
In Vitro Anti-Proliferative, and Kinase Inhibitory Activity of Phenanthroindolizidine Alkaloids Isolated from Tylophora indica

 ,
,  , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Kinase Receptors’ Structures and Their Roles in Cancers
2.1.1. Molecular Properties and QSPR
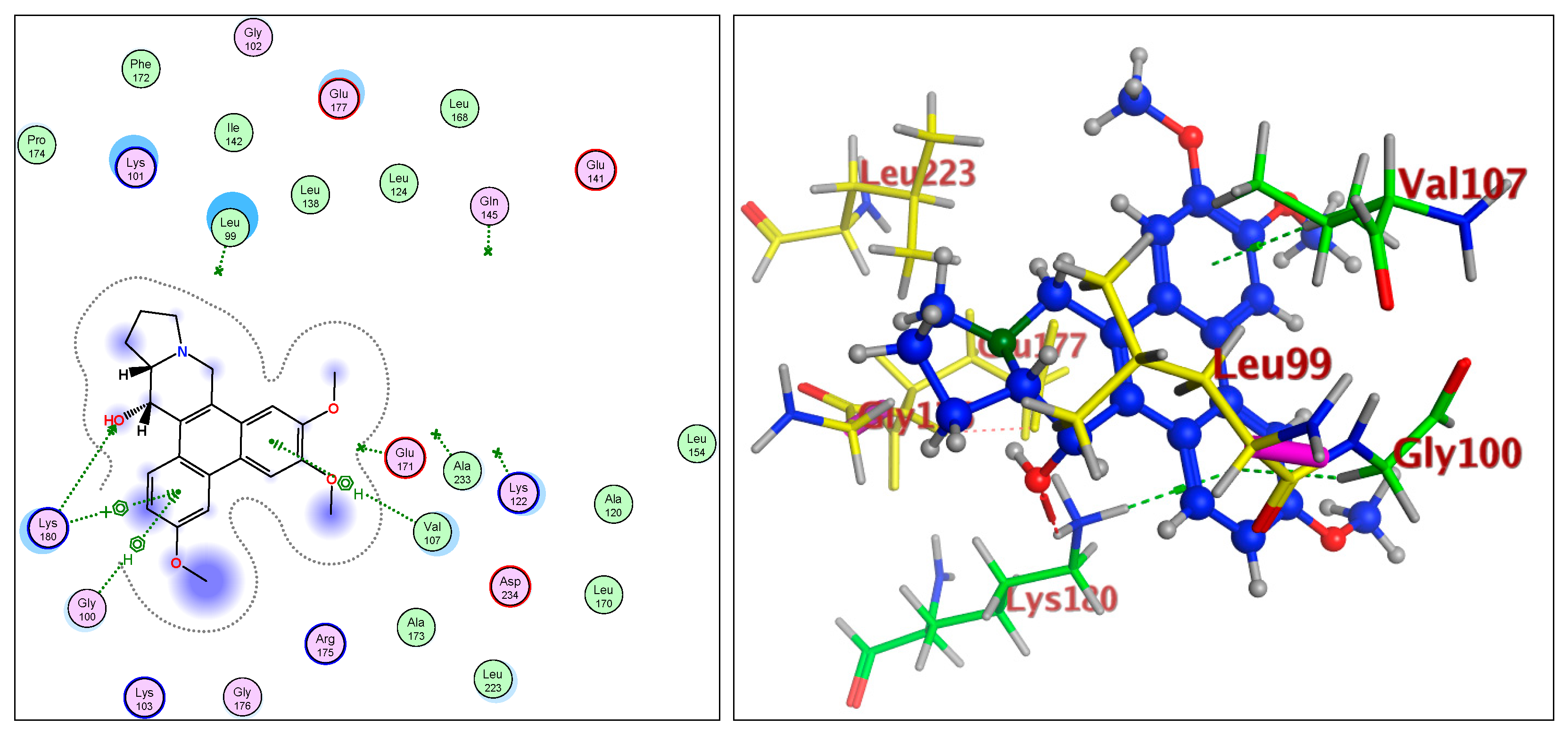
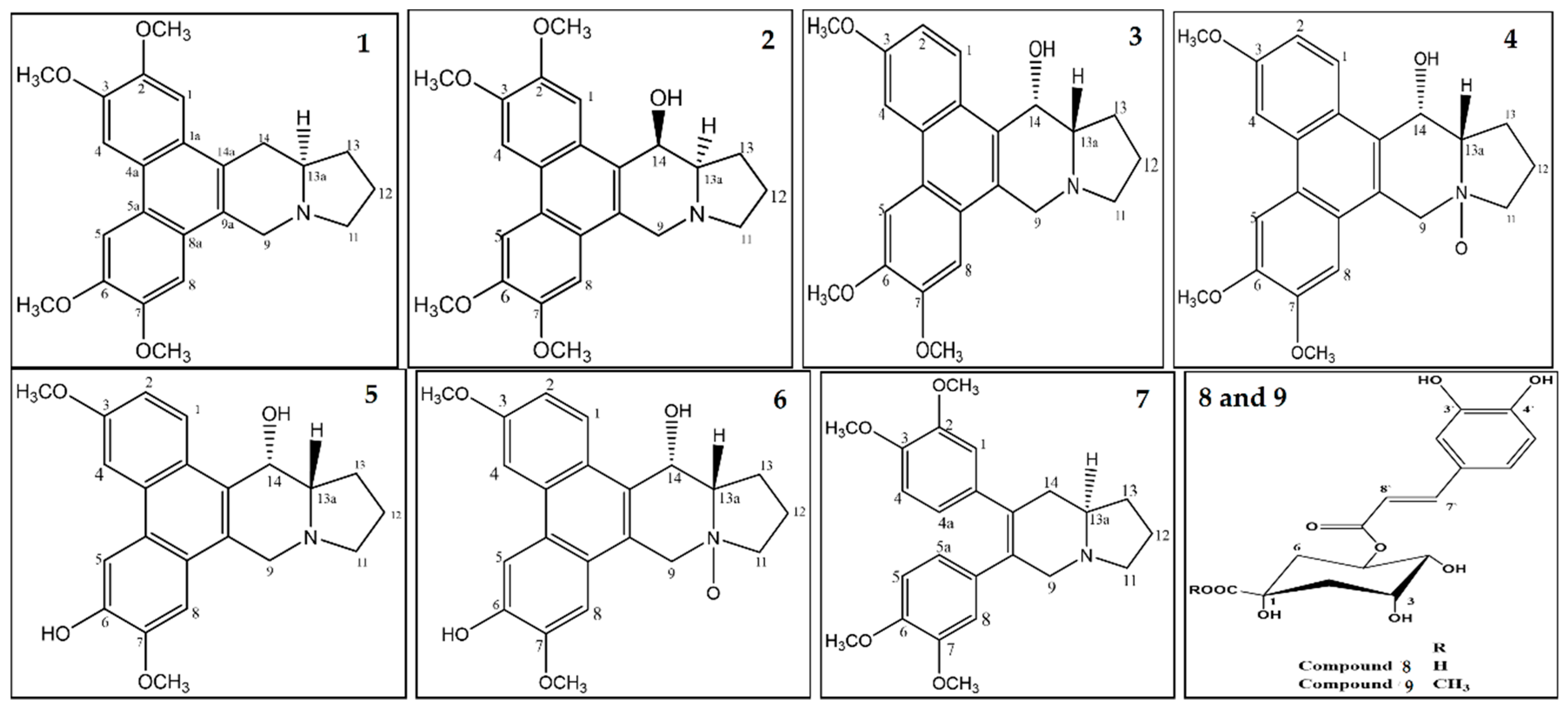
2.1.2. Cell Proliferation Assays, Structural Differentiations, Docking, and Energy Status of the Isolated Compounds from Tylophora indica
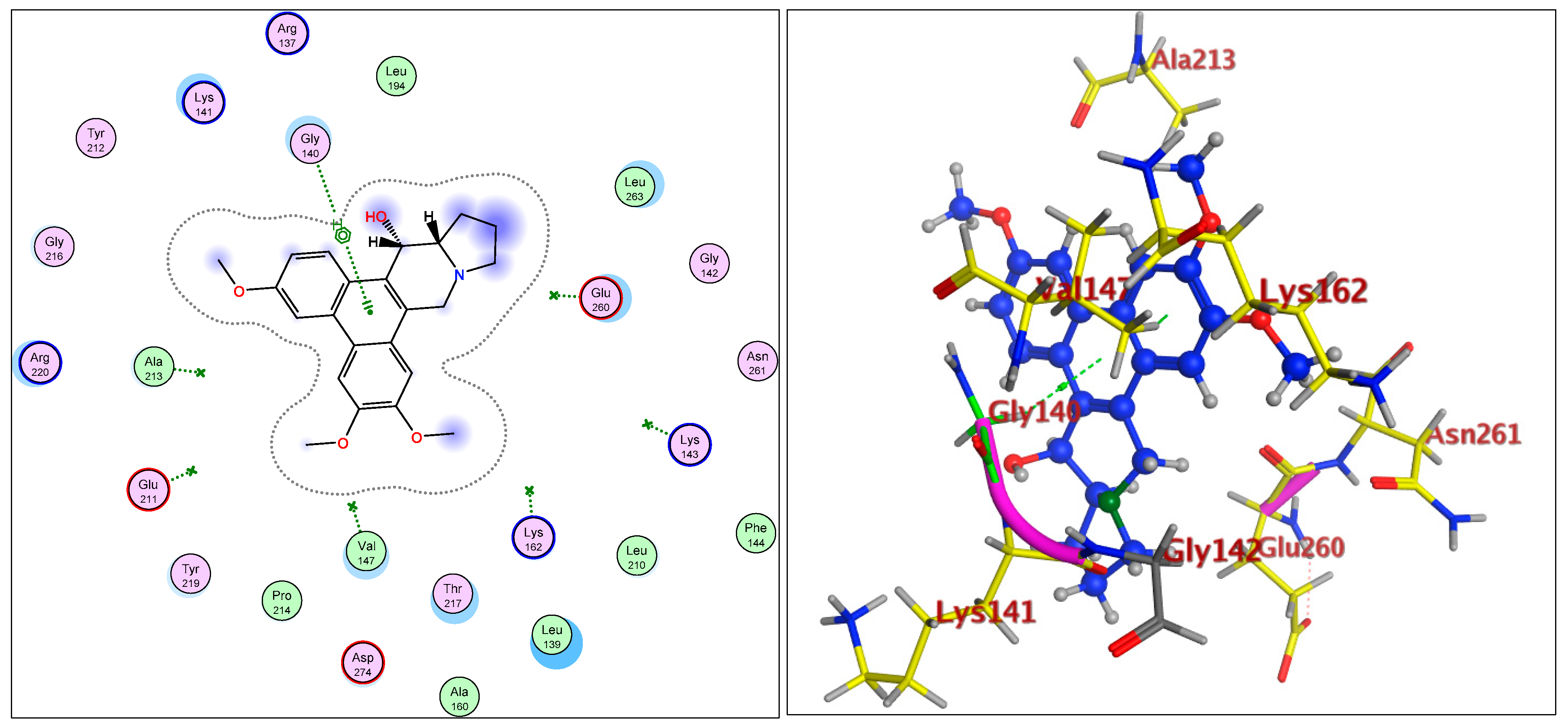
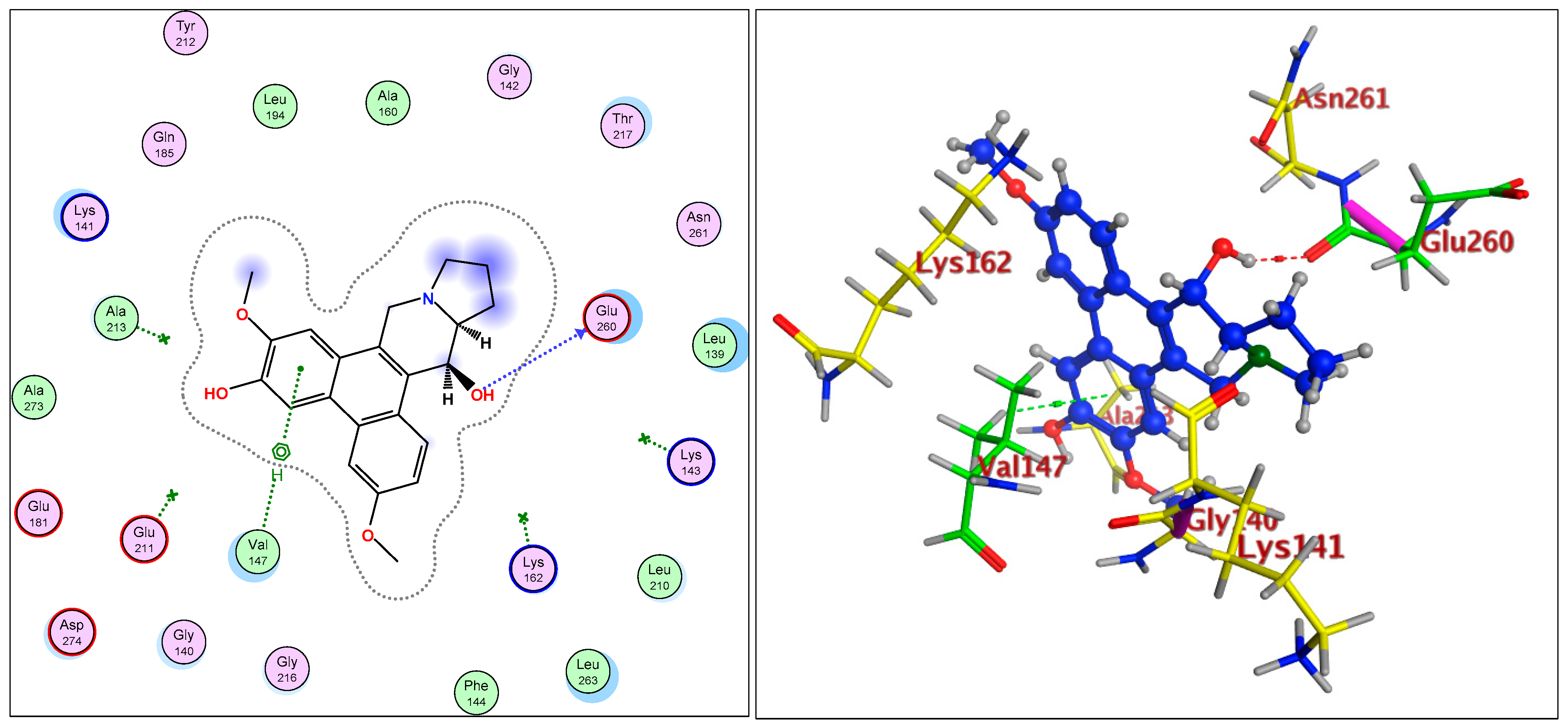
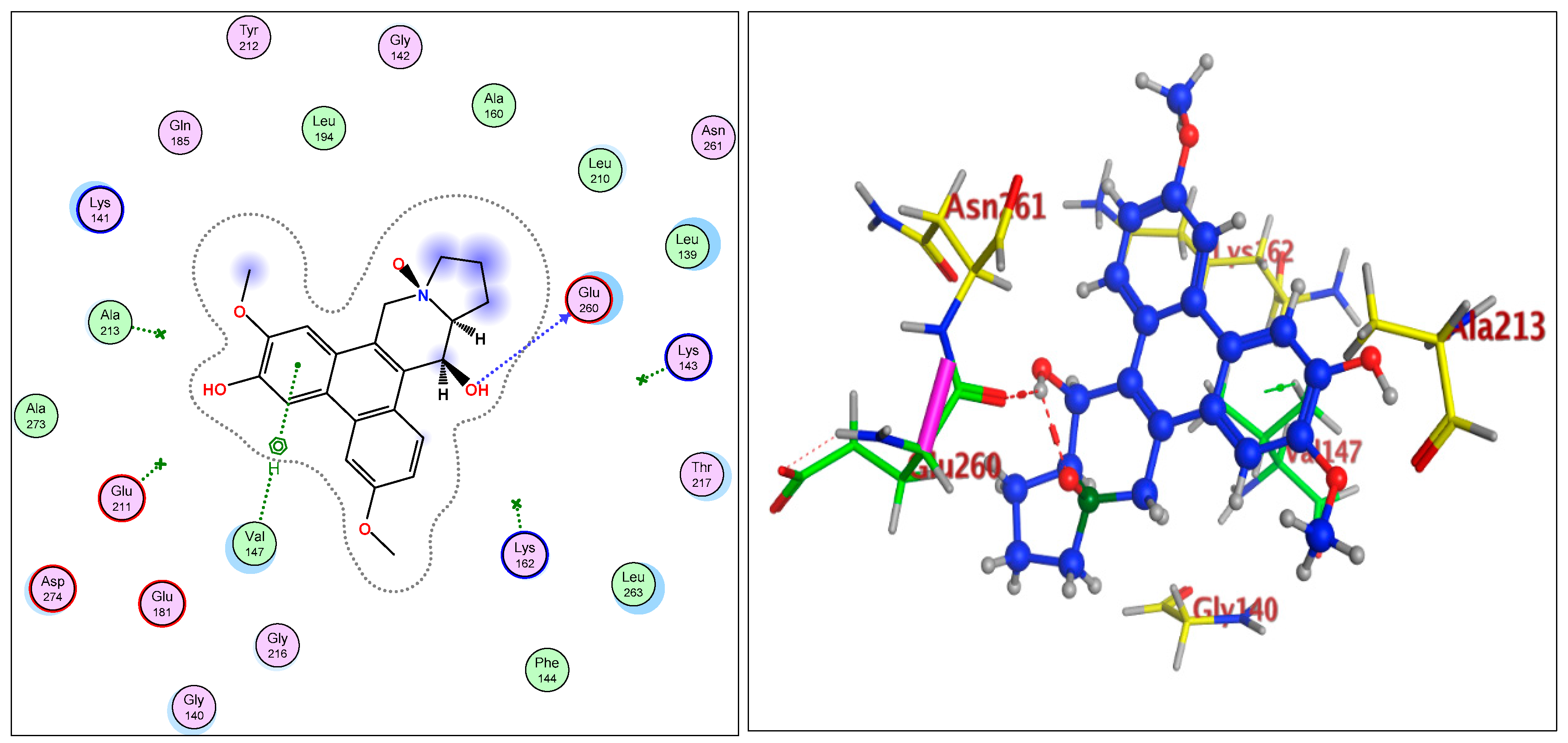
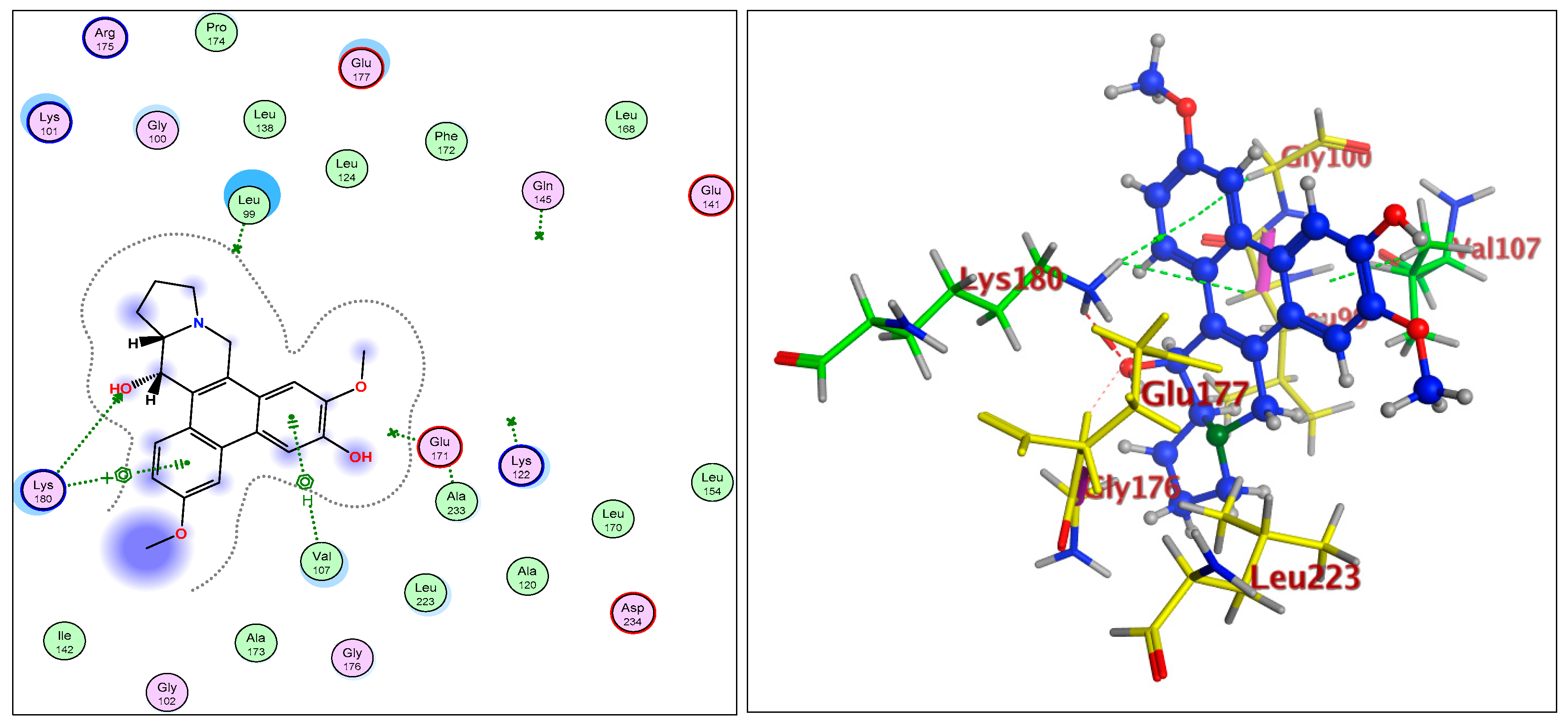
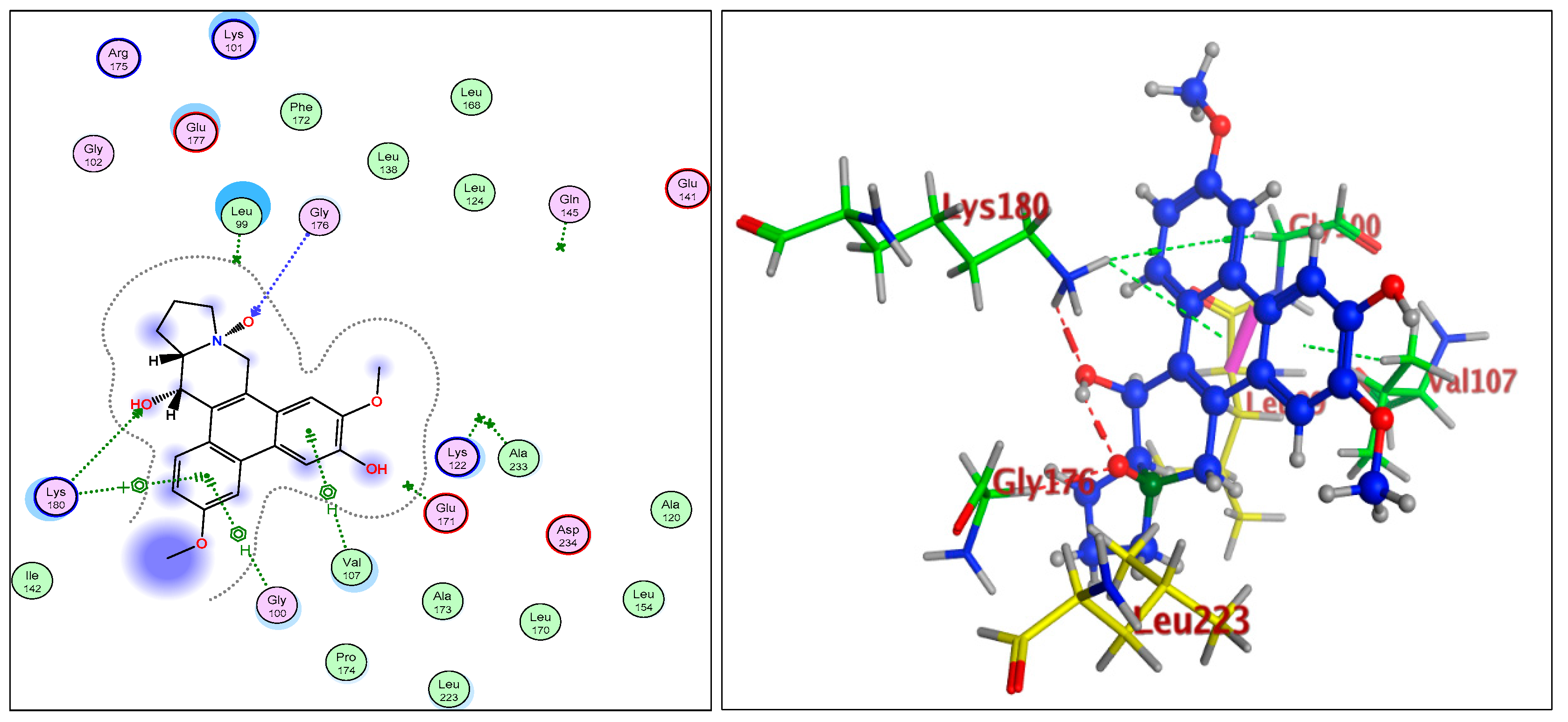
2.1.3. Radiometric Kinase Assay, Structural Differentiations, Docking, and Energy Status of the Isolated Compounds from Tylophora indica
Aurora Kinase-B: 4C2V
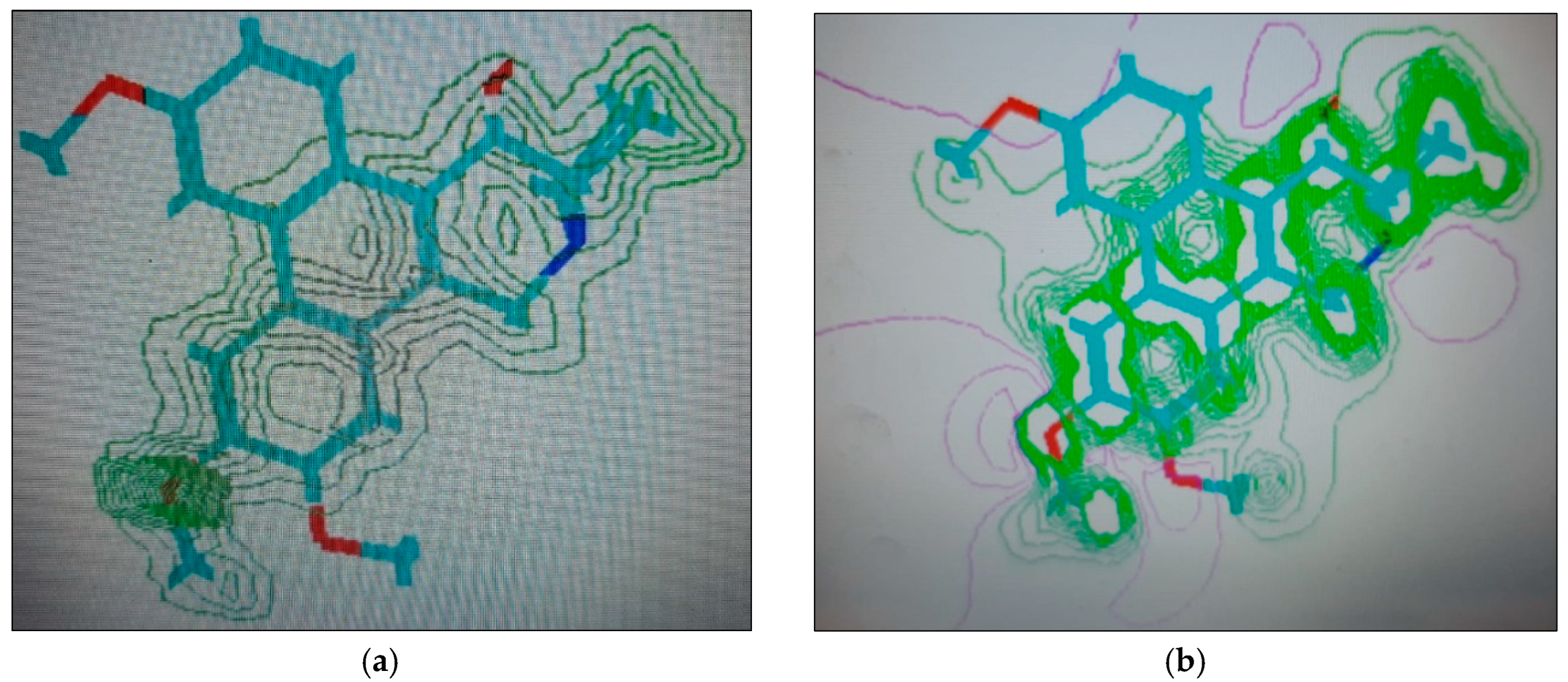
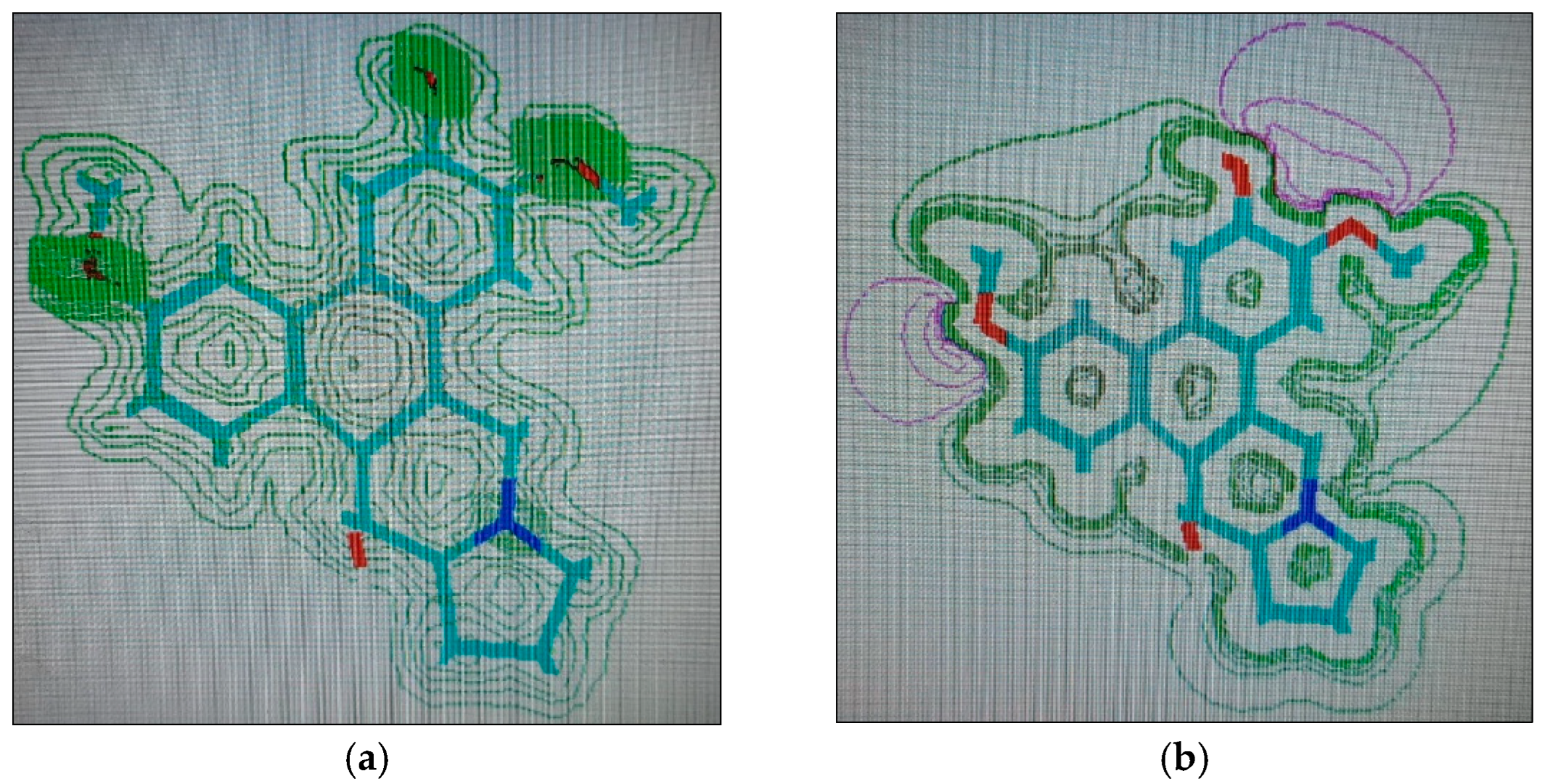
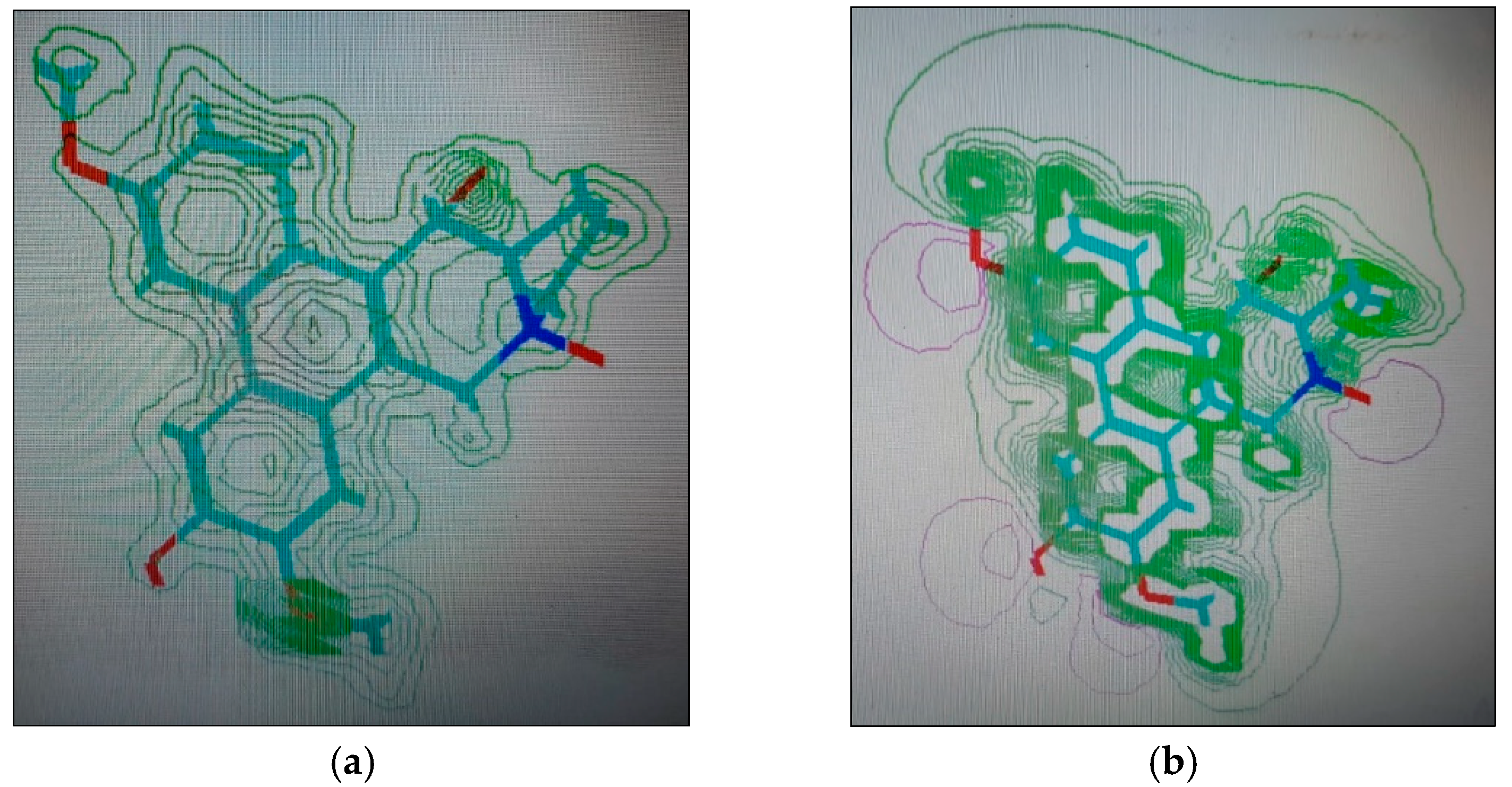
2.1.4. Molecular Computographics: Electrostatic Potential and Charge Density Distributions
3. Materials and Methods
3.1. Plant Material
3.2. HPLC Conditions and Isolation Procedures
3.3. Compounds Extractions and Purifications
3.4. Compounds’ Characterizations
3.5. Anti-Proliferative Assays
3.6. Kinase Inhibitory Assays
3.7. In Silico Studies
3.7.1. QSPR and Molecular Attributes Estimations
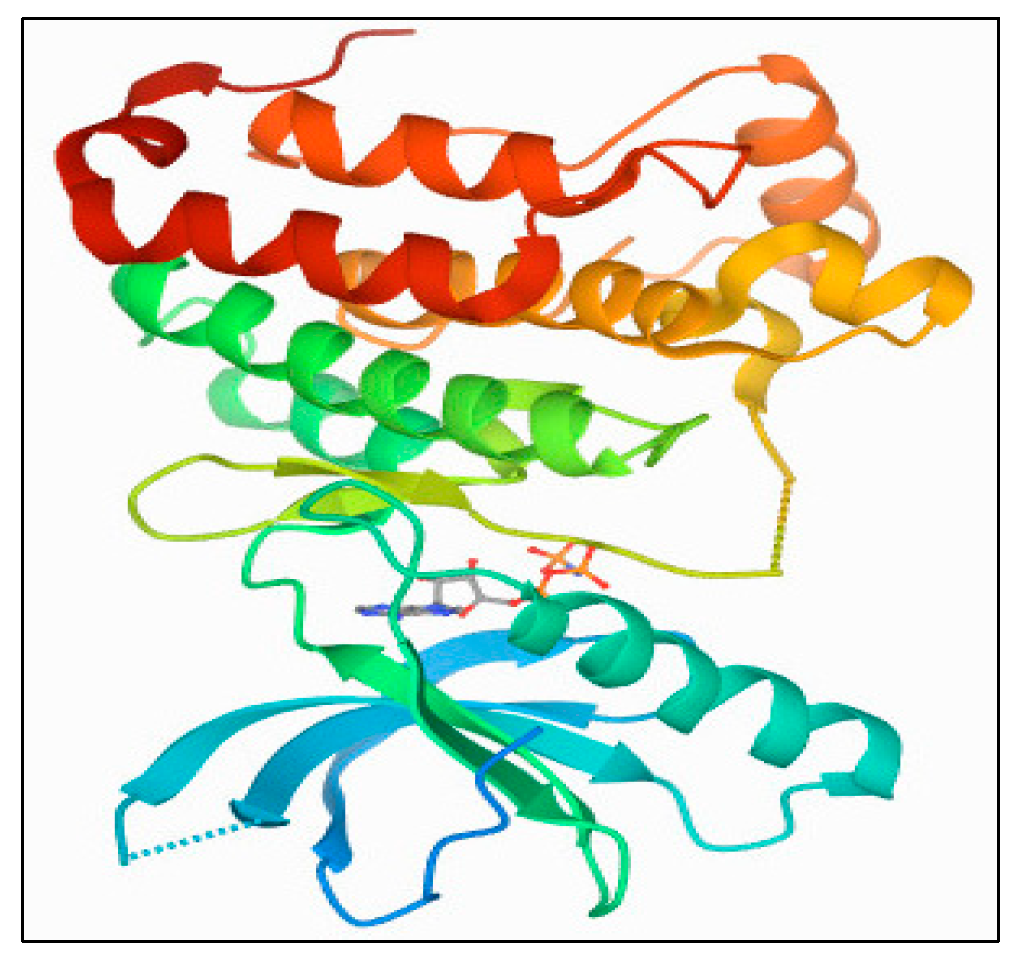
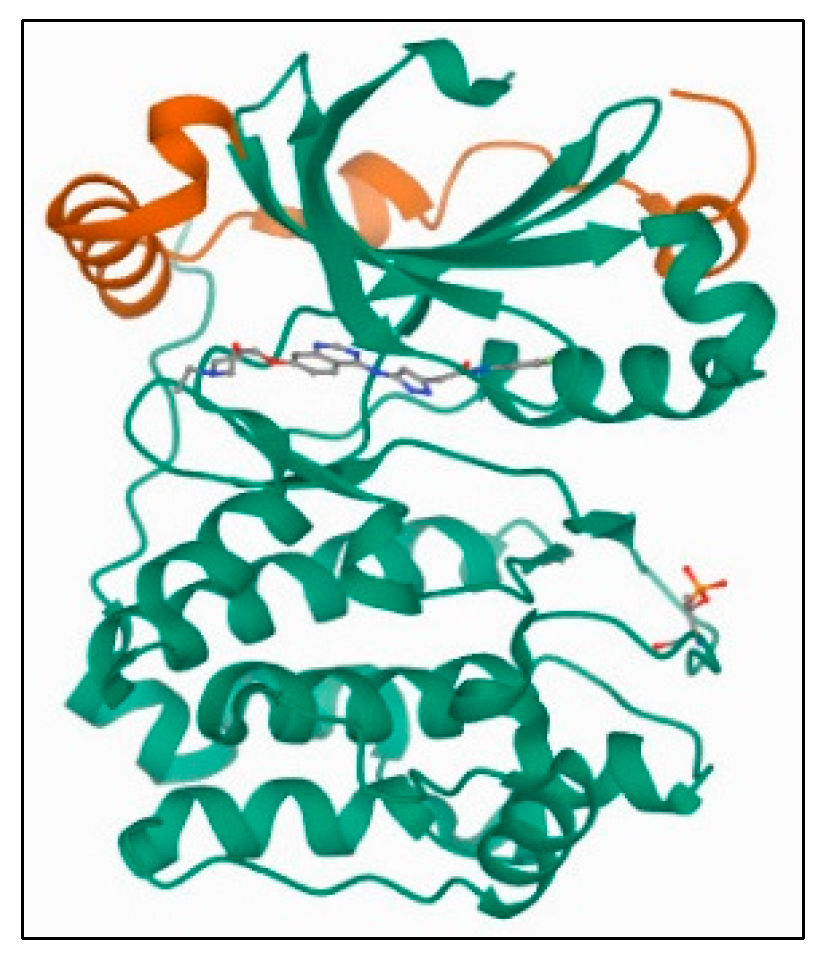
3.7.2. Receptors’ XRD Crystal Structures: Aurora-A and Aurora-B
Aurora-A Kinase: PDB 1MQ4
Aurora-B Kinase: PDB 4C2V
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poste, G.; Fidler, I.J. The pathogenesis of cancer metastasis. Nature 1980, 283, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institute, N.C. Type of Cancers. Available online: https://www.cancer.gov/types (accessed on 25 March 2022).
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar] [PubMed]
- Ji, H.; Li, X.; Zhang, H. Natural products and drug discovery: Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Rep. 2009, 10, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Gurib-Fakim, A. Medicinal plants: Traditions of yesterday and drugs of tomorrow. Mol. Asp. Med. 2006, 27, 1–93. [Google Scholar] [CrossRef]
- Safarzadeh, E.; Shotorbani, S.S.; Baradaran, B. Herbal medicine as inducers of apoptosis in cancer treatment. Adv. Pharm. Bull. 2014, 4, 421. [Google Scholar]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Kong, X.-Y.; Wang, J.-H.; Du, G.-H. Vinblastine and vincristine. In Natural Small Molecule Drugs from Plants; Springer: Berlin/Heidelberg, Germany, 2018; pp. 551–557. [Google Scholar]
- Ghesquieres, H.; Casasnovas, O.; Nicolas-Virelizier, E.; Laurent, D.G.; Delwail, V.; Touati, M.; Le Du, K.; Morschhauser, F.; Bonnet, C.; Waultier-Rascalou, A.; et al. Prednisone, vinblastine, doxorubicin and bendamustine (PVAB) regimen in first line therapy for older patients with advanced-stage classical Hodgkin lymphoma: Results of a Prospective Multicenter Phase II Trial of the Lymphoma Study Association (LYSA). Blood 2019, 134, 2832. [Google Scholar] [CrossRef]
- Vergote, I.; Scambia, G.; O’Malley, D.M.; Van Calster, B.; Park, S.-Y.; Del Campo, J.M.; Meier, W.; Bamias, A.; Colombo, N.; Wenham, R.M.; et al. Trebananib or placebo plus carboplatin and paclitaxel as first-line treatment for advanced ovarian cancer (TRINOVA-3/ENGOT-ov2/GOG-3001): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 862–876. [Google Scholar] [CrossRef]
- Bishop, J.F.; Dewar, J.; Toner, G.C.; Tattersall, M.H.; Olver, I.N.; Ackland, S.; Kennedy, I.; Goldstein, D.; Gurney, H.; Walpole, E.; et al. Paclitaxel as first-line treatment for metastatic breast cancer. The Taxol Investigational Trials Group, Australia and New Zealand. Oncology 1997, 11, 19–23. [Google Scholar]
- Broxterman, H.J.; Lankelma, J.; Hoekman, K. Resistance to cytotoxic and anti-angiogenic anticancer agents: Similarities and differences. Drug Resist. Update 2003, 6, 111–127. [Google Scholar] [CrossRef]
- Buyel, J.F. Plants as sources of natural and recombinant anti-cancer agents. Biotechnol. Adv. 2018, 36, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.A.; Khan, R.A.; Abdel-Hafez, A.A.; Abdel-Aziz, M.; Ahmed, E.; Enany, S.; Mahgoub, S.; Al-Rugaie, O.; Alsharidah, M.; Aly, M.S.A.; et al. Profiling, In Vitro and In Silico Anti-Microbial and Anti-Cancer Activity Evaluations and Staph GyraseB and h-TOP-IIβ Receptor-Docking Studies of Major Constituents of Zygophyllum coccineum L. Aqueous-Ethanolic Extract and Its Subsequent Fra. Molecules 2021, 26, 577. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.A.; Abd El-Wahab, M.F.; Shaheen, U.; Mohammed, A.E.-S.I.; Abdalla, A.N.; Ragab, E.A. Isolation, Characterization, Complete Structural Assignment, and Anticancer Activities of the Methoxylated Flavonoids from Rhamnus disperma Roots. Molecules 2021, 26, 5827. [Google Scholar] [CrossRef]
- Mohammed, H.A.; Al-Omar, M.S.; Khan, R.A.; Mohammed, S.A.A.; Qureshi, K.A.; Abbas, M.M.; Al Rugaie, O.; Abd-Elmoniem, E.; Ahmad, A.M.; Kandil, Y.I. Chemical Profile, Antioxidant, Antimicrobial, and Anticancer Activities of the Water-Ethanol Extract of Pulicaria undulata Growing in the Oasis of Central Saudi Arabian Desert. Plants 2021, 10, 1811. [Google Scholar] [CrossRef] [PubMed]
- Mileva, M.; Dimitrova, L.; Popova, M.; Bankova, V.; Krastev, D.; Najdenski, H.; Zhelev, Z.; Aoki, I.; Bakalova, R. Redox-modulation, Suppression of “Oncogenic” Superoxide and Induction of Apoptosis in Burkitt’s Lymphoma Cells Using Geum urbanum L. Extracts. Int. J. Bioautom. 2021, 25, 315. [Google Scholar] [CrossRef]
- Mohammed, H.A.; Ali, H.M.; Qureshi, K.A.; Alsharidah, M.; Kandil, Y.I.; Said, R.; Mohammed, S.A.A.; Al-Omar, M.S.; Al Rugaie, O.; Abdellatif, A.A.H.; et al. Comparative Phytochemical Profile and Biological Activity of Four Major Medicinal Halophytes from Qassim Flora. Plants 2021, 10, 2208. [Google Scholar] [CrossRef]
- Gezici, S.; Şekeroğlu, N. Current perspectives in the application of medicinal plants against cancer: Novel therapeutic agents. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem. Agents) 2019, 19, 101–111. [Google Scholar] [CrossRef]
- Twilley, D.; Rademan, S.; Lall, N. A review on traditionally used South African medicinal plants, their secondary metabolites and their potential development into anticancer. J. Ethnopharmacol. 2020, 261, 113101. [Google Scholar] [CrossRef]
- Rajabi, S.; Maresca, M.; Yumashev, A.V.; Choopani, R.; Hajimehdipoor, H. The most competent plant-derived natural products for targeting Apoptosis in cancer therapy. Biomolecules 2021, 11, 534. [Google Scholar] [CrossRef]
- Saraswati, S.; Kanaujia, P.K.; Kumar, S.; Kumar, R.; Alhaider, A.A. Tylophorine, a phenanthraindolizidine alkaloid isolated from Tylophora indica exerts antiangiogenic and antitumor activity by targeting vascular endothelial growth factor receptor 2–mediated angiogenesis. Mol. Cancer 2013, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.-W.; Lee, Y.-Z.; Hsu, H.-Y.; Wu, C.-M.; Chang, H.-Y.; Chao, Y.-S.; Lee, S.-J. c-Jun-mediated anticancer mechanisms of tylophorine. Carcinogenesis 2013, 34, 1304–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wei, L.; Bastow, K.; Zheng, W.; Brossi, A.; Lee, K.-H.; Tropsha, A. Antitumor agents 252. Application of validated QSAR models to database mining: Discovery of novel tylophorine derivatives as potential anticancer agents. J. Comput. Aided Mol. Des. 2007, 21, 97–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-M.; Yang, C.-W.; Lee, Y.-Z.; Chuang, T.-H.; Wu, P.-L.; Chao, Y.-S.; Lee, S.-J. Tylophorine arrests carcinoma cells at G1 phase by downregulating cyclin A2 expression. Biochem. Biophys. Res. Commun. 2009, 386, 140–145. [Google Scholar] [CrossRef]
- Joa, H.; Blažević, T.; Grojer, C.; Zeller, I.; Heiss, E.H.; Atanasov, A.G.; Feldler, I.; Gruzdaitis, P.; Czaloun, C.; Proksch, P.; et al. Tylophorine reduces protein biosynthesis and rapidly decreases cyclin D1, inhibiting vascular smooth muscle cell proliferation in vitro and in organ culture. Phytomedicine 2019, 60, 152938. [Google Scholar] [CrossRef]
- Lin, J.-C.; Yang, S.-C.; Hong, T.-M.; Yu, S.-L.; Shi, Q.; Wei, L.; Chen, H.; Yang, P.; Lee, K. Phenanthrene-based tylophorine-1 (PBT-1) inhibits lung cancer cell growth through the Akt and NF-κB pathways. J. Med. Chem. 2009, 52, 1903–1911. [Google Scholar] [CrossRef] [Green Version]
- Herbpathy. Tylophora Indica Herb Uses, Benefits, Cures, Side Effects, Nutrients. Available online: https://herbpathy.com/Uses-and-Benefits-of-Tylophora-Indica-Cid1640 (accessed on 25 March 2022).
- Tariq, A.; Mehmood, T.; Ahmad, M.; Sahar, U.; Mushtaq, S.; Zafar, M. Comparative assessment of genetic diversity among the Asclepiadaceous species using randomly amplified polymorphic DNA (RAPD) markers and numerical taxonomy system (NTSYS) cluster analysis. J. Med. Plants Res. 2014, 8, 88–94. [Google Scholar]
- Phillipson, J.D.; Tezcan, I.; Hylands, P.J. Alkaloids of Tylophora species from Sri Lanka. Planta Med. 1974, 25, 301–309. [Google Scholar] [CrossRef]
- Nazar, S.; Hussain, M.A.; Khan, A.; Muhammad, G.; Bukhari, S.N.A. Alkaloid-rich plant Tylophora indica; current trends in isolation strategies, chemical profiling and medicinal applications. Arab J. Chem. 2020, 13, 6348–6365. [Google Scholar] [CrossRef]
- Jeyachandran, R.; Bastin, M. In-vitro propagation of Tylophora ovata (Lind.) Hook. ex. Steud-an important medicinal plant. Int. J. Pharm. Life Sci. 2014, 5, 1083–1086. [Google Scholar]
- Devi, P.; Singh, R.; Kumar, R.; Kumar, M.; Awasthi, K.K.; Singh, T.; Awasthi, G. Alteration of Media Enables Efficient In-vitro Regeneration in Antamool (Tylophora indica): AThreatened Medicinal Plant. Gorteria 2021, 34, 50–60. [Google Scholar]
- Karpov, O.A.; Fearnley, G.W.; Smith, G.A.; Kankanala, J.; McPherson, M.J.; Tomlinson, D.C.; Harrison, M.A.; Ponnambalam, S. Receptor tyrosine kinase structure and function in health and disease. AIMS Biophys. 2015, 2, 476–502. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar]
- Dhiman, M.; Khanna, A.; Manju, S. A new phenanthroindolizidine alkaloid from Tylophora indica. Chem. Papers 2013, 67, 245–248. [Google Scholar] [CrossRef]
- Liu, J.; He, Y.; Zhang, D.; Cai, Y.; Zhang, C.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. In vitro anticancer effects of two novel phenanthroindolizidine alkaloid compounds on human colon and liver cancer cells. Mol. Med. Rep. 2017, 16, 2595–2603. [Google Scholar] [CrossRef] [Green Version]
- Ying, Z.Y.; Shi, H.X.; De Quan, Y.U.; Shan, Y.U.S. Antitumor alkaloids isolated from Tylophora ovata. J. Integr. Plant Biol. 2002, 44, 349. [Google Scholar]
- Comins, D.L.; Morgan, L.A. N-acyldihydropyridones as synthetic intermediates. Synthesis of (±)-septicine and (±)-tylophorine. Tetrahedron Lett. 1991, 32, 5919–5922. [Google Scholar] [CrossRef]
- Pauli, G.F.; Kuczkowiak, U.; Nahrstedt, A. Solvent effects in the structure dereplication of caffeoyl quinic acids. Magn. Reson. Chem. 1999, 37, 827–836. [Google Scholar] [CrossRef]
- Ida, Y.; Satoh, Y.; Ohtsuka, M.; Nagasao, M.; Shoji, J. Phenolic constituents of Phellodendron amurense bark. Phytochemistry 1993, 35, 209–215. [Google Scholar] [CrossRef]
- Musa, A.; Al-Sanea, M.M.; Alotaibi, N.H.; Alnusaire, T.S.; Ahmed, S.R.; Mostafa, E.M. In silico study, protein kinase inhibition and antiproliferative potential of flavonoids isolated from Bassia eriophora (Schrad.) growing in KSA. Indian J. Pharm. Educ. Res. 2021, 55, 483–490. [Google Scholar] [CrossRef]
- Mohammed, H.A.; Al-Omar, M.S.; El-Readi, M.Z.; Alhowail, A.H.; Aldubayan, M.A.; Abdellatif, A.A.H. Formulation of Ethyl Cellulose Microparticles Incorporated Pheophytin A Isolated from Suaeda vermiculata for Antioxidant and Cytotoxic Activities. Molecules 2019, 24, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, E.M. Exploration of aurora B and cyclin-dependent kinase 4 inhibitors isolated from Scorzonera tortuosissima boiss. and their docking studies. Pharmacogn. Mag. 2020, 16, 258. [Google Scholar]
- Musa, A.; Elmaidomy, A.H.; Sayed, A.M.; Alzarea, S.I.; Al-Sanea, M.M.; Mostafa, E.M.; Hendawy, O.M.; Abdelgawad, M.A.; Youssif, K.A.; Refaat, H.; et al. Cytotoxic Potential, Metabolic Profiling, and Liposomes of Coscinoderma sp. Crude Extract Supported by in silico Analysis. Int. J. Nanomed. 2021, 16, 3861. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial | Compound | Molecular Attributes and QSPR Properties * | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Molar Mass | Molar Volume | Log P | HBA | HBD | NRB | Hydration Energy | Polarizability | Refractivity | Surface Area | ||
| 1. | Tylophorine | 393.48 | 1083.60 | −2.59 | 5 | 0 | 4 | −4.73 | 43.50 | 120.39 | 620.10 |
| 2. | Tylophorinicine | 409.48 | 1099.16 | −3.21 | 6 | 1 | 4 | −6.72 | 44.14 | 121.60 | 614.94 |
| 3. | Tylophorinine | 379.46 | 969.52 | −2.22 | 5 | 1 | 3 | −6.35 | 41.66 | 115.23 | 569.52 |
| 4. | Tylophorinine-N-Oxide | 395.46 | 1047.52 | −1.03 | 5 | 1 | 3 | −7.40 | 41.22 | 114.27 | 597.33 |
| 5. | Tylophorinidine | 365.43 | 990.58 | −2.25 | 5 | 2 | 2 | −10.71 | 39.83 | 110.46 | 563.42 |
| 6. | Tylophorinidine-N-Oxide | 391.43 | 907.52 | −1.07 | 5 | 2 | 2 | −13.22 | 39.43 | 109.50 | 557.89 |
| 7. | Septicine | 395.50 | 1031.64 | −1.30 | 5 | 0 | 6 | −8.69 | 44.27 | 114.63 | 584.43 |
| 8. | Chlorogenic acid | 354.31 | 912.07 | −2.47 | 9 | 6 | 5 | −32.98 | 31.81 | 86.27 | 544.75 |
| 9. | Chlorogenic acid methyl ester | 368.34 | 977.05 | −2.19 | 9 | 5 | 6 | −26.65 | 34.28 | 91.39 | 585.77 |
| No. | Compounds | IC50 ± SD (μM) a | ||
|---|---|---|---|---|
| MCF-7 | HepG2 | HCT-116 | ||
| 1. | Tylophorine | 126.5 | 40.10 ± 2.94 | 142.20 ± 3.06 |
| 2. | Tylophorinicine | 50.71 ± 2.86 | 35.33 ± 2.97 | 75.55 |
| 3. | Tylophorinine | 31.96 ± 2.64 | 23.8 ± 3.02 | 86.95 ± 3.08 |
| 4. | Tylophorinine N-Oxide | ˃200 | 196.60 | ˃200 |
| 5. | Tylophorinidine | 6.45 ± 2.06 | 4.77 ± 2.11 | 20.08 ± 1.94 |
| 6. | Tylophorinidine-N-Oxide | 12.15 ± 1.81 | 15.31 ± 2.04 | 65.62 ± 2.24 |
| 7. | Septicine | ˃200 | ˃200 | ˃200 |
| 8. | Chlorogenic acid | 134.00 | 169.90 | ˃200 |
| 9. | Chlorogenic acid methyl ester | ˃200 | ˃200 | ˃200 |
| 10. | Cisplatin (Reference) | 5.70 ± 0.76 | 12.93 ± 0.91 | 14.42 ± 1.69 |
| Serial | Compounds | IC50 Values (µM) a | |
|---|---|---|---|
| Aurora-A | Aurora-B | ||
| 1. | Tylophorine | 21.4 ± 1.24 | 18.3 ± 1.32 |
| 2. | Tylophorinicine | 16.2 ± 1.43 | 10.2 ± 1.21 |
| 3. | Tylophorinine | 4.1 ± 1.08 | 3.6 ± 0.93 |
| 4. | Tylophorinine N-Oxide | >100 | >100 |
| 5. | Tylophorinidine | 0.6 ± 0.72 | 1.3 ± 0.58 |
| 6. | Tylophorinidine-N-Oxide | 2.6 ± 0.86 | 6.5 ± 0.76 |
| 7. | Septicine | >100 | >100 |
| 8. | Chlorogenic acid | ND | ND |
| 9. | Chlorogenic acid methyl ester | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostafa, E.M.; Mohammed, H.A.; Musa, A.; Abdelgawad, M.A.; Al-Sanea, M.M.; Almahmoud, S.A.; Ghoneim, M.M.; Gomaa, H.A.M.; Rahman, F.E.-Z.S.A.; Shalaby, K.; et al. In Vitro Anti-Proliferative, and Kinase Inhibitory Activity of Phenanthroindolizidine Alkaloids Isolated from Tylophora indica. Plants 2022, 11, 1295. https://doi.org/10.3390/plants11101295
Mostafa EM, Mohammed HA, Musa A, Abdelgawad MA, Al-Sanea MM, Almahmoud SA, Ghoneim MM, Gomaa HAM, Rahman FE-ZSA, Shalaby K, et al. In Vitro Anti-Proliferative, and Kinase Inhibitory Activity of Phenanthroindolizidine Alkaloids Isolated from Tylophora indica. Plants. 2022; 11(10):1295. https://doi.org/10.3390/plants11101295
Chicago/Turabian StyleMostafa, Ehab M., Hamdoon A. Mohammed, Arafa Musa, Mohamed A. Abdelgawad, Mohammad M. Al-Sanea, Suliman A. Almahmoud, Mohammed M. Ghoneim, Hesham A. M. Gomaa, Fatema El-Zahraa S. Abdel Rahman, Khaled Shalaby, and et al. 2022. "In Vitro Anti-Proliferative, and Kinase Inhibitory Activity of Phenanthroindolizidine Alkaloids Isolated from Tylophora indica" Plants 11, no. 10: 1295. https://doi.org/10.3390/plants11101295
APA StyleMostafa, E. M., Mohammed, H. A., Musa, A., Abdelgawad, M. A., Al-Sanea, M. M., Almahmoud, S. A., Ghoneim, M. M., Gomaa, H. A. M., Rahman, F. E. -Z. S. A., Shalaby, K., Selim, S., & Khan, R. A. (2022). In Vitro Anti-Proliferative, and Kinase Inhibitory Activity of Phenanthroindolizidine Alkaloids Isolated from Tylophora indica. Plants, 11(10), 1295. https://doi.org/10.3390/plants11101295