Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models


Abstract
:1. Introduction
2. Primary Cilium, Ciliopathy, and EVC
3. Overview of Human Signs
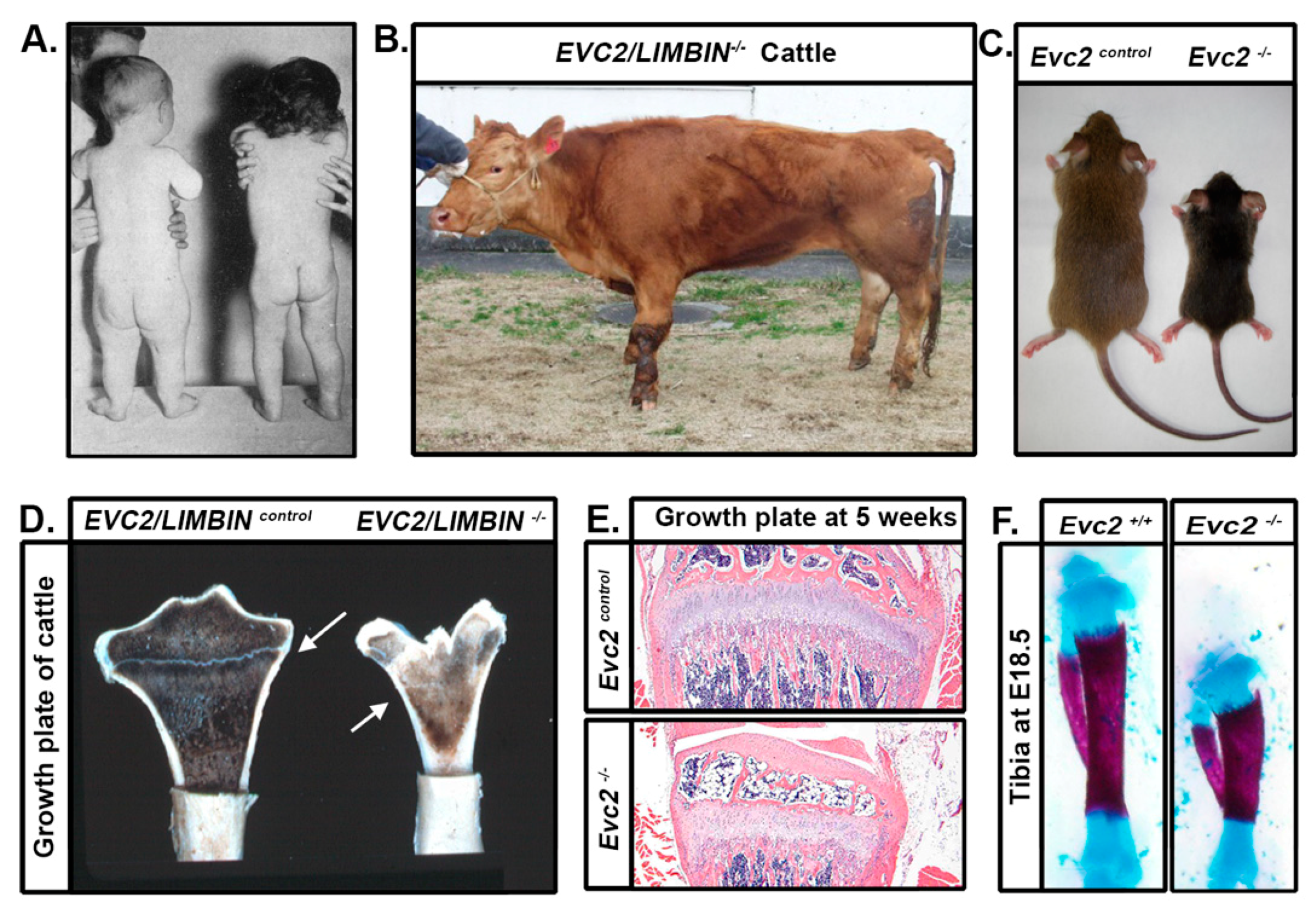
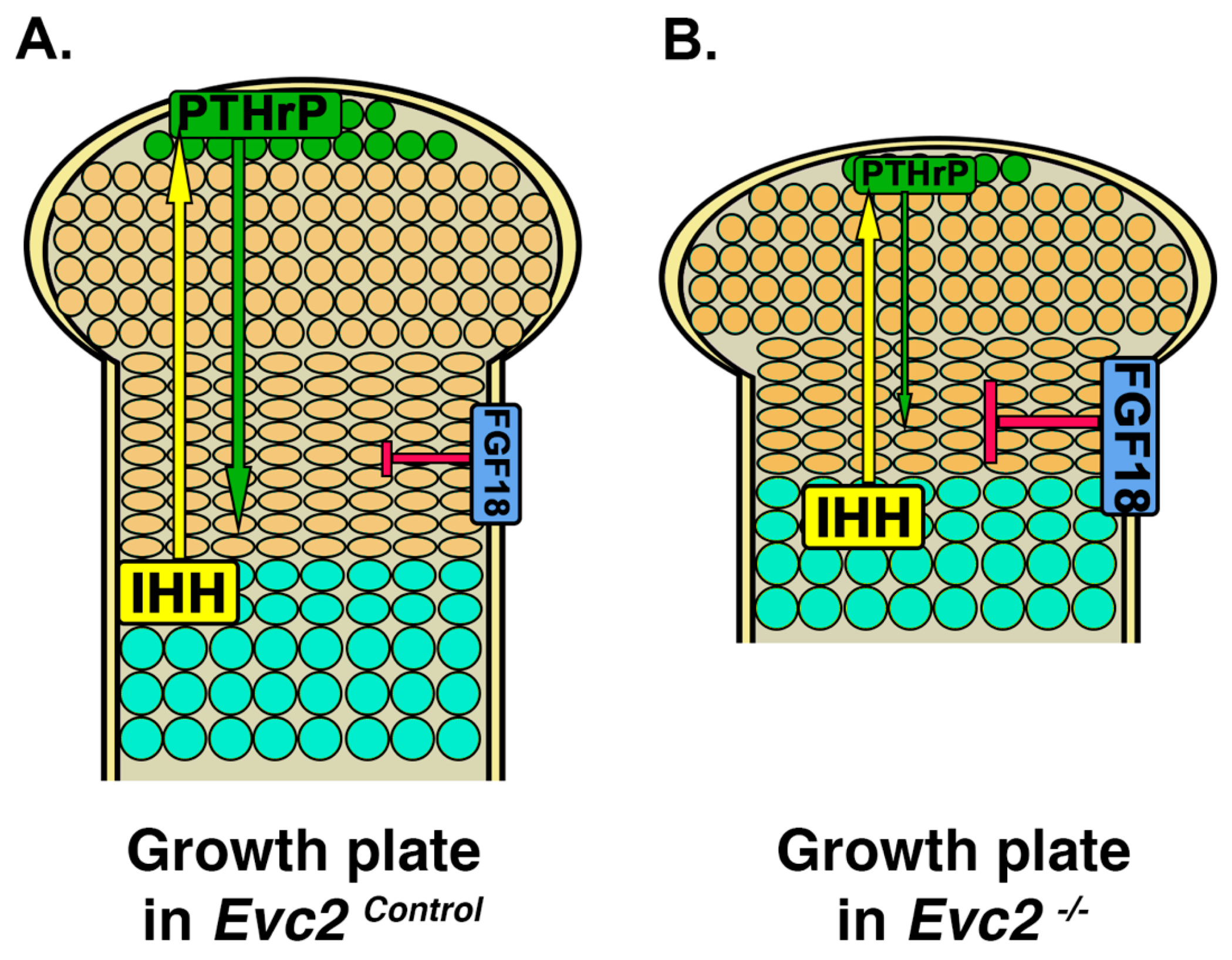
4. Limb Phenotypes
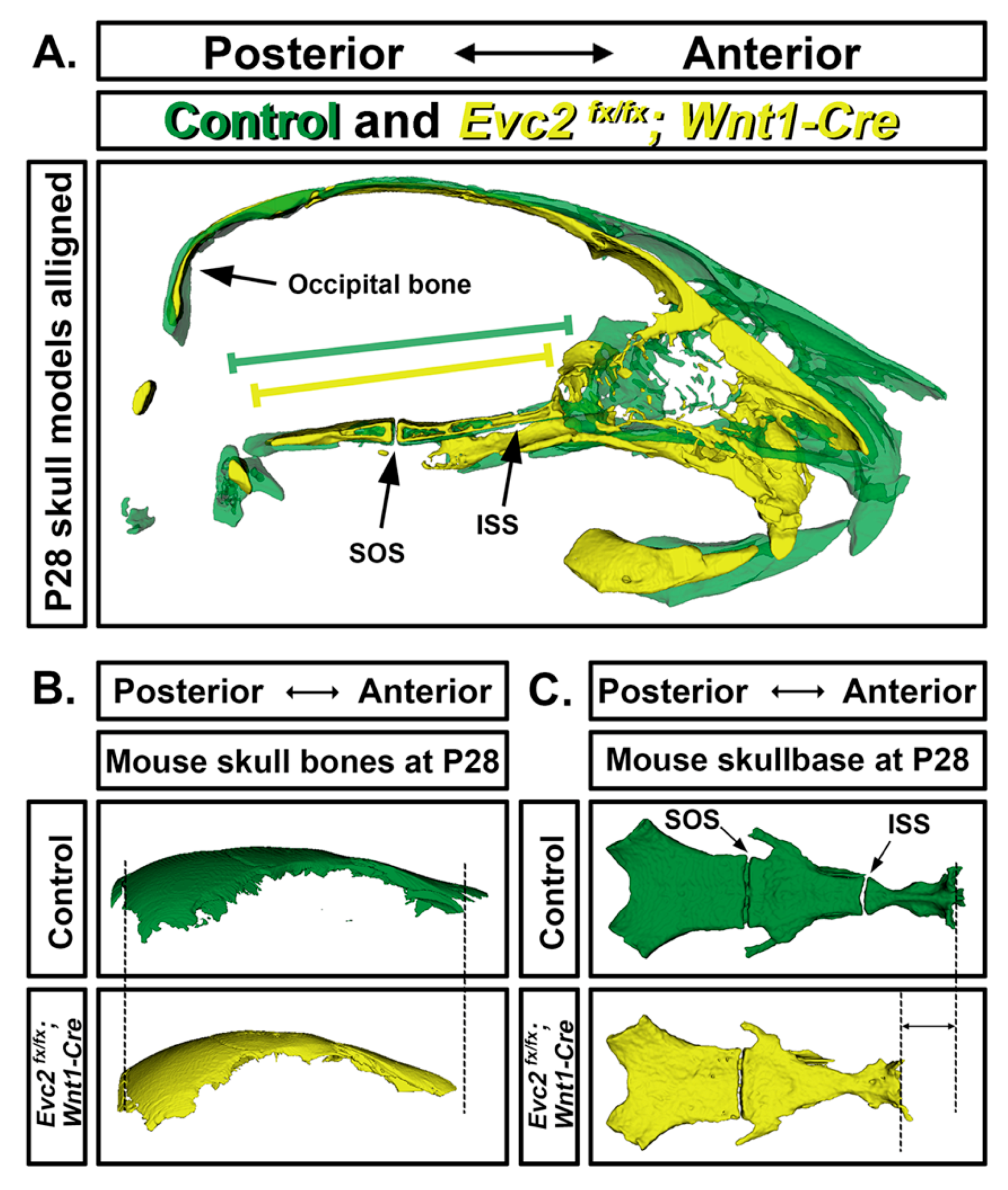
5. Craniofacial Phenotype
6. Tooth Phenotype
7. Tracheal Cartilage Phenotypes
8. EVC-Like Disorders in Other Non-Human Species
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ellis, R.W.B.; Van Creveld, S. A syndrome characterized by ectodermal dysplasia, polydactyly, chondro-dysplasia and congenital morbus cordis: Report of three cases. Arch. Dis. Child. 1940, 15, 65–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKusick, V.A. Dwarfism in the Amish I. The Ellis-van Creveld Syndrome. Bull. Johns Hopkins Hosp. 1964, 115, 306–336. [Google Scholar] [PubMed]
- Ruiz-Perez, V.L.; Ide, S.E.; Strom, T.M.; Lorenz, B.; Wilson, D.; Woods, K.; King, L.; Francomano, C.; Freisinger, P.; Spranger, S.; et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet. 2000, 24, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Baujat, G.; Merrer, M.L. Ellis-Van Creveld syndrome. Orphanet J. Rare Dis. 2007, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Perez, V.L.; Goodship, J.A. Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to Hedgehog ligands. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Tompson, S.W.; Ruiz-Perez, V.L.; Blair, H.J.; Barton, S.; Navarro, V.; Robson, J.L.; Wright, M.J.; Goodship, J.A. Sequencing EVC and EVC2 identifies mutations in two-thirds of Ellis-van Creveld syndrome patients. Hum. Genet. 2007, 120, 663–670. [Google Scholar] [CrossRef] [PubMed]
- D’Asdia, M.C.; Torrente, I.; Consoli, F.; Ferese, R.; Magliozzi, M.; Bernardini, L.; Guida, V.; Digilio, M.C.; Marino, B.; Dallapiccola, B.; et al. Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. Eur. J. Med. Genet. 2013, 56, 80–87. [Google Scholar] [CrossRef]
- Takeda, H.; Takami, M.; Oguni, T.; Tsuji, T.; Yoneda, K.; Sato, H.; Ihara, N.; Itoh, T.; Kata, S.R.; Mishina, Y.; et al. Positional cloning of the gene LIMBIN responsible for bovine chondrodysplastic dwarfism. Proc. Natl. Acad. Sci. USA 2002, 99, 10549–10554. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Takeda, H.; Tsuji, T.; Kamiya, N.; Rajderkar, S.; Louie, K.A.; Collier, C.; Scott, G.; Ray, M.; Mochida, Y.; et al. Generation of Evc2/Limbin global and conditional KO mice and its roles during mineralized tissue formation. Genesis 2015, 53, 612–626. [Google Scholar] [CrossRef]
- Ruiz-Perez, V.L.; Blair, H.J.; Rodrigues-Andres, M.E.; Blanco, M.J.; Wilson, A.; Liu, Y.N.; Miles, C.; Peters, H.; Goodship, J.A. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development 2007, 134, 2903–2912. [Google Scholar] [CrossRef] [Green Version]
- Caparrós-Martín, J.A.; Valencia, M.; Reytor, E.; Pacheco, M.; Fernandez, M.; Perez-Aytes, A.; Gean, E.; Lapunzina, P.; Peters, H.; Goodship, J.A.; et al. The ciliary EVC/EVC2 complex interacts with smo and controls hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet. 2013, 22, 124–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzelius, B.A. Cilia-related diseases. J. Pathol. 2004, 204, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Camner, P.; Mossberg, B.; Afzelius, B.A. Evidence of congenitally nonfunctioning cilia in the tracheobronchial tract in two subjects. Am. Rev. Respir. Dis. 1975, 112, 807–809. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef] [Green Version]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [Green Version]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Dorn, K.V.; Hughes, C.E.; Rohatgi, R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, H.J.; Tompson, S.; Liu, Y.N.; Campbell, J.; MacArthur, K.; Ponting, C.P.; Ruiz-Perez, V.L.; Goodship, J.A. Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biol. 2011, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valencia, M.; Lapunzina, P.; Lim, D.; Zannolli, R.; Bartholdi, D.; Wollnik, B.; Al-Ajlouni, O.; Eid, S.S.; Cox, H.; Buoni, S.; et al. Widening the mutation spectrum of EVC and EVC2: Ectopic expression of Weyer variants in NIH 3T3 fibroblasts disrupts Hedgehog signaling. Hum. Mutat. 2009, 30, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Ulucan, H.; Guel, D.; Sapp, J.C.; Cockerham, J.; Johnston, J.J.; Biesecker, L.G. Extending the spectrum of Ellis van Creveld syndrome: A large family with a mild mutation in the EVC gene. BMC Med. Genet. 2008, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Kamiya, N.; Tsuji, T.; Takeda, H.; Scott, G.; Rajderkar, S.; Ray, M.K.; Mochida, Y.; Allen, B.; Lefebvre, V.; et al. Elevated Fibroblast Growth Factor Signaling Is Critical for the Pathogenesis of the Dwarfism in Evc2/Limbin Mutant Mice. PLoS Genet. 2016, 12, e1006510. [Google Scholar] [CrossRef]
- Alvarez-Borja, A. Ellis-van Creveld Syndrome: Report of two cases. Pediatrics 1960, 26, 301–309. [Google Scholar] [CrossRef]
- Husson, G.S.; Parkman, P. Chondroectodermal dysplasia (Ellis-Van Creveld syndrome) with a complex cardiac malformation. Pediatrics 1961, 28, 285–292. [Google Scholar]
- Lynch, J.I. Congenital Heart Disease and Chondroectodermal Dysplasia. Am. J. Dis. Child. 1968, 115, 80. [Google Scholar] [CrossRef]
- Goor, D.; Rotem, Y.; Friedman, A.; Neufeld, H.N. ELLIS-VAN CREVELD SYNDROME IN IDENTICAL TWINS * In 1933 McIntosh (Holt and McIntosh, 1933) described abnormal findings in a girl who had. Br. Heart J. 1965, 27, 797–804. [Google Scholar] [CrossRef] [Green Version]
- Metrakos, J.D.; Fraser, F.C. Evidence for a hereditary factor in chondroectodermal dysplasia (Ellis-van Creveld syndrome). Am. J. Hum. Genet. 1954, 6, 260–269. [Google Scholar]
- Smith, H.L.; Hand, A.M. Chondroectodermal Dysplasia (Ellis-van Creveld Syndrome): Report of Two Cases. Pediatrics 1958, 21, 298–307. [Google Scholar] [PubMed]
- Weiss, H.; Crosett, A.D. Chondroectodermal dysplasia: Report of a case and review of the literature. J. Pediatrics 1955, 46, 268–275. [Google Scholar] [CrossRef]
- Mitchell, F.N.; Waddell, W.W. Ellis-van Creveld syndrome: Report of two cases in siblings. Acta Paediatr. 1958, 47, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.D. Two cases of Ellis van Creveld syndrome in a small island population. J. Med. Genet. 1977, 14, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.O.; Janovitz, D.; Cavalcanti De Albuquerque, S. Ellis-van Creveld syndrome: Report of 15 cases in an inbred kindred. J. Med. Genet. 1980, 17, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Cahuana, A.; Palma, C.; Gonzáles, W.; Geán, E. Oral manifestations in Ellis-van Creveld syndrome: Report of five cases. Pediatr. Dent. 2004, 26, 277–282. [Google Scholar]
- Hanemann, J.A.C.; de Carvalho, B.C.F.; Franco, E.C. Oral manifestations in Ellis-van Creveld syndrome: Report of a case and review of the literature. J. Oral Maxillofac. Surg. 2010, 68, 456–460. [Google Scholar] [CrossRef]
- Naqash, T.A.; Alshahrani, I.; Simasetha, S. Ellis-van creveld syndrome: A rare clinical report of oral rehabilitation by interdisciplinary approach. Case Rep. Dent. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Bohnsack, B.L.; Gallina, D.; Thompson, H.; Kasprick, D.S.; Lucarelli, M.J.; Dootz, G.; Nelson, C.; McGonnell, I.M.; Kahana, A. Development of extraocular muscles requires early signals from periocular neural crest and the developing eye. Arch. Ophthalmol. 2011, 129, 1030–1041. [Google Scholar] [CrossRef] [Green Version]
- Versteegh, F.G.; Buma, S.A.; Costin, G.; de Jong, W.C.; Hennekam, R.C.; Ev, C.W.P. Growth hormone analysis and treatment in Ellis-van Creveld syndrome. Am. J. Med. Genet. A 2007, 143A, 2113–2121. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.C.; Kitagawa, M.; Xue, N.; Xie, B.; Garofalo, S.; Cho, J.; Deng, C.; Horton, W.A.; Fu, X.Y. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 1997, 386, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Sahni, M.; Ambrosetti, D.C.; Mansukhani, A.; Gertner, R.; Levy, D.; Basilico, C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999, 13, 1361–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Balmes, G.; McKinney, S.; Zhang, Z.; Givol, D.; de Crombrugghe, B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004, 18, 290–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef]
- Garcia, S.; Dirat, B.; Tognacci, T.; Rochet, N.; Mouska, X.; Bonnafous, S.; Patouraux, S.; Tran, A.; Gual, P.; Le Marchand-Brustel, Y.; et al. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci. Transl. Med. 2013, 5, 203ra124. [Google Scholar] [CrossRef]
- Pacheco, M.; Valencia, M.; Caparrós-Martín, J.A.; Mulero, F.; Goodship, J.A.; Ruiz-Perez, V.L. Evc works in chondrocytes and osteoblasts to regulate multiple aspects of growth plate development in the appendicular skeleton and cranial base. Bone 2012, 50, 28–41. [Google Scholar] [CrossRef]
- Badri, M.K.; Zhang, H.; Ohyama, Y.; Venkitapathi, S.; Alamoudi, A.; Kamiya, N.; Takeda, H.; Ray, M.; Scott, G.; Tsuji, T.; et al. Expression of Evc2 in craniofacial tissues and craniofacial bone defects in Evc2 knockout mouse. Arch. Oral Biol. 2016, 68, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Badri, M.K.; Zhang, H.; Ohyama, Y.; Venkitapathi, S.; Kamiya, N.; Takeda, H.; Ray, M.; Scott, G.; Tsuji, T.; Kunieda, T.; et al. Ellis Van Creveld2 is Required for Postnatal Craniofacial Bone Development. Anat. Rec. 2016, 299, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Mishina, Y.; Snider, T.N. Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp. Cell Res. 2015, 325, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.K.; Louie, K.a.W.; Yatabe, M.; De Oliveira Ruellas, A.C.; Mochida, Y.; Cevidanes, L.H.S.; Mishina, Y.; Zhang, H. A Ciliary Protein EVC2/LIMBIN Plays a Critical Role in the Skull Base for Mid-Facial Development. Front. Physiol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, E.K.; Louie, K.a.W.; Kulkarni, A.; Yatabe, M.; Carlos, A.; Ruellas, D.E.O.; Snider, T.N.; Mochida, Y.; Cevidanes, L.H.S.; Mishina, Y. The Role of Ellis-Van Creveld 2 ( EVC2 ) in Mice During Cranial Bone Development. Anat. Rec. 2018, 301, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thesleff, I.; Hurmerinta, K. Tissue Interactions in Tooth Development. Differentiation 1981, 18, 75–88. [Google Scholar] [CrossRef]
- Zhang, H.; Takeda, H.; Tsuji, T.; Kamiya, N.; Kunieda, T.; Mochida, Y.; Mishina, Y. Loss of Function of Evc2 in Dental Mesenchyme Leads to Hypomorphic Enamel. J. Dent. Res. 2017, 96, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, Y.; Ishibashi, T.; Ashizawa, H.; Shibata, T. Chondrodysplastic Dwarfism in Japanese Brown Cattle. J. Jpn. Vet. Med. Assoc. 1989, 42, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, K.; Moritomo, Y.; Takami, M.; Hirata, S.; Kikukawa, Y.; Kunieda, T. Localization of a locus responsible for the bovine chondrodysplastic dwarfism (bcd) on chromosome 6. Mamm. Genome 1999, 10, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Murgiano, L.; Jagannathan, V.; Benazzi, C.; Bolcato, M.; Brunetti, B.; Muscatello, L.V.; Dittmer, K.; Piffer, C.; Gentile, A.; Drögemüller, C. Deletion in the EVC2 gene causes chondrodysplastic dwarfism in Tyrolean grey cattle. PLoS ONE 2014, 9, e0094861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, T.; Nishimura, T.; Kuchida, K.; Mannen, H. The Japanese Wagyu beef industry: Current situation and future prospects—A review. Asian-Australas. J. Anim. Sci. 2018, 31, 933–950. [Google Scholar] [CrossRef] [Green Version]
- Muscatello, L.V.; Benazzi, C.; Dittmer, K.E.; Thompson, K.G.; Murgiano, L.; Drögemüller, C.; Avallone, G.; Gentile, A.; Edwards, J.F.; Piffer, C.; et al. Ellis–van Creveld Syndrome in Grey Alpine Cattle: Morphologic, Immunophenotypic, and Molecular Characterization. Vet. Pathol. 2015, 52, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhang, Y.; Li, J.; Kong, L.; Hu, H.; Pan, H.; Xu, L.; Deng, Y.; Li, Q.; Jin, L.; et al. Two Antarctic penguin genomes reveal insights into their evolutionary history and molecular changes related to the Antarctic environment. GigaScience 2014, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anatomical Locations | EVC | Weyers Acrodental Dysostosis | Evc Mutant Mice | Evc2/Limbin Mutant Mice | Hedgehog Signaling Defects in Mice |
|---|---|---|---|---|---|
| Limb | Short | Short (mild) | Short | Short | Short |
| Craniofacial | Normal | Normal | Normal | Normal | Cleft lips, cleft Palate |
| Neural tube | Normal | Normal | Normal | Normal | Open neural tube |
| Digit | Postaxial polydactyly | Postaxial polydactyly | Postaxial polydactyly | Postaxial polydactyly | Preaxial polydactyly (?) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louie, K.W.; Mishina, Y.; Zhang, H. Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models. J. Dev. Biol. 2020, 8, 25. https://doi.org/10.3390/jdb8040025
Louie KW, Mishina Y, Zhang H. Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models. Journal of Developmental Biology. 2020; 8(4):25. https://doi.org/10.3390/jdb8040025
Chicago/Turabian StyleLouie, Ke’ale W., Yuji Mishina, and Honghao Zhang. 2020. "Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models" Journal of Developmental Biology 8, no. 4: 25. https://doi.org/10.3390/jdb8040025
APA StyleLouie, K. W., Mishina, Y., & Zhang, H. (2020). Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models. Journal of Developmental Biology, 8(4), 25. https://doi.org/10.3390/jdb8040025