome of Lowe. PLoS ONE 2013, 8, e66727. [Google Scholar] [CrossRef]
- Shrimpton, A.E.; Hoopes, R.R., Jr.; Knohl, S.J.; Hueber, P.; Reed, A.A.; Christie, P.T.; Igarashi, T.; Lee, P.; Lehman, A.; White, C.; et al. OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron. Physiol. 2009, 112, p27–p36. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, M.A.; Staiano, L.; Emma, F.; Devuyst, O. The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nat. Rev. Nephrol. 2017, 13, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Dumic, K.K.; Anticevic, D.; Petrinovic-Doresic, J.; Zigman, T.; Zarkovic, K.; Rokic, F.; Vugrek, O. Lowe syndrome—Old and new evidence of secondary mitochondrial dysfunction. Eur. J. Med. Genet. 2020, 63, 104022. [Google Scholar] [CrossRef]
- Gianesello, L.; Arroyo, J.; Del Prete, D.; Priante, G.; Ceol, M.; Harris, P.C.; Lieske, J.C.; Anglani, F. Genotype Phenotype Correlation in Dent Disease 2 and Review of the Literature: OCRL Gene Pleiotropism or Extreme Phenotypic Variability of Lowe Syndrome? Genes 2021, 12, 1597. [Google Scholar] [CrossRef]
- Mehta, Z.B.; Pietka, G.; Lowe, M. The Cellular and Physiological Functions of the Lowe Syndrome Protein OCRL1. Traffic 2014, 15, 471–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hichri, H.; Rendu, J.; Monnier, N.; Coutton, C.; Dorseuil, O.; Poussou, R.V.; Baujat, G.; Blanchard, A.; Nobili, F.; Ranchin, B.; et al. From Lowe syndrome to Dent disease: Correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. Mutat. 2011, 32, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janne, P.A.; Suchy, S.F.; Bernard, D.; MacDonald, M.; Crawley, J.; Grinberg, A.; Wynshaw-Boris, A.; Westphal, H.; Nussbaum, R.L. Functional overlap between murine Inpp5b and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J. Clin. Investig. 1998, 101, 2042–2053. [Google Scholar] [CrossRef] [Green Version]
- Bothwell, S.P.; Chan, E.; Bernardini, I.M.; Kuo, Y.M.; Gahl, W.A.; Nussbaum, R.L. Mouse model for Lowe syndrome/Dent Disease 2 renal tubulopathy. J. Am. Soc. Nephrol. 2011, 22, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Balkin, D.M.; Liu, L.; Nandez, R.; Wu, Y.; Tian, X.; Wang, T.; Nussbaum, R.; De Camilli, P.; Ishibe, S. Kidney Tubular Ablation of Ocrl/Inpp5b Phenocopies Lowe Syndrome Tubulopathy. J. Am. Soc. Nephrol. 2017, 28, 1399–1407. [Google Scholar] [CrossRef] [Green Version]
- Bothwell, S.P.; Farber, L.W.; Hoagland, A.; Nussbaum, R.L. Species-specific difference in expression and splice-site choice in Inpp5b, an inositol polyphosphate 5-phosphatase paralogous to the enzyme deficient in Lowe Syndrome. Mamm. Genome 2010, 21, 458–466. [Google Scholar] [CrossRef]
- Gaidarov, I.; Smith, M.E.K.; Domin, J.; Keen, J.H. The class II phosphoinositide 3-kinase C2 alpha is activated by clathrin and regulates clathrin-mediated membrane trafficking. Mol. Cell 2001, 7, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schoneberg, J.; Ullrich, A.; Lampe, A.; Muller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Marsland, R., III; Upadhyayula, S.; Song, E.; Dang, S.; Capraro, B.R.; Wang, W.; Skillern, W.; Gaudin, R.; Ma, M.; et al. Dynamics of phosphoinositide conversion in clathrin-mediated endocytic traffic. Nature 2017, 552, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Maffucci, T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem. J. 2012, 443, 587–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulluni, F.; Martini, M.; De Santis, M.C.; Campa, C.C.; Ghigo, A.; Margaria, J.P.; Ciraolo, E.; Franco, I.; Ala, U.; Annaratone, L.; et al. Mitotic Spindle Assembly and Genomic Stability in Breast Cancer Require PI3K-C2 alpha Scaffolding Function. Cancer Cell 2017, 32, 444–459.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, N.M.; Schipper, J.L.; Karnes, J.B.; Kauffman, A.L.; Martin, K.R.; MacKeigan, J.P. PI3K-C2alpha knockdown decreases autophagy and maturation of endocytic vesicles. PLoS ONE 2017, 12, e0184909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukhalfa, A.; Nascimbeni, A.C.; Ramel, D.; Dupont, N.; Hirsch, E.; Gayral, S.; Laffargue, M.; Codogno, P.; Morel, E. PI3KC2alpha-dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat. Commun. 2020, 11, 294. [Google Scholar] [CrossRef] [Green Version]
- Knodler, A.; Feng, S.S.; Zhang, J.; Zhang, X.Y.; Das, A.; Peranen, J.; Guo, W. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6346–6351. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K.; Yoshida, K.; Cui, H.; Wakayama, T.; Takuwa, N.; Okamoto, Y.; Du, W.; Qi, X.; Asanuma, K.; Sugihara, K.; et al. Endothelial PI3K-C2 alpha, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat. Med. 2012, 18, 1560–1569. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.M.; Ramalho-Santos, M.; McMahon, A.P. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell 2001, 105, 781–792. [Google Scholar] [CrossRef]
- Sasai, N.; Toriyama, M.; Kondo, T. Hedgehog Signal and Genetic Disorders. Front. Genet. 2019, 10, 1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiosano, D.; Baris, H.N.; Chen, A.; Hitzert, M.M.; Schueler, M.; Gulluni, F.; Wiesener, A.; Bergua, A.; Mory, A.; Copeland, B.; et al. Mutations in PIK3C2A cause syndromic short stature, skeletal abnormalities, and cataracts associated with ciliary dysfunction. PLoS Genet. 2019, 15, e1008088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Zhang, Y.; Xiong, X.; Huang, Y.; Salisbury, J.L.; Hu, J.; Ling, K. PIPKIgamma targets to the centrosome and restrains centriole duplication. J. Cell Sci. 2014, 127, 1293–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Xu, Q.; Zhang, Y.; Davies, B.A.; Huang, Y.; Katzmann, D.J.; Harris, P.C.; Hu, J.; Ling, K. Ciliopathy protein HYLS1 coordinates the biogenesis and signaling of primary cilia by activating the ciliary lipid kinase PIPKIgamma. Sci. Adv. 2021, 7, eabe3401. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.; Doughman, R.L.; Firestone, A.J.; Bunce, M.W.; Anderson, R.A. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 2002, 420, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.; Bairstow, S.F.; Carbonara, C.; Turbin, D.A.; Huntsman, D.G.; Anderson, R.A. Type I gamma phosphatidylinositol phosphate kinase modulates adherens junction and E-cadherin trafficking via a direct interaction with mu 1B adaptin. J. Cell Biol. 2007, 176, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Xu, Q.; Huang, Y.; Singh, R.D.; Anderson, R.; Leof, E.; Hu, J.; Ling, K. An association between type Igamma PI4P 5-kinase and Exo70 directs E-cadherin clustering and epithelial polarization. Mol. Biol. Cell 2012, 23, 87–98. [Google Scholar] [CrossRef]
- Schill, N.J.; Hedman, A.C.; Choi, S.; Anderson, R.A. Isoform 5 of PIPKIgamma regulates the endosomal trafficking and degradation of E-cadherin. J. Cell Sci. 2014, 127, 2189–2203. [Google Scholar] [CrossRef] [Green Version]
- Narkis, G.; Ofir, R.; Landau, D.; Manor, E.; Volokita, M.; Hershkowitz, R.; Elbedour, K.; Birk, O.S. Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKI gamma of the phophatidylinsitol pathway. Am. J. Hum. Genet. 2007, 81, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.P.; Brennan, K.; Scheidel, N.; McGettigan, P.; Lavin, P.T.; Carter, S.; Ennis, S.; Dorkins, H.; Ghali, N.; Blacque, O.E.; et al. Recessive NEK9 mutation causes a lethal skeletal dysplasia with evidence of cell cycle and ciliary defects. Hum. Mol. Genet. 2016, 25, 1824–1835. [Google Scholar] [CrossRef]
- Wang, Y.; Lian, L.; Golden, J.A.; Morrisey, E.E.; Abrams, C.S. PIP5KI gamma is required for cardiovascular and neuronal development. Proc. Natl. Acad. Sci. USA 2007, 104, 11748–11753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paolo, G.; Moskowitz, H.S.; Gipson, K.; Wenk, M.R.; Voronov, S.; Obayashi, M.; Flavell, R.; Fitzsimonds, R.M.; Ryan, T.A.; De Camilli, P. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 2004, 431, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Legate, K.R.; Montag, D.; Bottcher, R.T.; Takahashi, S.; Fassler, R. Comparative phenotypic analysis of the two major splice isoforms of phosphatidylinositol phosphate kinase type Igamma in vivo. J. Cell Sci. 2012, 125, 5636–5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shnitsar, I.; Bashkurov, M.; Masson, G.R.; Ogunjimi, A.A.; Mosessian, S.; Cabeza, E.A.; Hirsch, C.L.; Trcka, D.; Gish, G.; Jiao, J.; et al. PTEN regulates cilia through Dishevelled. Nat. Commun. 2015, 6, 8388. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Yu, P.; Li, J.; Cao, Y.; Zhang, J. Phosphatidylinositol 4-kinase beta is required for the ciliogenesis of zebrafish otic vesicle. J. Genet. Genom. 2020, 47, 627–636. [Google Scholar] [CrossRef]
- Hamze-Komaiha, O.; Sarr, S.; Arlot-Bonnemains, Y.; Samuel, D.; Gassama-Diagne, A. SHIP2 Regulates Lumen Generation, Cell Division, and Ciliogenesis through the Control of Basolateral to Apical Lumen Localization of Aurora A and HEF 1. Cell Rep. 2016, 17, 2738–2752. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Idevall-Hagren, O.; De Camilli, P. Detection and manipulation of phosphoinositides. Biochim. Biophys. Acta 2015, 1851, 736–745. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.R.; Schiavo, G.; Irvine, R.F. Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem. J. 2009, 422, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T.; Takatori, S.; Fujimoto, T. Definition of phosphoinositide distribution in the nanoscale. Curr. Opin. Cell Biol. 2019, 57, 33–39. [Google Scholar] [CrossRef]
- Hertel, F.; Li, S.; Chen, M.; Pott, L.; Mehta, S.; Zhang, J. Fluorescent Biosensors for Multiplexed Imaging of Phosphoinositide Dynamics. ACS Chem. Biol. 2020, 15, 33–38. [Google Scholar] [CrossRef]
- Chu, N.; Viennet, T.; Bae, H.; Salguero, A.; Boeszoermenyi, A.; Arthanari, H.; Cole, P.A. The structural determinants of PH domain-mediated regulation of Akt revealed by segmental labeling. eLife 2020, 9, e59151. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Streets, A.J.; Hounslow, A.M.; Tran, U.; Jean-Alphonse, F.; Needham, A.J.; Vilardaga, J.P.; Wessely, O.; Williamson, M.P.; Ong, A.C. The Polycystin-1, Lipoxygenase, and alpha-Toxin Domain Regulates Polycystin-1 Trafficking. J. Am. Soc. Nephrol. 2016, 27, 1159–1173. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Disease | Disease-Causal Mutations | Domain |
|---|---|---|
| Joubert Syndrome (INPP5E-associated) | R345S, R378C, T426N, R435Q, R435W, T436N, W474R, R512W, R515W, Y534D/R621Q, Y543X, G552A, R563H, K580E, V303M/R585C, Y588C | phosphatase domain |
| G286R, R621Q/L234Pfs*56, C641R | Others 1 | |
| MORM Syndrome (INPP5E-associated) | Q627X, Q633X | Others 1 |
| Lowe Syndrome (OCRL-associated) | F242S, W247C/X, W261C, Y272H, F276S, Q277R, Q295X, W297X, E302X, A328P, R334P/X, Y335X, R337C, G357E, R361I, T367del, V372G, N373Y, S374F, H414R, G421E, D422N, N424D, L448G, D451N/G, Q452R, R457G, K460X, F463S, E468K/G, P475H, R476W, Y477X, P478L, P495L, W497G/X, C498Y, D499H, R500G/Q/X, W503R, V508D, Y513C, S522R, H524R/Q, P526T/L, V527D, I533S | phosphatase domain |
| V777E, E585del, N597K, L634P, F668V, C679W, L687P | ASH domain | |
| I768N, A797P, P801L, A861T, L891R | RhoGAP domain | |
| Q215X | Others 1 | |
| Dent-2 disease (OCRL-associated) | C87X, L56DfsX1, Q70RfsX18, E85FfsX26, | PH domain |
| I257T, R301C, R301H, G304E, G321E, N354H, Y462C, R476W, P478L, Y479C, R493W | 5-phosphatase | |
| E737D, P799L | RhoGAP domain | |
| T121NfsX1, I147KfsX1, S149X, P161PfsX3, M170IfsX1, F226S | Others 1 | |
| Lowe’s syndrome and Dent 2 disease (OCRL-associated) | I274T, F287S, R318C/H, D523N/G | 5-phosphatase domain |
| Human Disease | Clinical Features |
|---|---|
| Joubert Syndrome (INPP5E-associated) | MTS (molar tooth sign), hypotonia, abnormal breathing, retinal dystrophy, cystic echogenic kidneys |
| MORM Syndrome (INPP5E-associated) | mental retardation, obesity, congenital retinal dystrophy, and micropenis in males |
| Lowe Syndrome (OCRL-associated) | mental retardation, congenital cataracts, maladaptive behavior, renal dysfunction, postnatal growth retardation, areflexia, and arthropathy |
| Dent-2 disease (OCRL-associated) | renal dysfunction, mild mental retardation, hypotonia, cataracts, and rickets |
| Human Disease | Disease-Causing Mutations |
|---|---|
| PI3K-C2α-associated | skeletal malformations, short stature, cataracts, secondary glaucoma, and developmental delay |
| LCCS3 (PIPKIγ-associated) | arthrogryposis with multiple joint contractures, muscle wasting, and atrophy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Hu, J.; Ling, K. The Role of Primary Cilia-Associated Phosphoinositide Signaling in Development. J. Dev. Biol. 2022, 10, 51. https://doi.org/10.3390/jdb10040051
Chen C, Hu J, Ling K. The Role of Primary Cilia-Associated Phosphoinositide Signaling in Development. Journal of Developmental Biology. 2022; 10(4):51. https://doi.org/10.3390/jdb10040051
Chicago/Turabian StyleChen, Chuan, Jinghua Hu, and Kun Ling. 2022. "The Role of Primary Cilia-Associated Phosphoinositide Signaling in Development" Journal of Developmental Biology 10, no. 4: 51. https://doi.org/10.3390/jdb10040051
APA StyleChen, C., Hu, J., & Ling, K. (2022). The Role of Primary Cilia-Associated Phosphoinositide Signaling in Development. Journal of Developmental Biology, 10(4), 51. https://doi.org/10.3390/jdb10040051