
Transcriptome Analysis Revealed the Potential Molecular Mechanism of Anthocyanidins’ Improved Salt Tolerance in Maize Seedlings

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
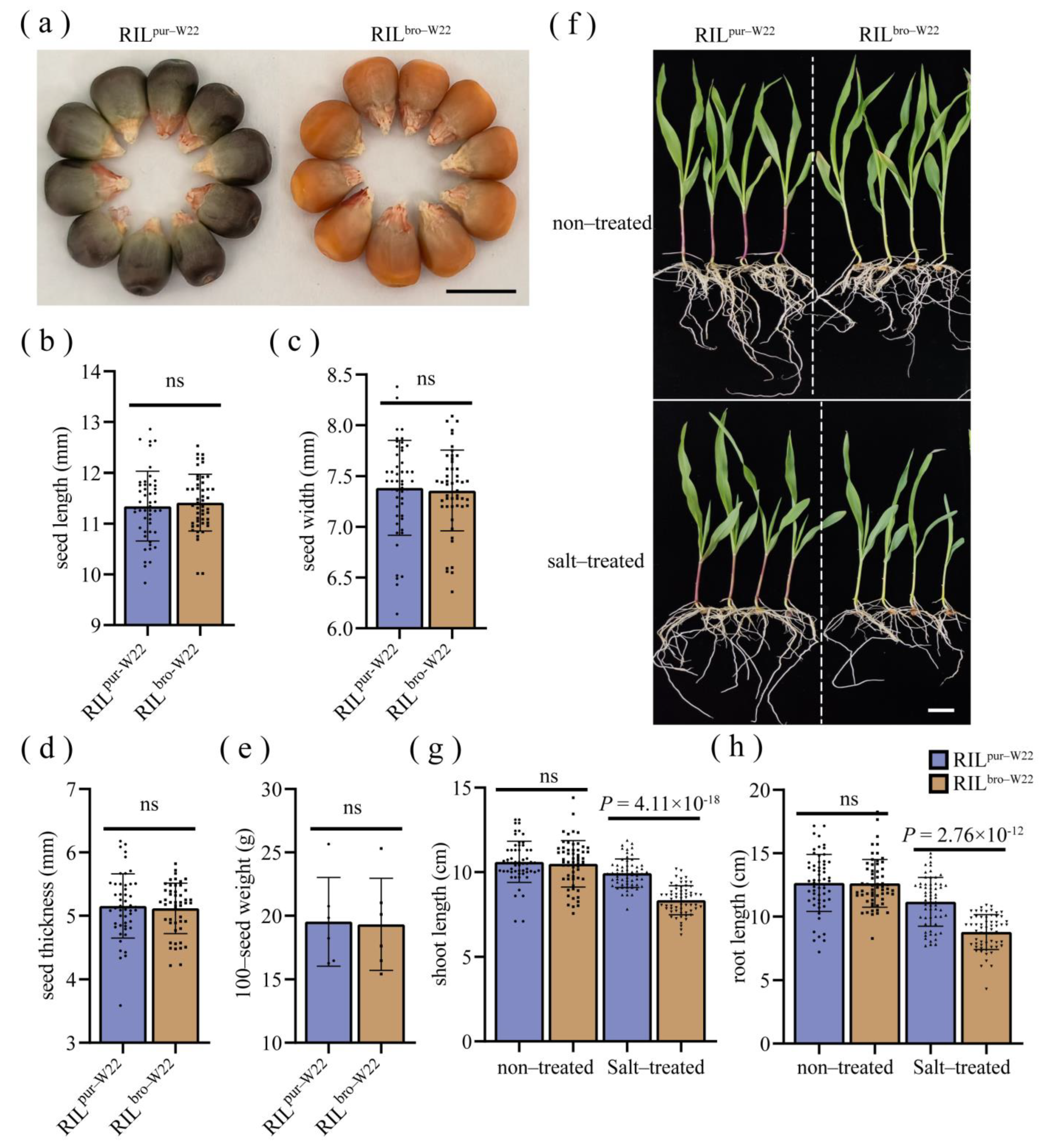
2.1. The Purple–Colored W22 and Bronze–Colored W22 Show Different Seedling Growth under Salt Stress
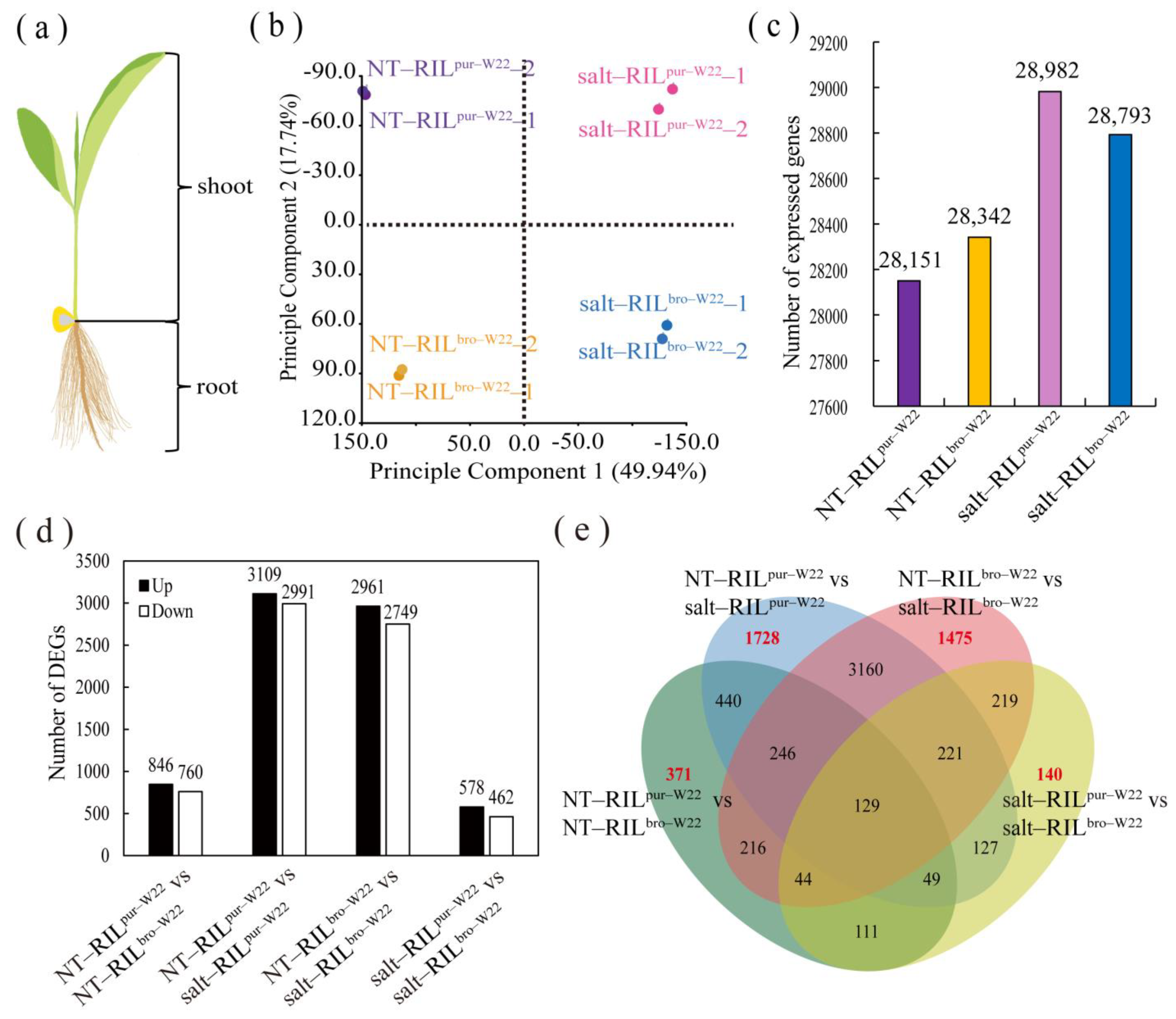
2.2. Transcriptome Analysis of RILs undergoing Salt Treatment
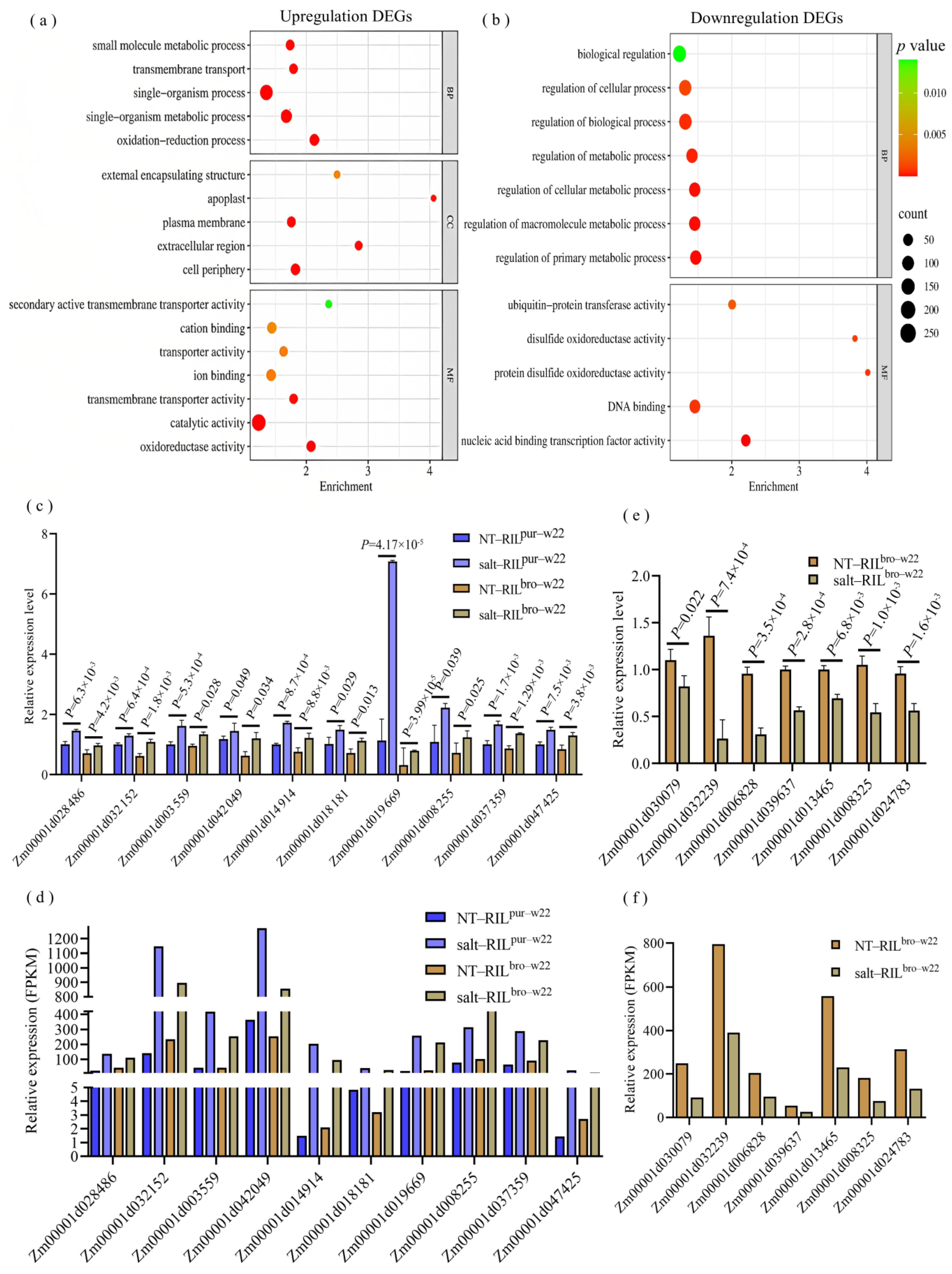
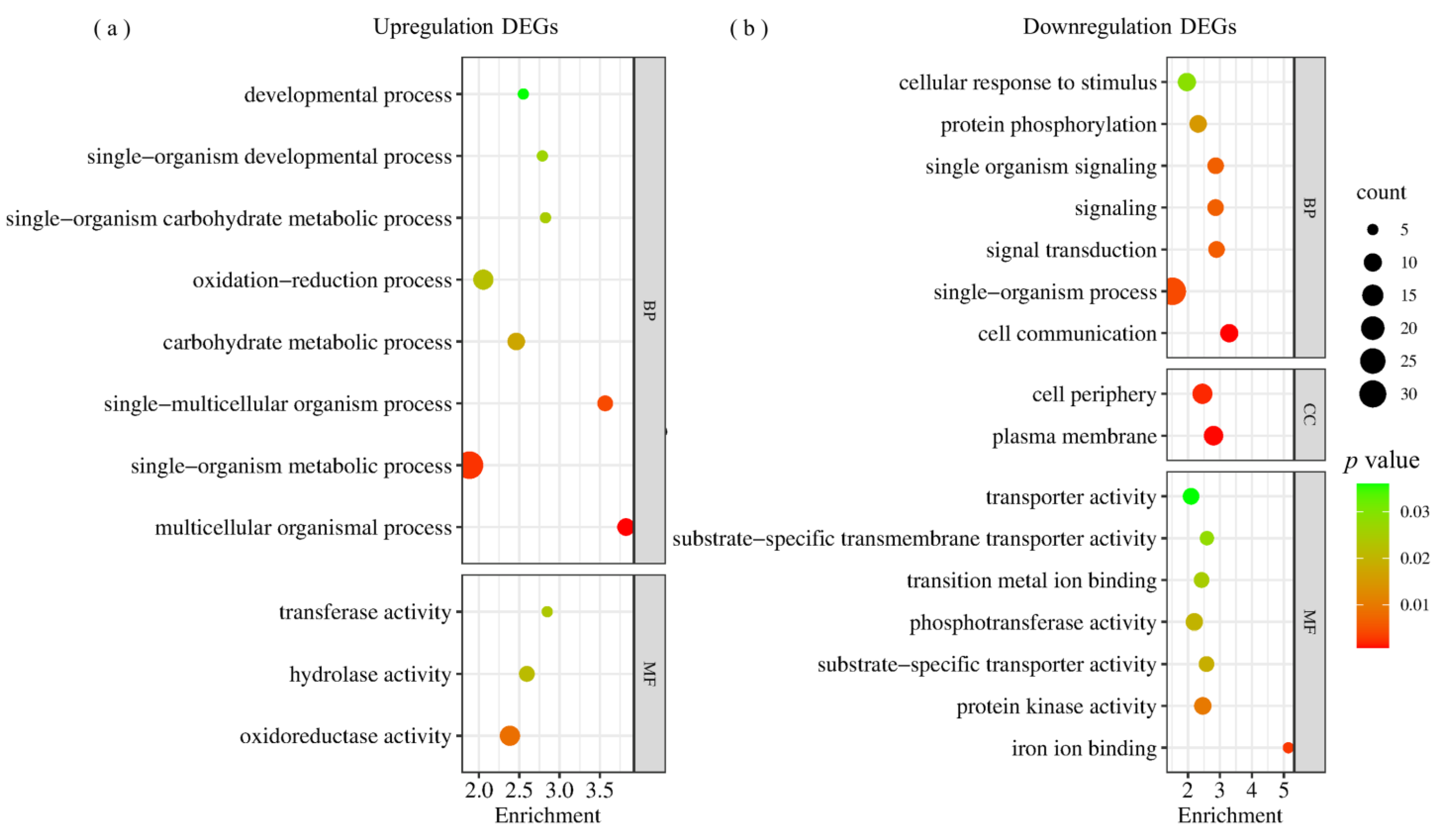
2.3. Differentially Expressed Genes in RILpur–W22 and RILbro–W22 Seedlings
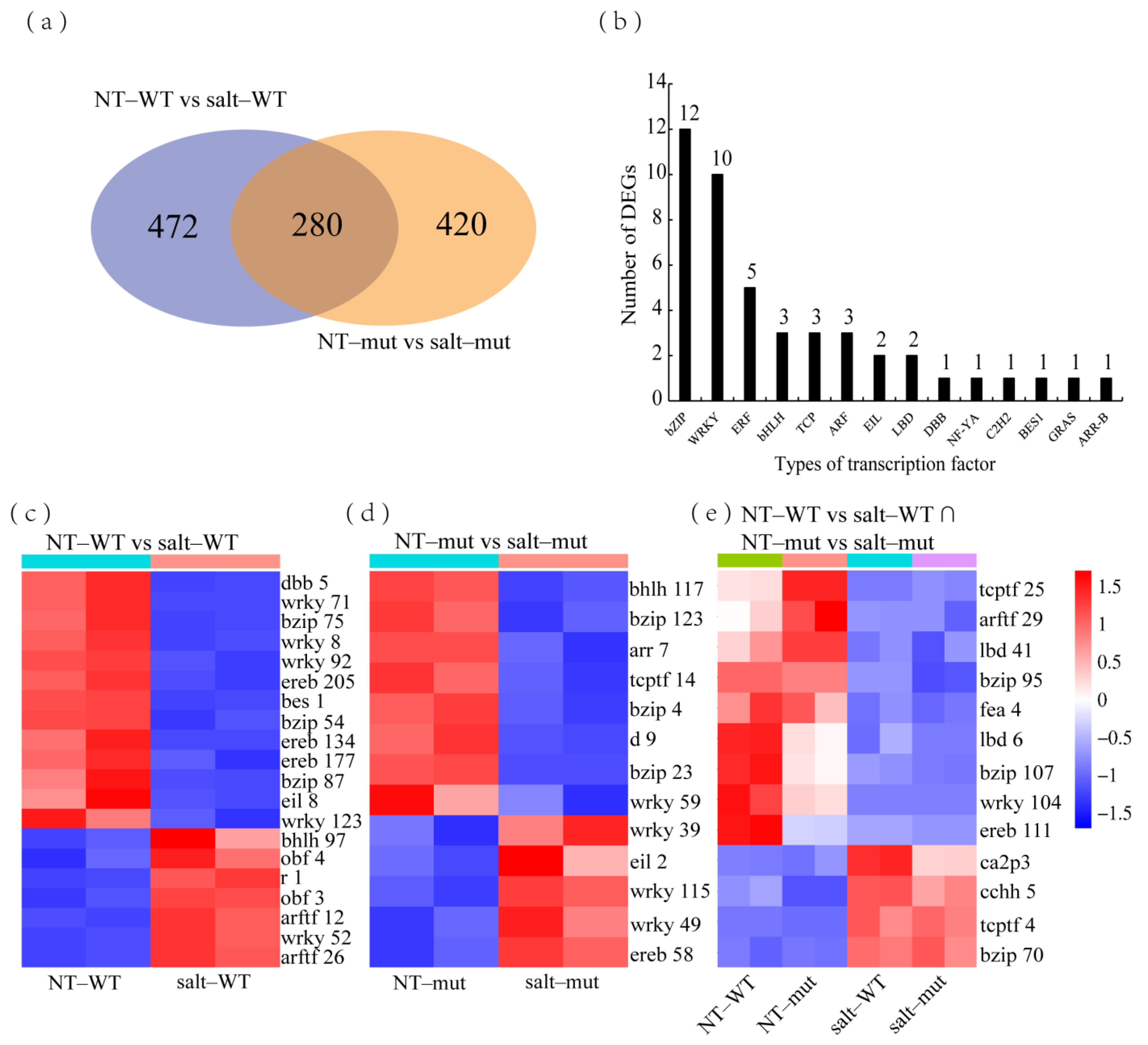
2.4. Common Salt–Induced DEGs in RILpur–W22 and RILbro–W22
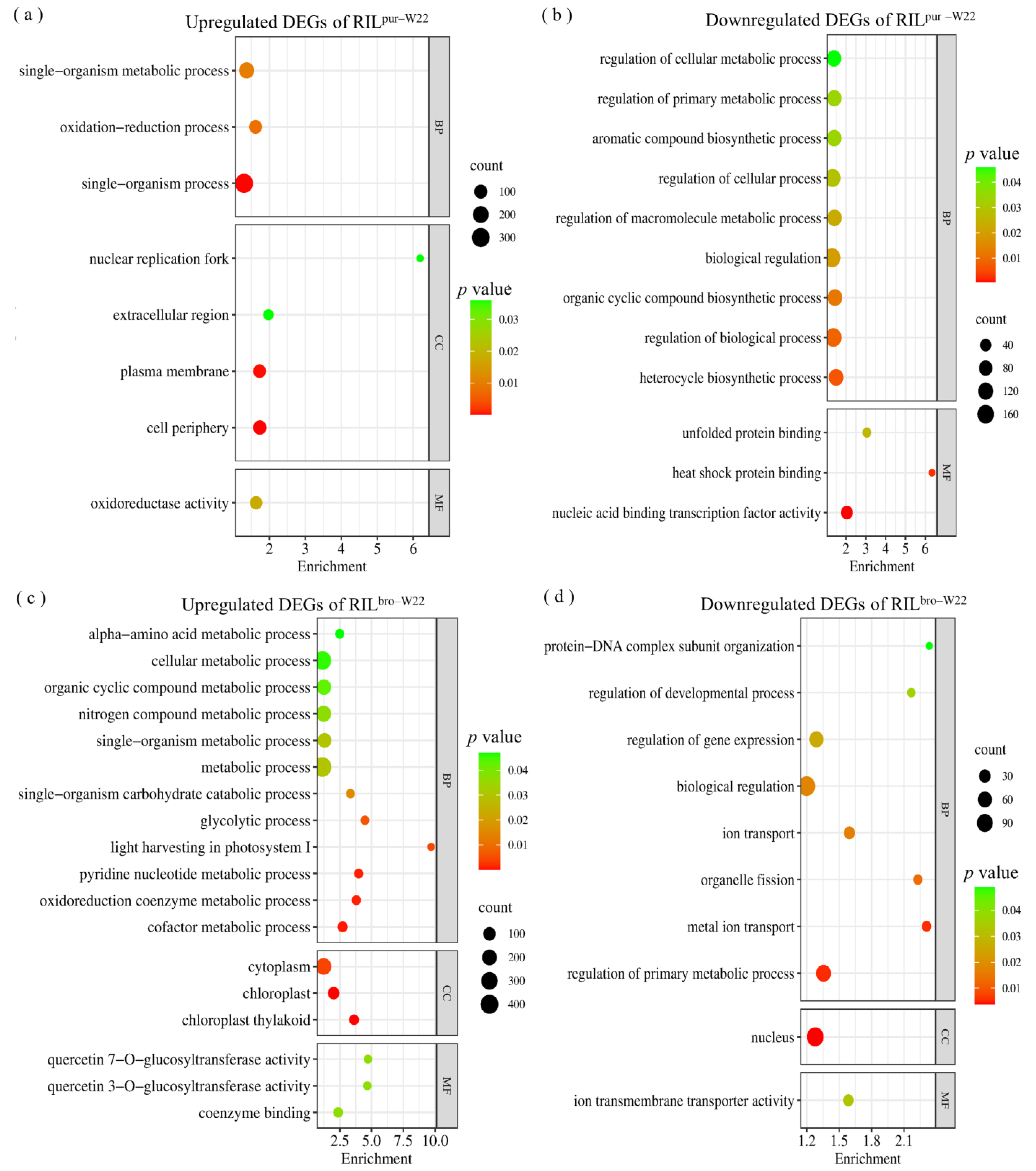
2.5. Specific Salt–Induced DEGs in RILpur–W22 and RILbro–W22
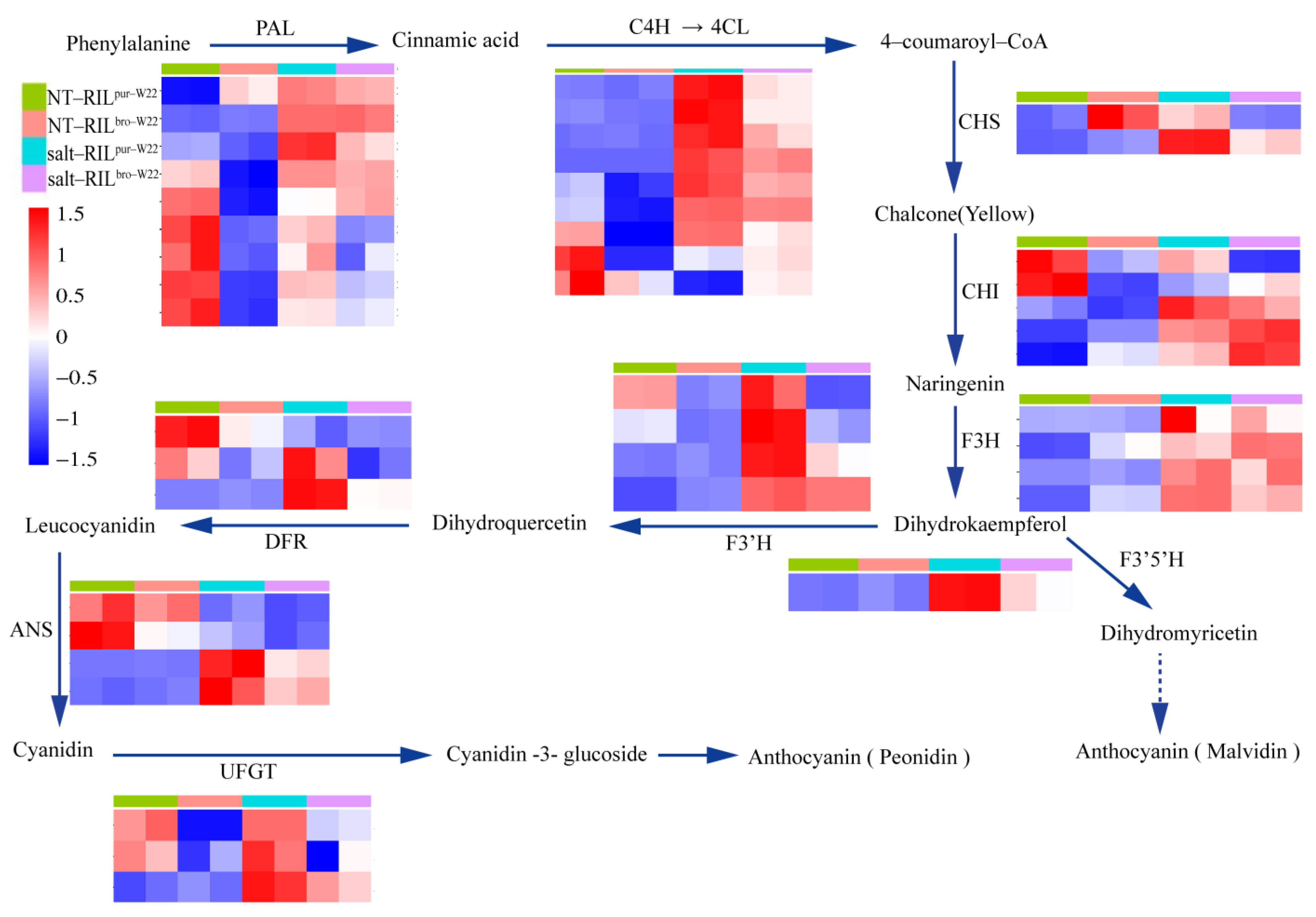
2.6. Analysis of DEGs Related to Anthocyanin Biosynthesis in RILs
3. Discussions
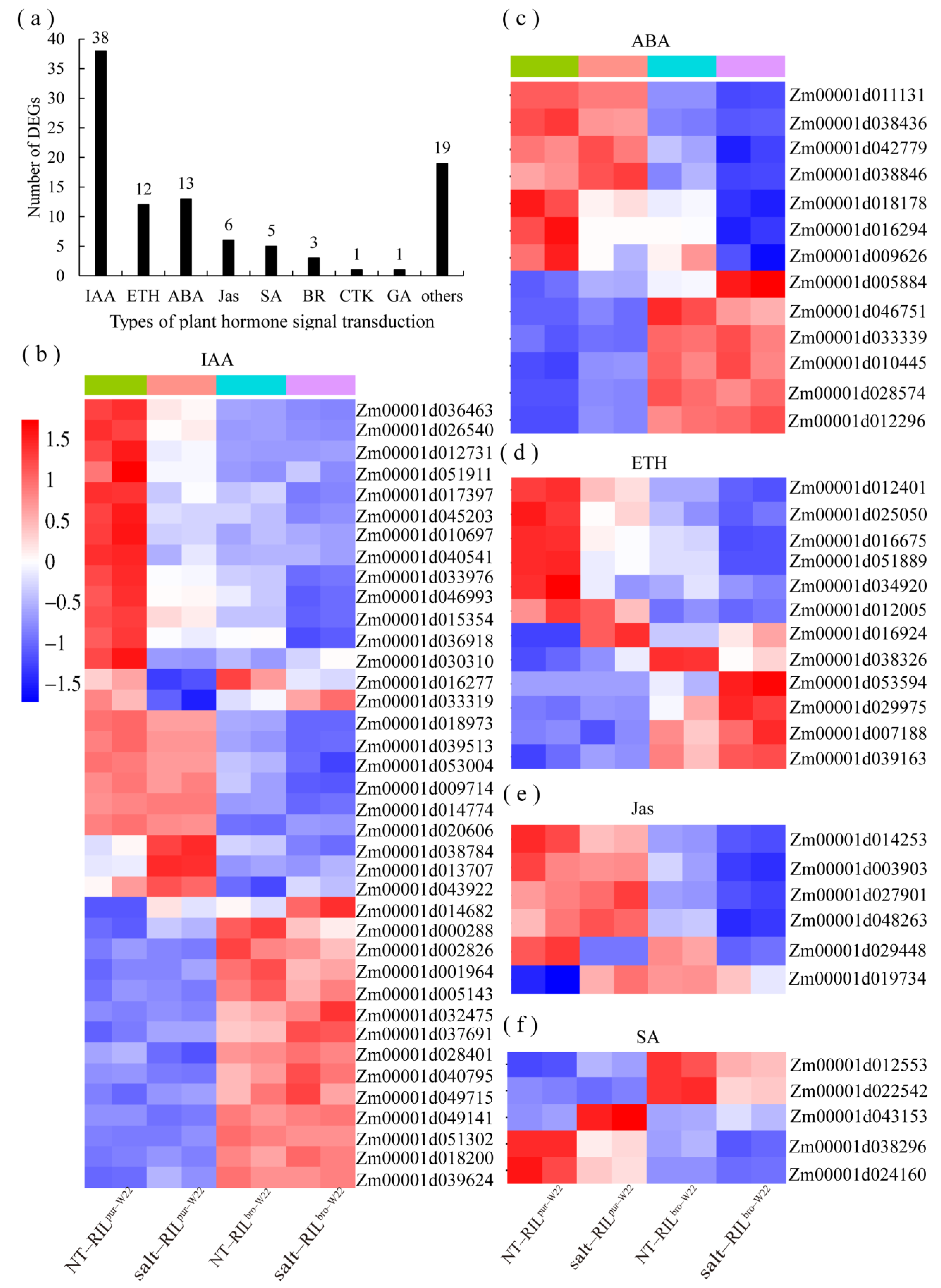
3.1. The Salt Stress Tolerance in Maize Seedlings May Be Mediated by Plant Hormones
3.2. Mining Transcription Factors That Potentially Regulate Salt Stress Tolerance in Maize Seedlings
4. Materials and Methods
4.1. Evaluation of Plant Materials and Traits
4.2. Phenotypic Analysis of Seed Morphology
4.3. RNA Extraction and Sequencing
4.4. Sequence Data Analysis
4.5. Cluster Analysis and Functional Annotation Enrichment Analysis
4.6. qRT–PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prasanna, B.M.; Palacios–Rojas, N.; Hossain, F.; Muthusamy, V.; Menkir, A.; Dhliwayo, T.; Ndhlela, T.; San Vicente, F.; Nair, S.K.; Vivek, B.S.; et al. Molecular breeding for nutritionally enriched maize: Status and prospects. Front. Genet. 2020, 10, 1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, R.; Ferron, L.; Papetti, A. Colored corn: An up–date on metabolites extraction, health implication, and potential use. Molecules 2021, 26, 199. [Google Scholar] [CrossRef] [PubMed]
- Suriano, S.; Balconi, C.; Valoti, P.; Redaelli, R. Comparison of total polyphenols, profile anthocyanins, color analysis, carotenoids and tocols in pigmented maize. LWT–Food Sci. Technol. 2021, 144, 111257. [Google Scholar] [CrossRef]
- Chatham, L.A.; Paulsmeyer, M.; Juvik, J.A. Prospects for economical natural colorants: Insights from maize. Theor. Appl. Genet. 2019, 132, 2927–2946. [Google Scholar] [CrossRef] [PubMed]
- Paulaneyer, M.; Chatham, L.; Becker, T.; West, M.; West, L.; Juvik, J. Survey of anthocyanin composition and concentration in diverse maize germplasms. J. Agric. Food Chem. 2017, 65, 4341–4350. [Google Scholar] [CrossRef]
- Zilic, S.; Serpen, A.; Akillioglu, G.; Gokmen, V.; Vancetovic, J. Phenolic compounds, carotenoids, anthocyanins, and antioxidant capacity of colored maize (Zea mays L.) kernels. J. Agric. Food Chem. 2012, 60, 1224–1231. [Google Scholar] [CrossRef]
- Mannino, G.; Di Stefano, V.; Lauria, A.; Pitonzo, R.; Gentile, C. Vaccinium macrocarpon (Cranberry)–based dietary supplements: Variation in mass uniformity, proanthocyanidin dosage and anthocyanin profile demonstrates quality control standard needed. Nutrients 2020, 12, 992. [Google Scholar] [CrossRef] [Green Version]
- Pojer, E.; Mattivi, F.; Johnson, D.; Stockley, C.S. The case for anthocyanin consumption to promote human health: A review. Compr. Rev. Food Sci. Food Saf. 2013, 12, 483–508. [Google Scholar] [CrossRef]
- Cruz, L.; Basilio, N.; Mateus, N.; de Freitas, V.; Pina, F. Natural and synthetic flavylium–based dyes: The chemistry behind the color. Chem. Rev. 2022, 122, 1416–1481. [Google Scholar] [CrossRef]
- Kou, M.; Liu, Y.J.; Li, Z.Y.; Zhang, Y.G.; Tang, W.; Yan, H.; Wang, X.; Chen, X.G.; Su, Z.X.; Arisha, M.H.; et al. A novel glutathione S–transferase gene from sweet potato, IbGSTF4, is involved in anthocyanin sequestration. Plant Physiol. Bioch. 2019, 135, 395–403. [Google Scholar] [CrossRef]
- Graham, T.L. Flavonoid and flavonol glycoside metabolism in Arabidopsis. Plant Physiol. Bioch. 1998, 36, 135–144. [Google Scholar] [CrossRef]
- Anderson, D.W.; Gueffroy, D.E.; Webb, A.D.; Kepner, R.E. Identification of acetic acid as an acylating agent of anthocyanin pigments in grapes. Phytochemistry 1970, 9, 1579–1583. [Google Scholar] [CrossRef]
- Jez, J.M.; Noel, J.P. Reaction mechanism of chalcone isomerase: PH–dependence, diffusion control, and product binding differences. J. Biol. Chem. 2002, 277, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Kovinich, N.; Saleem, A.; Arnason, J.T.; Miki, B. Functional characterization of a UDP–glucose: Flavonoid 3–O–glucosyltransferase from the seed coat of black soybean (Glycine max (L.) Merr.). Phytochemistry 2010, 71, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013, 18, 477–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Huo, J.; Liu, J.; Yu, J.; Zhou, J.; Sun, C.; Wang, Y.; Leng, F. Anthocyanins distribution, transcriptional regulation, epigenetic and post–translational modification in fruits. Food Chem. 2023, 411, 135540. [Google Scholar] [CrossRef]
- Li, X.L.; Qian, X.K.; Lu, X.; Wang, X.H.; Jia, N.; Zhang, M.S.; Ren, M.J. Upregulated structural and regulatory genes involved in anthocyanin biosynthesis for coloration of purple grains during the middle and late grain–filling stages. Plant Physiol. Bioch. 2018, 130, 235–247. [Google Scholar] [CrossRef]
- Ming, H.N.; Wang, Q.; Wu, Y.; Liu, H.M.; Zheng, L.M.; Zhang, G.F. Transcriptome analysis reveals the mechanism of anthocyanidins biosynthesis during grains development in purple corn (Zea mays L.). J. Plant Physiol. 2021, 257, 153328. [Google Scholar] [CrossRef]
- Li, T.C.; Zhang, W.; Yang, H.Y.; Dong, Q.; Ren, J.; Fan, H.H.; Zhang, X.; Zhou, Y.B. Comparative transcriptome analysis reveals differentially expressed genes related to the tissue–specific accumulation of anthocyanins in pericarp and aleurone layer for maize. Sci. Rep. 2019, 9, 2485. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, D.L.; Peng, Y.X.; Cai, M.H.; Liang, Z.; Yuan, Z.P.; Du, X.M.; Wang, J.H.; Schnable, P.S.; Gu, R.L.; et al. The anthocyanin accumulation related ZmBZ1, facilitates seedling salinity stress tolerance via ROS scavenging. Int. J. Mol. Sci. 2022, 23, 16123. [Google Scholar] [CrossRef]
- Shomali, A.; Das, S.; Arif, N.; Sarraf, M.; Zahra, N.; Yadav, V.; Aliniaeifard, S.; Chauhan, D.K.; Hasanuzzaman, M. Diverse physiological roles of flavonoids in plant environmental stress responses and tolerance. Plants 2022, 11, 3158. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Zhao, D.M.; Akram, M.A.; Guo, C.X.; Jin, H.X.; Hu, W.G.; Zhang, Y.H.; Wang, X.T.; Ma, A.; Xiong, J.L.; et al. Effects of environmental factors on anthocyanin accumulation in the fruits of Lycium ruthenicum Murray across different desert grasslands. J. Plant Physiol. 2022, 279, 153828. [Google Scholar] [CrossRef] [PubMed]
- Kruger, E.; Will, F.; Kumar, K.; Celejewska, K.; Chartier, P.; Masny, A.; Mott, D.; Petit, A.; Savini, G.; Sonsteby, A. Influence of Post–Flowering Climate Conditions on Anthocyanin Profile of Strawberry Cultivars Grown from North to South Europe. Appl. Sci. 2021, 11, 1326. [Google Scholar] [CrossRef]
- Gunther, C.S.; Plunkett, B.J.; Cooney, J.M.; Jensen, D.; Trower, T.M.; Elborough, C.; Nguyen, H.M.; Deng, C.H.; Lafferty, D.J.; Albert, N.W.; et al. Biotic stress–induced and ripening–related anthocyanin biosynthesis are regulated by alternate phytohormone signals in blueberries. Environ. Exp. Bot. 2022, 203, 105065. [Google Scholar] [CrossRef]
- van Zelm, E.; Zhang, Y.X.; Testerink, C. Salt tolerance mechanisms of plants. Annu. Rev. Plant Biol. 2020, 71, 403–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrao, S.; Schmockel, S.M.; Tester, M. Evaluating physiological responses of plants to salinity stress. Ann. Bot. 2017, 119, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Konapala, G.; Mishra, A.K.; Wada, Y.; Mann, M.E. Climate change will affect global water availability through compounding changes in seasonal precipitation and evaporation. Nat. Commun. 2020, 11, 3044. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, S.; Zhao, Z.; You, L.; Harrison, M.D.; Zhang, Z. Enzymatic acylation of cyanidin–3–glucoside with fatty acid methyl esters improves stability and antioxidant activity. Food Chem. 2021, 343, 128482. [Google Scholar] [CrossRef]
- Wang, F.B.; Kong, W.L.; Wong, G.; Fu, L.F.; Peng, R.H.; Li, Z.J.; Yao, Q.H. AtMYB12 regulates flavonoids accumulation and abiotic stress tolerance in transgenic Arabidopsis thaliana. Mol. Genet. Genom. 2016, 291, 1545–1559. [Google Scholar] [CrossRef]
- Wang, D.R.; Yang, K.; Wang, X.; Lin, X.L.; Rui, L.; Liu, H.F.; Liu, D.D.; You, C.X. Overexpression of MdZAT5, an C2H2–Type Zinc Finger Protein, Regulates Anthocyanin Accumulation and Salt Stress Response in Apple Calli and Arabidopsis. Int. J. Mol. Sci. 2022, 23, 1897. [Google Scholar] [CrossRef]
- Kim, J.; Lee, W.J.; Vu, T.T.; Jeong, C.Y.; Hong, S.W.; Lee, H. High accumulation of anthocyanins via the ectopic expression of AtDFR confers significant salt stress tolerance in Brassica napus L. Plant Cell Rep. 2017, 36, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.N.; Chen, M.; Zheng, N.N.; Xu, Z.Y.; Zhang, J.; Hu, X.M.; Li, L.; Gu, R.L.; Du, X.M.; Wang, J.H. Transcriptome analysis identifies novel genes associated with low–temperature seed germination in sweet corn. Plants 2023, 12, 159. [Google Scholar] [CrossRef]
- Li, X.H.; Hu, H.R.; Hu, X.M.; Wang, G.H.; Du, X.M.; Li, L.; Wang, F.; Fu, J.J.; Wang, G.Y.; Wang, J.H.; et al. Transcriptome analysis of near–isogenic lines provides novel insights into genes associated with seed low–temperature germination ability in maize (Zea mays L.). Plants 2022, 11, 887. [Google Scholar] [CrossRef]
- Liu, W.W.; Zhang, Y.L.; He, H.; He, G.M.; Deng, X.W. From hybrid genomes to heterotic trait output: Challenges and opportunities. Curr. Opin. Plant Biol. 2022, 66, 102193. [Google Scholar] [CrossRef]
- Zheng, H.X.; Yang, Z.; Wang, W.Q.; Guo, S.J.; Li, Z.X.; Liu, K.C.; Sui, N. Transcriptome analysis of maize inbred lines differing in drought tolerance provides novel insights into the molecular mechanisms of drought responses in roots. Plant Physiol. Bioch. 2020, 149, 11–26. [Google Scholar] [CrossRef]
- Ma, S.Q.; Lv, L.; Meng, C.; Zhang, C.S.; Li, Y.Q. Integrative analysis of the metabolome and transcriptome of Sorghum bicolor reveals dynamic changes in flavonoids accumulation under saline–alkali stress. J. Agric. Food Chem. 2020, 68, 14781–14789. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Su, N.N.; Jia, L.; Tian, J.Y.; Li, H.; Huang, L.S.; Shen, Z.G.; Cui, J. Transcriptome analysis of radish sprouts hypocotyls reveals the regulatory role of hydrogen–rich water in anthocyanin biosynthesis under UV–A. BMC Plant Biol. 2018, 18, 227. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.R.; Wang, P.P.; Chen, J.Q.; Li, C.J.; Wang, Y.Z.; Yuan, Y.B.; Fang, J.G.; Leng, X.P. Genome–wide identification and analysis of B–BOX gene family in grapevine reveal its potential functions in berry development. BMC Plant Biol. 2020, 20, 72. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.J.; Fang, S.Z.; Liu, Y.; Shang, X.L. Integrative analysis of metabolome and transcriptome reveals molecular regulatory mechanism of flavonoid biosynthesis in Cyclocarya paliurus under salt stress. Ind. Crop. Prod. 2021, 170, 113823. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, B.G.; Park, Y.; Ko, J.H.; Lim, C.E.; Lim, J.; Lim, Y.; Ahn, J.H. Characterization of flavonoid 7–O–glucosyltransferase from Arabidopsis thaliana. Biosci. Biotechnol. Biochem. 2006, 70, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Arif, Y.; Bajguz, A.; Hayat, S. The role of quercetin in plants. Plant Physiol. Bioch. 2021, 166, 10–19. [Google Scholar] [CrossRef]
- Naing, A.H.; Kim, C.K. Abiotic stress–induced anthocyanins in plants: Their role in tolerance to abiotic stresses. Physiol. Plant. 2021, 172, 1711–1723. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, W.J.; Le, Q.T.; Hong, S.W.; Lee, H. Growth performance can be increased under high nitrate and high salt stress through enhanced nitrate reductase activity in Arabidopsis anthocyanin over–producing mutant plants. Front. Plant Sci. 2021, 12, 644455. [Google Scholar] [CrossRef]
- Sharma, A.; Shahzad, B.; Kumar, V.; Kohli, S.K.; Sidhu, G.; Bali, A.S.; Handa, N.; Kapoor, D.; Bhardwaj, R.; Zheng, B.S. Phytohormones regulate accumulation of osmolytes under abiotic stress. Biomolecules 2019, 9, 285. [Google Scholar] [CrossRef] [Green Version]
- Rai, K.K. Revisiting the critical role of ROS and RNS in plant defense. J. Plant Growth Regul. 2022. [Google Scholar] [CrossRef]
- Yu, Z.P.; Duan, X.B.; Luo, L.; Dai, S.J.; Ding, Z.J.; Xia, G.M. How plant hormones mediate salt stress responses. Trends Plant Sci. 2020, 25, 1117–1130. [Google Scholar] [CrossRef]
- Gao, H.N.; Jiang, H.; Cui, J.Y.; You, C.X.; Li, Y.Y. Review: The effects of hormones and environmental factors on anthocyanin biosynthesis in apple. Plant Sci. 2021, 312, 111024. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.T.; Hu, X.Q.; Wang, P.; Gao, L.Y.; Pei, Y.K.; Ge, Z.Y.; Ge, X.Y.; Li, F.G.; Hou, Y.X. GhPLP2 positively regulates cotton resistance to verticillium wilt by modulating fatty acid accumulation and jasmonic acid signaling pathway. Front. Plant Sci. 2021, 12, 749630. [Google Scholar] [CrossRef]
- Li, X.Y.; Liao, M.M.; Huang, J.Y.; Xu, Z.; Lin, Z.Q.; Ye, N.H.; Zhang, Z.S.; Peng, X.X. Glycolate oxidase–dependent H2O2 production regulates IAA biosynthesis in rice. BMC Plant Biol. 2021, 21, 326. [Google Scholar] [CrossRef]
- Jia, K.P.; Mi, J.N.; Ali, S.; Ohyanagi, H.; Moreno, J.C.; Ablazov, A.; Balakrishna, A.; Berqdar, L.; Fiore, A.; Diretto, G.; et al. An alternative, zeaxanthin epoxidase–independent abscisic acid biosynthetic pathway in plants. Mol. Plant 2022, 15, 151–166. [Google Scholar] [CrossRef]
- Fu, B.L.; Wang, W.Q.; Liu, X.F.; Duan, X.W.; Allan, A.C.; Grierson, D.; Yin, X.R. An ethylene–hypersensitive methionine sulfoxide reductase regulated by NAC transcription factors increases methionine pool size and ethylene production during kiwifruit ripening. New Phytol. 2021, 232, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Postiglione, A.E.; Muday, G.K. Abscisic acid increases hydrogen peroxide in mitochondria to facilitate stomatal closure. Plant Physiol. 2022, 192, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.K.; Khan, Z.; Wu, H.H.; Khan, M.N.; Hu, L.Y. Gibberellic acid priming improved rapeseed drought tolerance by modulating root morphology, ROS homeostasis, and chloroplast autophagy. J. Plant Growth Regul. 2022. [Google Scholar] [CrossRef]
- Zhang, A.Y.; Yang, X.; Lu, J.; Song, F.Y.; Sun, J.H.; Wang, C.; Lian, J.; Zhao, L.L.; Zhao, B.C. OsIAA20, an Aux/IAA protein, mediates abiotic stress tolerance in rice through an ABA pathway. Plant Sci. 2021, 308, 110903. [Google Scholar] [CrossRef]
- Wang, C.K.; Han, P.L.; Zhao, Y.W.; Ji, X.L.; Yu, J.Q.; You, C.X.; Hu, D.G.; Hao, Y.J. Auxin regulates anthocyanin biosynthesis through the auxin repressor protein MdIAA26. Biochem. Biophys. Res. Commun. 2020, 533, 717–722. [Google Scholar] [CrossRef]
- Ye, H.; Qiao, L.Y.; Guo, H.Y.; Guo, L.P.; Ren, F.; Bai, J.F.; Wang, Y.K. Genome–wide identification of wheat WRKY gene family reveals that TaWRKY75–A is referred to drought and salt resistances. Front. Plant Sci. 2021, 12, 663118. [Google Scholar] [CrossRef]
- Liu, L.; DU, H.; Tang, X.; Wu, Y.; Huang, Y.; Tang, Y. The roles of MYB transcription factors on plant defense responses and its molecular mechanism. Yi Chuan = Hereditas 2008, 30, 1265–1271. [Google Scholar] [CrossRef]
- Li, M.; Wu, Z.Y.; Gu, H.; Cheng, D.W.; Guo, X.Z.; Li, L.; Shi, C.Y.; Xu, G.Y.; Gu, S.C.; Abid, M.; et al. AvNAC030, a NAC domain transcription factor, enhances salt stress tolerance in kiwifruit. Int. J. Mol. Sci. 2021, 22, 11897. [Google Scholar] [CrossRef]
- Huang, J.J.; Liu, F.H.; Chao, D.; Xin, B.N.; Liu, K.; Cao, S.L.; Chen, X.X.; Peng, L.Y.; Zhang, B.L.; Fu, S.; et al. The WRKY transcription factor OsWRKY54 is involved in salt tolerance in rice. Int. J. Mol. Sci. 2022, 23, 11999. [Google Scholar] [CrossRef]
- Lee, Y.H.; Song, S.I. OsbZIP62 positively regulates drought and salt stress tolerance and ABA signaling in rice. J. Plant Biol. 2022, 66, 123–133. [Google Scholar] [CrossRef]
- Huang, S.Z.; Hu, L.J.; Zhang, S.H.; Zhang, M.X.; Jiang, W.Z.; Wu, T.; Du, X.L. Rice OsWRKY50 mediates ABA–dependent seed germination and seedling growth, and ABA–independent salt stress tolerance. Int. J. Mol. Sci. 2021, 22, 8625. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.L.; Cheng, Y.; Ning, C.L.; Yang, C.M.; Dong, X.H.; Cao, H.L.; Sun, Q. Non–destructive viability discrimination for individual scutellaria baicalensis seeds based on high–throughput phenotyping and machine learning. Agriculture 2022, 12, 1616. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph–based genome alignment and genotyping with hisat2 and hisat–genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA–seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.Y.; You, Q.; Yi, X.; Du, Z.; Xu, W.Y.; Su, Z. Agrigo v2.0: A go analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, D.C.; Luo, H.T.; Huo, P.P.; Wang, Z.H.; Zhang, S.; He, Z.H.; Wu, Y.; Zhao, L.H.; Liu, J.J.; Guo, J.C.; et al. Kobas–i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lines | Rep | Total Reads | Rate of Total Mapped Reads (%) | Num. of Expressed Genes | Rate of Expressed Genes (%) |
|---|---|---|---|---|---|
| NT–RILpur–W22 | 1 | 47,540,654 | 96.8 | 28,187 | 60.72 |
| 2 | 47,933,524 | 97.04 | 28,115 | 60.56 | |
| NT–RILbro–W22 | 1 | 44,597,506 | 97.33 | 28,317 | 61.00 |
| 2 | 45,014,472 | 96.00 | 28,367 | 61.10 | |
| salt–RILpur–W22 | 1 | 47,815,598 | 96.41 | 28,991 | 62.45 |
| 2 | 46,306,714 | 95.15 | 28,974 | 62.41 | |
| salt–RILbro–W22 | 1 | 45,334,562 | 96.63 | 28,886 | 62.22 |
| 2 | 45,274,946 | 95.58 | 28,701 | 61.82 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Yuan, Z.; Li, D.; Cai, M.; Liang, Z.; Chen, Q.; Du, X.; Wang, J.; Gu, R.; Li, L. Transcriptome Analysis Revealed the Potential Molecular Mechanism of Anthocyanidins’ Improved Salt Tolerance in Maize Seedlings. Plants 2023, 12, 2793. https://doi.org/10.3390/plants12152793
Wang J, Yuan Z, Li D, Cai M, Liang Z, Chen Q, Du X, Wang J, Gu R, Li L. Transcriptome Analysis Revealed the Potential Molecular Mechanism of Anthocyanidins’ Improved Salt Tolerance in Maize Seedlings. Plants. 2023; 12(15):2793. https://doi.org/10.3390/plants12152793
Chicago/Turabian StyleWang, Jie, Zhipeng Yuan, Delin Li, Minghao Cai, Zhi Liang, Quanquan Chen, Xuemei Du, Jianhua Wang, Riliang Gu, and Li Li. 2023. "Transcriptome Analysis Revealed the Potential Molecular Mechanism of Anthocyanidins’ Improved Salt Tolerance in Maize Seedlings" Plants 12, no. 15: 2793. https://doi.org/10.3390/plants12152793
APA StyleWang, J., Yuan, Z., Li, D., Cai, M., Liang, Z., Chen, Q., Du, X., Wang, J., Gu, R., & Li, L. (2023). Transcriptome Analysis Revealed the Potential Molecular Mechanism of Anthocyanidins’ Improved Salt Tolerance in Maize Seedlings. Plants, 12(15), 2793. https://doi.org/10.3390/plants12152793