

Annotation of the Turnera subulata (Passifloraceae) Draft Genome Reveals the S-Locus Evolved after the Divergence of Turneroideae from Passifloroideae in a Stepwise Manner
 and
and
Abstract
:1. Introduction
2. Results
2.1. Genome Assembly and Annotation
2.2. Comparison of Orthologous Groups between Members of Malpighiales
2.3. Identification of the BAHD Acyltransferase (BAHD) Gene Family
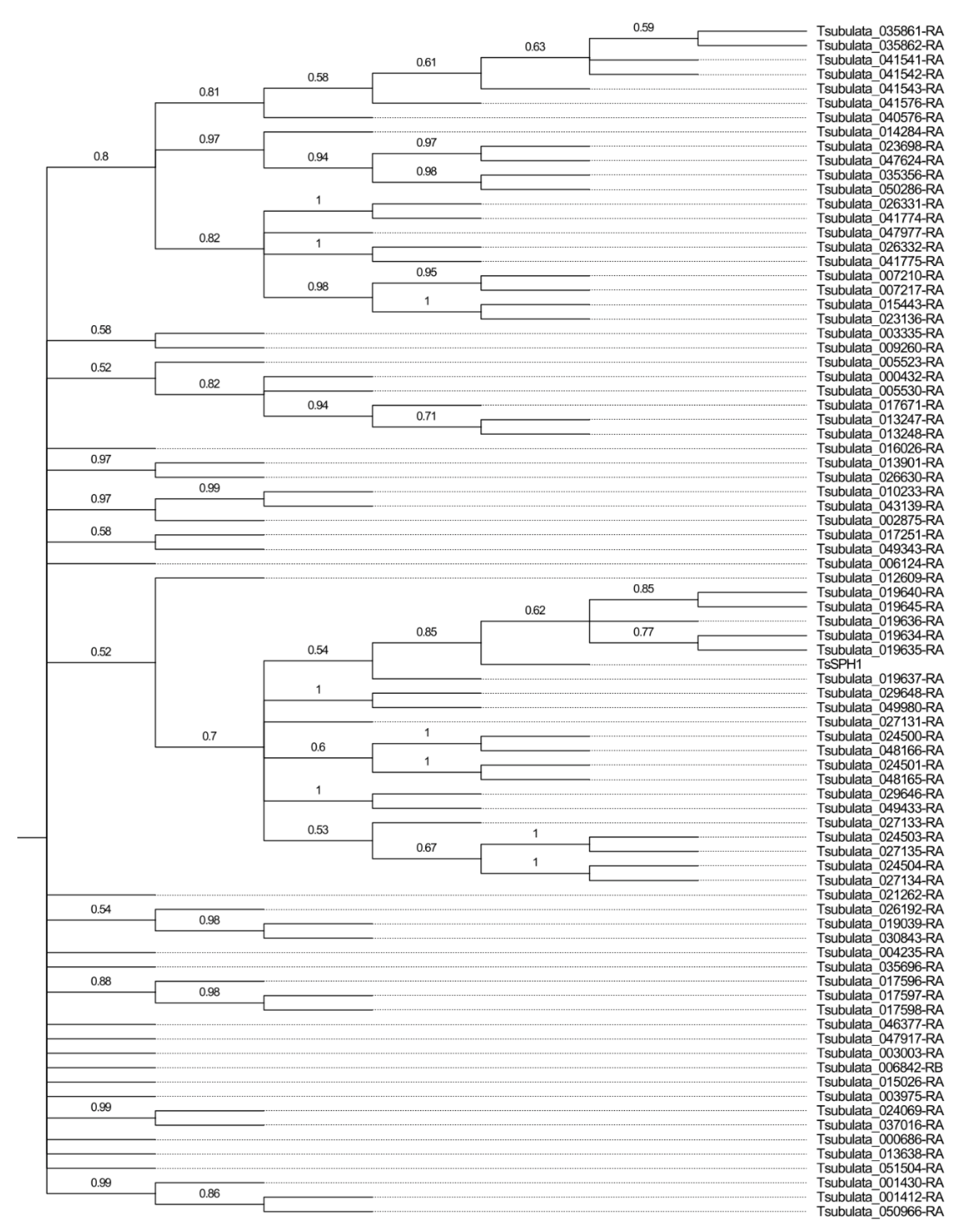
2.4. Identification of the S-Protein Homolog (SPH) Gene Family
2.5. Identification of the YUCCA (YUC) Gene Family
2.6. Possible Origin of the S-Genes
2.7. Expression Patterns of the BAHD and YUCCA Family
2.8. Identifying Hypothetical Neofunctionalization of the S-Genes
3. Discussion
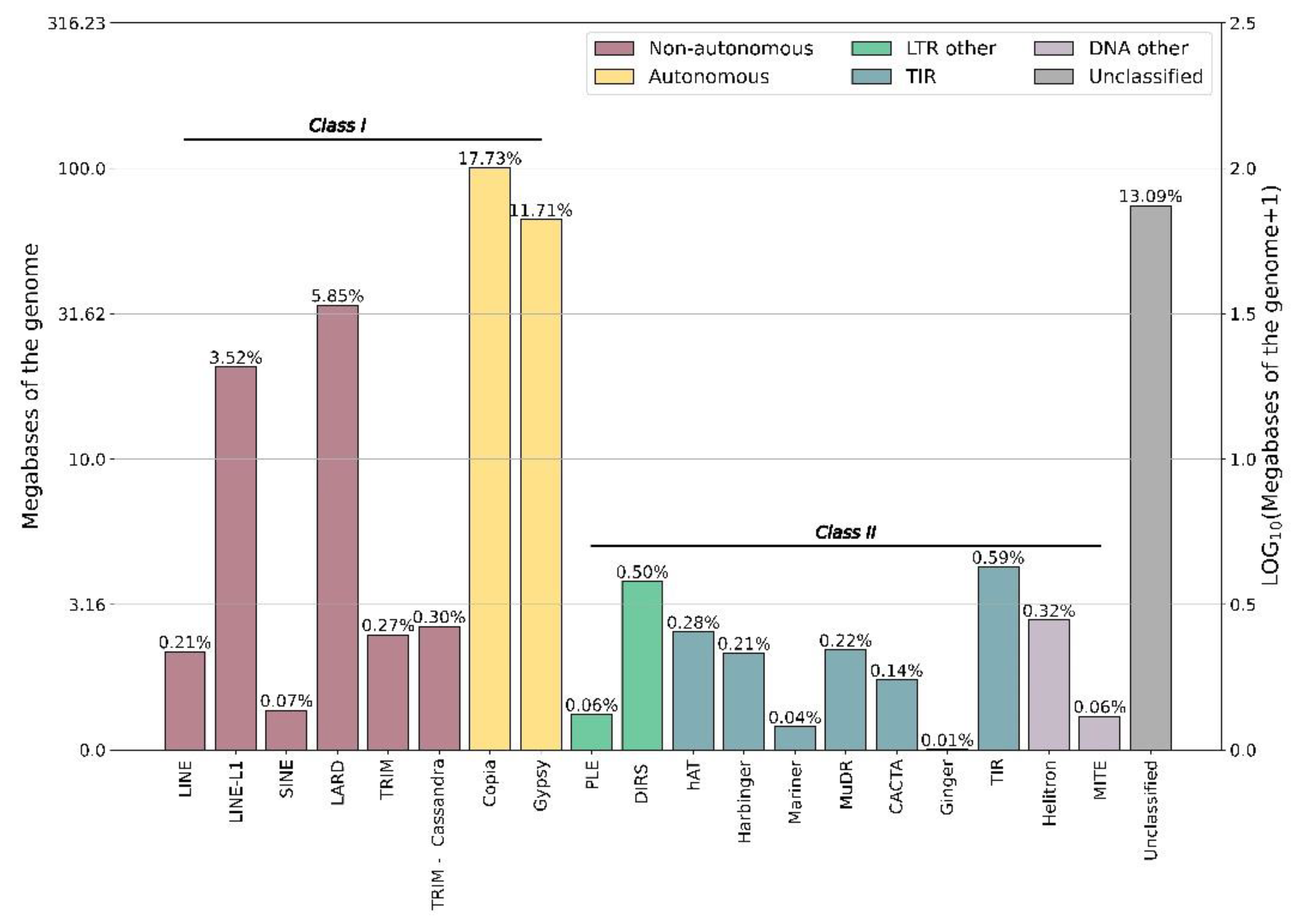
3.1. Repetitive Elements in the Turnera subulata Genome
3.2. The S-Protein Homolog Gene Family Is Present in some Monocots
3.3. The S-Locus Likely Evolved in Turneroideae via a Step-By-Step Manner Similar to the S-Loci from Primula and Linum
3.4. TsBAHD and TsSPH1 Likely Retained Their Ancestral Function, While TsYUC6 Likely Has Undergone Alteration of Function
4. Materials and Methods
4.1. Genome Assembly
4.2. Genome Annotation
4.3. Putative BAHD, SPH, and YUCCA Identification in Turnera subulata
4.4. Data retrieval, Alignment, and Phylogenetic Analysis
4.5. Expression Comparison across Turnera, Populus, and Arabidopsis
4.6. RT-qPCR Analysis of Putative YUCCA Family Members
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrett, S.C.H. ‘A Most Complex Marriage Arrangement’: Recent Advances on Heterostyly and Unresolved Questions. New Phytol. 2019, 224, 1051–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cocker, J.M.; Wright, J.; Webster, M.A.; McMullan, M.; Dyer, S.; Swarbreck, D.; Caccamo, M.; van Oosterhout, C.; Gilmartin, P.M. Genetic Architecture and Evolution of the S Locus Supergene in Primula vulgaris. Nat. Plants 2016, 2, 16188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, J.S.; Hamam, H.J.; Chafe, P.D.J.; Labonne, J.D.J.; Henning, P.M.; McCubbin, A.G. The Long and Short of the S-locus in Turnera (Passifloraceae). New Phytol. 2019, 224, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Valencia, J.; Fracassetti, M.; Berdan, E.L.; Bunikis, I.; Soler, L.; Dainat, J.; Kutschera, V.E.; Losvik, A.; Désamoré, A.; Hughes, P.W.; et al. Genomic Analyses of the Linum Distyly Supergene Reveal Convergent Evolution at the Molecular Level. Curr. Biol. 2022, 32, S0960982222013641. [Google Scholar] [CrossRef]
- Yasui, Y.; Mori, M.; Aii, J.; Abe, T.; Matsumoto, D.; Sato, S.; Hayashi, Y.; Ohnishi, O.; Ota, T. S-LOCUS EARLY FLOWERING 3 Is Exclusively Present in the Genomes of Short-Styled Buckwheat Plants That Exhibit Heteromorphic Self-Incompatibility. PLoS ONE 2012, 7, e31264. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.D.; Russo, G.; Schlapbach, R.; Huu, C.N.; Lenhard, M.; Conti, E. The Draft Genome of Primula veris Yields Insights into the Molecular Basis of Heterostyly. Genome Biol. 2015, 16, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, B.A.; McCubbin, A.G. Sequencing the Genomic Regions Flanking S-Linked PvGLO Sequences Confirms the Presence of Two GLO Loci, One of Which Lies Adjacent to the Style-Length Determinant Gene CYP734A50. Plant Reprod. 2017, 30, 53–67. [Google Scholar] [CrossRef]
- Huu, C.N.; Keller, B.; Conti, E.; Kappel, C.; Lenhard, M. Supergene Evolution via Stepwise Duplications and Neofunctionalization of a Floral-Organ Identity Gene. Proc. Natl. Acad. Sci. USA 2020, 117, 23148–23157. [Google Scholar] [CrossRef]
- Huu, C.N.; Kappel, C.; Keller, B.; Sicard, A.; Takebayashi, Y.; Breuninger, H.; Nowak, M.D.; Bäurle, I.; Himmelbach, A.; Burkart, M.; et al. Presence versus Absence of CYP734A50 Underlies the Style-Length Dimorphism in Primroses. eLife 2016, 5, e17956. [Google Scholar] [CrossRef]
- Huu, C.N.; Plaschil, S.; Himmelbach, A.; Kappel, C.; Lenhard, M. Female Self-Incompatibility Type in Heterostylous Primula Is Determined by the Brassinosteroid-Inactivating Cytochrome P450 CYP734A50. Curr. Biol. 2021, 32, S0960982221016055. [Google Scholar] [CrossRef]
- Potente, G.; Léveillé-Bourret, É.; Yousefi, N.; Choudhury, R.R.; Keller, B.; Diop, S.I.; Duijsings, D.; Pirovano, W.; Lenhard, M.; Szövényi, P.; et al. Comparative Genomics Elucidates the Origin of a Supergene Controlling Floral Heteromorphism. Mol. Biol. Evol. 2022, 39, msac035. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, K.; Nakano, R.; Bando, M.; Shigezane, Y.; Ikeda, K.; Namba, Y.; Kume, S.; Kitabata, T.; Mori, H.; Kubo, Y. Isolation of the Floral Morph-related Genes in Heterostylous Flax (Linum grandiflorum): The Genetic Polymorphism and the Transcriptional and Post-transcriptional Regulations of the S Locus. Plant J. 2012, 69, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Matzke, C.M.; Shore, J.S.; Neff, M.M.; McCubbin, A.G. The Turnera Style S-Locus Gene TsBAHD Possesses Brassinosteroid-Inactivating Activity When Expressed in Arabidopsis thaliana. Plants 2020, 9, 1566. [Google Scholar] [CrossRef] [PubMed]
- Matzke, C.M.; Hamam, H.J.; Henning, P.M.; Dougherty, K.; Shore, J.S.; Neff, M.M.; McCubbin, A.G. Pistil Mating Type and Morphology Are Mediated by the Brassinosteroid Inactivating Activity of the S-Locus Gene BAHD in Heterostylous Turnera Species. Int. J. Mol. Sci. 2021, 22, 10603. [Google Scholar] [CrossRef] [PubMed]
- Henning, P.M.; Shore, J.S.; McCubbin, A.G. The S-Gene YUC6 Pleiotropically Determines Male Mating Type and Pollen Size in Heterostylous Turnera (Passifloraceae): A Novel Neofunctionalization of the YUCCA Gene Family. Plants 2022, 11, 2640. [Google Scholar] [CrossRef]
- Henning, P.M.; Shore, J.S.; McCubbin, A.G. Transcriptome and Network Analyses of Heterostyly in Turnera subulata Provide Mechanistic Insights: Are S-Loci a Red-Light for Pistil Elongation? Plants 2020, 9, 713. [Google Scholar] [CrossRef]
- Stern, D.L. The Genetic Causes of Convergent Evolution. Nat. Rev. Genet. 2013, 14, 751–764. [Google Scholar] [CrossRef]
- Hao, Y.; Qu, Y.; Song, G.; Lei, F. Genomic Insights into the Adaptive Convergent Evolution. Curr. Genom. 2019, 20, 81–89. [Google Scholar] [CrossRef]
- Sackton, T.B.; Clark, N. Convergent Evolution in the Genomics Era: New Insights and Directions. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190102. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Chan, A.P.; Crabtree, J.; Zhao, Q.; Lorenzi, H.; Orvis, J.; Puiu, D.; Melake-Berhan, A.; Jones, K.M.; Redman, J.; Chen, G.; et al. Draft Genome Sequence of the Oilseed Species Ricinus communis. Nat. Biotechnol. 2010, 28, 951–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motamayor, J.C.; Mockaitis, K.; Schmutz, J.; Haiminen, N.; Iii, D.L.; Cornejo, O.; Findley, S.D.; Zheng, P.; Utro, F.; Royaert, S.; et al. The Genome Sequence of the Most Widely Cultivated Cacao Type and Its Use to Identify Candidate Genes Regulating Pod Color. Genome Biol. 2013, 14, r53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Cassave Genetic Map Consortium. High-resolution linage map and chromosome-scale genome assembly for cassava (Manihot esculenta Crantz) from 10 populations. G3 Genes Genomes Genet. 2014, 5, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Tuskan, G.A.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, Z.P.; Varani, A.M.; Cauz-Santos, L.A.; Sader, M.A.; Giopatto, H.A.; Zirpoli, B.; Callot, C.; Cauet, S.; Marande, W.; Souza Cardoso, J.L.; et al. A Genome Sequence Resource for the Genus Passiflora, the Genome of the Wild Diploid Species Passiflora organensis. Plant Genome 2021, 14, e20117. [Google Scholar] [CrossRef]
- Sader, M.; Vaio, M.; Cauz-Santos, L.A.; Dornelas, M.C.; Vieira, M.L.C.; Melo, N.; Pedrosa-Harand, A. Large vs Small Genomes in Passiflora: The Influence of the Mobilome and the Satellitome. Planta 2021, 253, 86. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined, Comprehensive Pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.-H.; Gou, J.-Y.; Liu, C.-J. BAHD Superfamily of Acyl-CoA Dependent Acyltransferases in Populus and Arabidopsis: Bioinformatics and Gene Expression. Plant Mol. Biol. 2009, 70, 421–442. [Google Scholar] [CrossRef]
- Tuominen, L.K.; Johnson, V.E.; Tsai, C.-J. Differential Phylogenetic Expansions in BAHD Acyltransferases across Five Angiosperm Taxa and Evidence of Divergent Expression among Populus Paralogues. BMC Genom. 2011, 12, 236. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Ye, X.; Kang, B.; Osburn, L.D.; Li, Y.; Zong-Ming (Max), C. Identification of the Flavin-Dependent Monooxygenase-Encoding YUCCA Gene Family in Populus trichocarpa and Their Expression in Vegetative Tissues and in Response to Hormone and Environmental Stresses. Plant Cell Tissue Organ Cult. PCTOC 2009, 97, 271–283. [Google Scholar] [CrossRef]
- Kakeda, K.; Jordan, N.D.; Conner, A.; Ride, J.P.; Franklin-Tong, V.E.; Franklin, F.C.H. Identification of Residues in a Hydrophilic Loop of the Papaver rhoeas S Protein That Play a Crucial Role in Recognition of Incompatible Pollen. Plant Cell 1998, 10, 1723–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekar, K.V.; Ji, S.; Coulthard, R.J.; Ride, J.P.; Reynolds, G.L.; Winn, P.J.; Wheeler, M.J.; Hyde, E.I.; Smith, L.J. Structure of SPH (Self-Incompatibility Protein Homologue) Proteins: A Widespread Family of Small, Highly Stable, Secreted Proteins. Biochem. J. 2019, 476, 809–826. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin Biosynthesis by the YUCCA Flavin Monooxygenases Controls the Formation of Floral Organs and Vascular Tissues in Arabidopsis. Genes Dev. 2006, 20, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Cecchetti, V.; Celebrin, D.; Napoli, N.; Ghelli, R.; Brunetti, P.; Costantino, P.; Cardarelli, M. An Auxin Maximum in the Middle Layer Controls Stamen Development and Pollen Maturation in Arabidopsis. New Phytol. 2017, 213, 1194–1207. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y. Essential Roles of Local Auxin Biosynthesis in Plant Development and in Adaptation to Environmental Changes. Annu. Rev. Plant Biol. 2018, 69, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Blakeslee, J.J.; Rossi, T.S.; Kriechbaumer, V. Auxin Biosynthesis: Spatial Regulation and Adaptation to Stress. J. Exp. Bot. 2019, 70, 5041–5049. [Google Scholar] [CrossRef]
- Matthes, M.S.; Best, N.B.; Robil, J.M.; Malcomber, S.; Gallavotti, A.; McSteen, P. Auxin EvoDevo: Conservation and Diversification of Genes Regulating Auxin Biosynthesis, Transport, and Signaling. Mol. Plant 2019, 12, 298–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin Synthesized by the YUCCA Flavin Monooxygenases Is Essential for Embryogenesis and Leaf Formation in Arabidopsis. Plant Cell 2007, 19, 2430–2439. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Dai, X.; De-Paoli, H.; Cheng, Y.; Takebayashi, Y.; Kasahara, H.; Kamiya, Y.; Zhao, Y. Auxin Overproduction in Shoots Cannot Rescue Auxin Deficiencies in Arabidopsis Roots. Plant Cell Physiol. 2014, 55, 1072–1079. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A Web Server for Whole-Genome Comparison and Annotation of Orthologous Clusters across Multiple Species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, X.; Paterson, A.H. Genome and Gene Duplications and Gene Expression Divergence: A View from Plants: Gene Duplication and Expression Divergence. Ann. N. Y. Acad. Sci. 2012, 1256, 1–14. [Google Scholar] [CrossRef]
- D’Auria, J.C. Acyltransferases in Plants: A Good Time to Be BAHD. Curr. Opin. Plant Biol. 2006, 9, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein Localization Predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescot, M. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and Interpreting Transposable Element Expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Wang, K.; Huang, G.; Zhu, Y. Transposable Elements Play an Important Role during Cotton Genome Evolution and Fiber Cell Development. Sci. China Life Sci. 2016, 59, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Ferguson, A.A.; Jiang, N. What Makes up Plant Genomes: The Vanishing Line between Transposable Elements and Genes. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2016, 1859, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Zhang, W. Characterization of Transposon-Derived Accessible Chromatin Regions in Rice (Oryza sativa). Int. J. Mol. Sci. 2022, 23, 8947. [Google Scholar] [CrossRef]
- Noshay, J.M.; Marand, A.P.; Anderson, S.N.; Zhou, P.; Mejia Guerra, M.K.; Lu, Z.; O’Connor, C.H.; Crisp, P.A.; Hirsch, C.N.; Schmitz, R.J.; et al. Assessing the Regulatory Potential of Transposable Elements Using Chromatin Accessibility Profiles of Maize Transposons. Genetics 2021, 217, 1–13. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Yrjälä, K.; Satheesh, V.; Zhou, M.-B. Bamboo Transposon Research: Current Status and Perspectives. In Plant Transposable Elements; Methods in Molecular Biology; Cho, J., Ed.; Springer: New York, NY, USA, 2021; Volume 2250, pp. 257–270. ISBN 978-1-07-161133-3. [Google Scholar]
- Niu, S.; Li, J.; Bo, W.; Yang, W.; Zuccolo, A.; Giacomello, S.; Chen, X.; Han, F.; Yang, J.; Song, Y.; et al. The Chinese Pine Genome and Methylome Unveil Key Features of Conifer Evolution. Cell 2022, 185, 204–217. [Google Scholar] [CrossRef]
- Liao, N.; Hu, Z.; Miao, J.; Hu, X.; Lyu, X.; Fang, H.; Zhou, Y.-M.; Mahmoud, A.; Deng, G.; Meng, Y.-Q.; et al. Chromosome-Level Genome Assembly of Bunching Onion Illuminates Genome Evolution and Flavor Formation in Allium Crops. Nat. Commun. 2022, 13, 6690. [Google Scholar] [CrossRef] [PubMed]
- Ågren, J.; Wang, W.; Koenig, D.; Neuffer, B.; Weigel, D.; Wright, S.I. Mating System Shifts and Transposable Element Evolution in the Plant Genus Capsella. BMC Genom. 2014, 15, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, A.; Panseri, A.F.; Poggio, L.; Fernández, A. Nuclear DNA Content in the Polyploid Complex Turnera ulmifolia (Turnera L., Passifloraceae). Plant Syst. Evol. 2011, 296, 225–230. [Google Scholar] [CrossRef]
- Lease, K.A.; Walker, J.C. The Arabidopsis Unannotated Secreted Peptide Database, a Resource for Plant Peptidomics. Plant Physiol. 2006, 142, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Tian, L.; Liu, H.; Li, X.; Zhang, J.; Chen, X.; Jia, X.; Zheng, X.; Wu, S.; Chen, Y.; et al. Large-Scale Discovery of Non-Conventional Peptides in Maize and Arabidopsis through an Integrated Peptidogenomic Pipeline. Mol. Plant 2020, 13, 1078–1093. [Google Scholar] [CrossRef]
- Pei, M.-S.; Liu, H.-N.; Wei, T.-L.; Yu, Y.-H.; Guo, D.-L. Large-Scale Discovery of Non-Conventional Peptides in Grape (Vitis vinifera L.) through Peptidogenomics. Hortic. Res. 2022, 9, uhac023. [Google Scholar] [CrossRef] [PubMed]
- Ride, J.P.; Davies, E.M.; Franklin, F.C.H.; Marshall, D.F. Analysis of Arabidopsis Genome Sequence Reveals a Large New Gene Family in Plants. Plant Mol. Biol. 1999, 39, 927–932. [Google Scholar] [CrossRef]
- Kappel, C.; Huu, C.N.; Lenhard, M. A Short Story Gets Longer: Recent Insights into the Molecular Basis of Heterostyly. J. Exp. Bot. 2017, 68, 5719–5730. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Huang, D.; Zhang, S.; Wang, W.; Ma, F.; Wu, B.; Xu, Y.; Xu, B.; Chen, D.; Zou, M.; et al. Chromosome-Scale Genome Assembly Provides Insights into the Evolution and Flavor Synthesis of Passion Fruit (Passiflora edulis Sims). Hortic. Res. 2021, 8, 14. [Google Scholar] [CrossRef]
- Muschner, V.C.; Zamberlan, P.M.; Bonatto, S.L.; Freitas, L.B. Phylogeny, Biogeography and Divergence Times in Passiflora (Passifloraceae). Genet. Mol. Biol. 2012, 35, 1036–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thulin, M.; Razafimandimbison, S.G.; Chafe, P.; Heidari, N.; Kool, A.; Shore, J.S. Phylogeny of the Turneraceae Clade (Passifloraceae s.l.): Trans-Atlantic Disjunctions and Two New Genera in Africa. TAXON 2012, 61, 308–323. [Google Scholar] [CrossRef]
- Wang, M.; Liu, X.; Wang, R.; Li, W.; Rodermel, S.; Yu, F. Overexpression of a Putative Arabidopsis BAHD Acyltransferase Causes Dwarfism That Can Be Rescued by Brassinosteroid. J. Exp. Bot. 2012, 63, 5787–5801. [Google Scholar] [CrossRef] [Green Version]
- Kiba, T.; Naitou, T.; Koizumi, N.; Yamashino, T.; Sakakibara, H.; Mizuno, T. Combinatorial Microarray Analysis Revealing Arabidopsis Genes Implicated in Cytokinin Responses through the His→Asp Phosphorelay Circuitry. Plant Cell Physiol. 2005, 46, 339–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.J.; Park, J.-Y.; Ku, S.-J.; Ha, Y.-M.; Kim, S.; Kim, M.D.; Oh, M.-H.; Kim, J. Genome-Wide Expression Profiling of ARABIDOPSIS RESPONSE REGULATOR 7(ARR7) Overexpression in Cytokinin Response. Mol. Genet. Genom. 2007, 277, 115–137. [Google Scholar] [CrossRef]
- Černý, M.; Kuklová, A.; Hoehenwarter, W.; Fragner, L.; Novák, O.; Rotková, G.; Jedelský, P.L.; Žáková, K.; Šmehilová, M.; Strnad, M.; et al. Proteome and Metabolome Profiling of Cytokinin Action in Arabidopsis Identifying Both Distinct and Similar Responses to Cytokinin Down- and up-Regulation. J. Exp. Bot. 2013, 64, 4193–4206. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.; Sharma, I.; Pati, P.K. Versatile Roles of Brassinosteroid in Plants in the Context of Its Homoeostasis, Signaling and Crosstalks. Front. Plant Sci. 2015, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.X.; Malitsky, S.; De Oliveira, S.; Branigan, C.; Franke, R.B.; Schreiber, L.; Aharoni, A. SHINE Transcription Factors Act Redundantly to Pattern the Archetypal Surface of Arabidopsis Flower Organs. PLoS Genet. 2011, 7, e1001388. [Google Scholar] [CrossRef] [Green Version]
- Janda, T.; Szalai, G.; Pál, M. Salicylic Acid Signalling in Plants. Int. J. Mol. Sci. 2020, 21, 2655. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Qualley, A.; Fan, B.; Dudareva, N.; Chen, Z. An Important Role of a BAHD Acyl Transferase-like Protein in Plant Innate Immunity. Plant J. 2009, 57, 1040–1053. [Google Scholar] [CrossRef]
- Williamson, R.J.; Josephs, E.B.; Platts, A.E.; Hazzouri, K.M.; Haudry, A.; Blanchette, M.; Wright, S.I. Evidence for Widespread Positive and Negative Selection in Coding and Conserved Noncoding Regions of Capsella grandiflora. PLoS Genet. 2014, 10, e1004622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.J.; Doyle, J.L. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Weisenfeld, N.I.; Yin, S.; Sharpe, T.; Lau, B.; Hegarty, R.; Holmes, L.; Sogoloff, B.; Tabbaa, D.; Williams, L.; Russ, C.; et al. Comprehensive Variation Discovery in Single Human Genomes. Nat. Genet. 2014, 46, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggett, R.M.; Clavijo, B.J.; Clissold, L.; Clark, M.D.; Caccamo, M. NextClip: An Analysis and Read Preparation Tool for Nextera Long Mate Pair Libraries. Bioinformatics 2014, 30, 566–568. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding Pre-Assembled Contigs Using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Goodman, L.; Edmunds, S.C.; Basford, A.T. Large and Linked in Scientific Publishing. GigaScience 2012, 1, 1–2047. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.M.; Stajich, J. Nextgenusfs/Funannotate: Funannotate v1.7.4 2020. Goodman L, Edmunds SC, Basford AT. Large and linked in scientific publishing. Gigascience 2012, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Holt, C.; Yandell, M. MAKER2: An Annotation Pipeline and Genome-Database Management Tool for Second-Generation Genome Projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.F.A.; Hubley, R. RepeatModeler Open-1.0 2008–2015. Available online: http://www.repeatmasker.org (accessed on 1 November 2017).
- Merchant, N.; Lyons, E.; Goff, S.; Vaughn, M.; Ware, D.; Micklos, D.; Antin, P. The IPlant Collaborative: Cyberinfrastructure for Enabling Data to Discovery for the Life Sciences. PLoS Biol. 2016, 14, e1002342. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0 2013–2015. Available online: http://www.repeatmasker.org (accessed on 1 January 2020).
- Haas, B.J. Improving the Arabidopsis Genome Annotation Using Maximal Transcript Alignment Assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Korf, I. Gene Finding in Novel Genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS: Table 1. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab Initio Prediction of Alternative Transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinforma. 2014, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.P.; Lowe, T.M. TRNAscan-SE: Searching for TRNA Genes in Genomic Sequences. In Gene Prediction; Methods in Molecular Biology; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; Volume 1962, pp. 1–14. ISBN 978-1-4939-9172-3. [Google Scholar]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis Information Resource: Making and Mining the “Gold Standard” Annotated Reference Plant Genome: Tair: Making and Mining the “Gold Standard” Plant Genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein Domains Identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H. Predicting Secretory Proteins with SignalP. In Protein Function Prediction; Methods in Molecular Biology; Kihara, D., Ed.; Springer: New York, NY, USA, 2017; Volume 1611, pp. 59–73. ISBN 978-1-4939-7013-1. [Google Scholar]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic Annotation of Eukaryotic Genes, Pseudogenes and Promoters. Genome Biol. 2006, 7, S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelier, J.; Casse, N.; Daccord, N.; Jamilloux, V.; Saint-Jean, B.; Carrier, G. A transposable element annotation pipeline and expression analysis reveal potentially active elements in the microalga Tisochrysis lutea. Genomics. 2018, 19(1), 378. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for Occurrences of a given Motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Expósito-Rodríguez, M.; Borges, A.A.; Borges-Pérez, A.; Pérez, J.A. Gene Structure and Spatiotemporal Expression Profile of Tomato Genes Encoding YUCCA-like Flavin Monooxygenases: The ToFZY Gene Family. Plant Physiol. Biochem. 2011, 49, 782–791. [Google Scholar] [CrossRef]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved Prediction of Protein Structural Features by Integrated Deep Learning. Proteins Struct. Funct. Bioinforma. 2019, 87, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A Comparative Platform for Green Plant Genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Morales-Quintana, L.; Moya-León, M.A.; Herrera, R. Computational Study Enlightens the Structural Role of the Alcohol Acyltransferase DFGWG Motif. J. Mol. Model. 2015, 21, 216. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Grover, J.W.; Bomhoff, M.; Davey, S.; Gregory, B.D.; Mosher, R.A.; Lyons, E. CoGe LoadExp+: A Web-based Suite That Integrates Next-generation Sequencing Data Analysis Workflows and Visualization. Plant Direct 2017, 1, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waese, J.; Fan, J.; Pasha, A.; Yu, H.; Fucile, G.; Shi, R.; Cumming, M.; Kelley, L.A.; Sternberg, M.J.; Krishnakumar, V.; et al. EPlant: Visualizing and Exploring Multiple Levels of Data for Hypothesis Generation in Plant Biology. Plant Cell 2017, 29, 1806–1821. [Google Scholar] [CrossRef] [Green Version]
- Cecchetti, V.; Altamura, M.M.; Falasca, G.; Costantino, P.; Cardarelli, M. Auxin Regulates Arabidopsis Anther Dehiscence, Pollen Maturation, and Filament Elongation. Plant Cell 2008, 20, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Veltri, D.; Wight, M.M.; Crouch, J.A. SimpleSynteny: A Web-Based Tool for Visualization of Microsynteny across Multiple Species. Nucleic Acids Res. 2016, 44, W41–W45. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An Upgraded Gene Feature Visualization Server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Predicted Subcellular Location |
|---|---|
| TsBAHD | nucl: 8, cyto: 5 |
| Tsubulata_042462-RA | nucl: 6, nucl_plas: 5, cyto: 2, mito: 2, plas: 2, chlo: 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henning, P.M.; Roalson, E.H.; Mir, W.; McCubbin, A.G.; Shore, J.S. Annotation of the Turnera subulata (Passifloraceae) Draft Genome Reveals the S-Locus Evolved after the Divergence of Turneroideae from Passifloroideae in a Stepwise Manner. Plants 2023, 12, 286. https://doi.org/10.3390/plants12020286
Henning PM, Roalson EH, Mir W, McCubbin AG, Shore JS. Annotation of the Turnera subulata (Passifloraceae) Draft Genome Reveals the S-Locus Evolved after the Divergence of Turneroideae from Passifloroideae in a Stepwise Manner. Plants. 2023; 12(2):286. https://doi.org/10.3390/plants12020286
Chicago/Turabian StyleHenning, Paige M., Eric H. Roalson, Wali Mir, Andrew G. McCubbin, and Joel S. Shore. 2023. "Annotation of the Turnera subulata (Passifloraceae) Draft Genome Reveals the S-Locus Evolved after the Divergence of Turneroideae from Passifloroideae in a Stepwise Manner" Plants 12, no. 2: 286. https://doi.org/10.3390/plants12020286
APA StyleHenning, P. M., Roalson, E. H., Mir, W., McCubbin, A. G., & Shore, J. S. (2023). Annotation of the Turnera subulata (Passifloraceae) Draft Genome Reveals the S-Locus Evolved after the Divergence of Turneroideae from Passifloroideae in a Stepwise Manner. Plants, 12(2), 286. https://doi.org/10.3390/plants12020286