
Proteomic Approach during the Induction of Somatic Embryogenesis in Coffea canephora

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
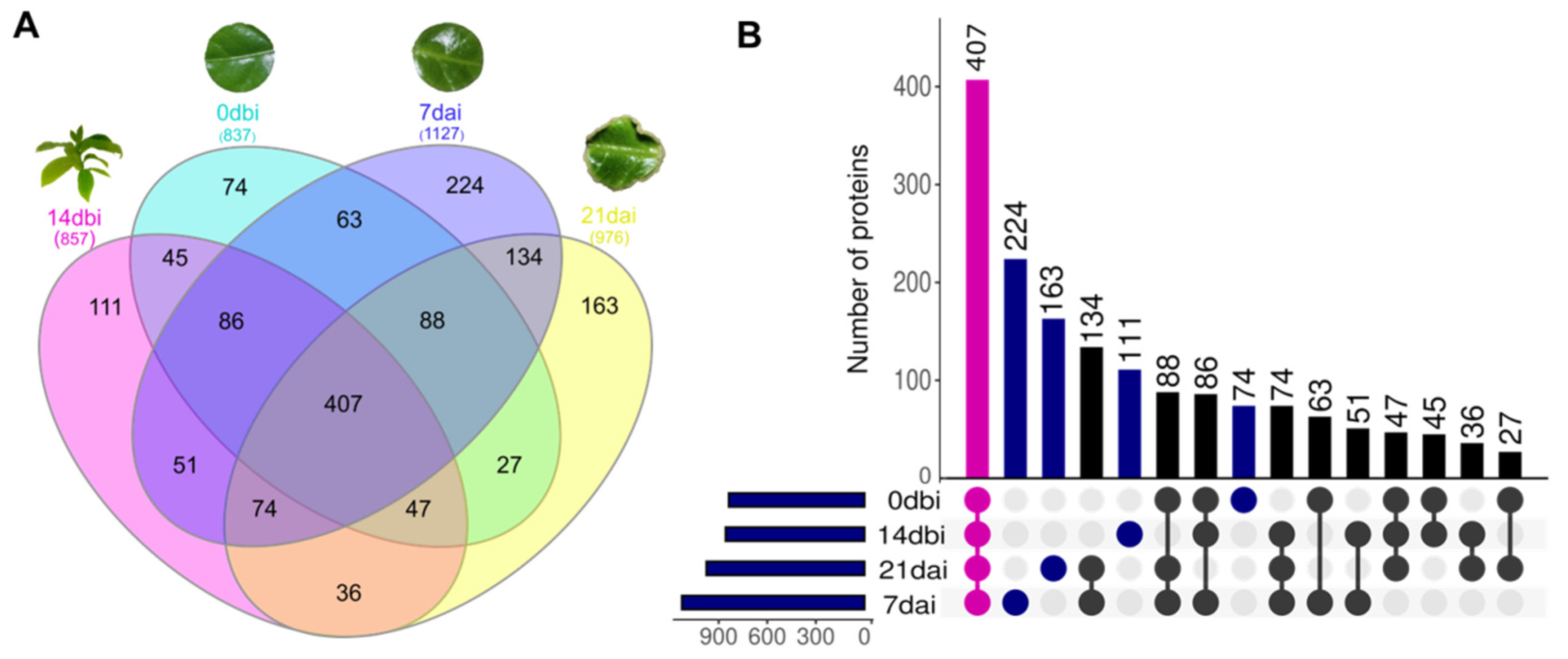
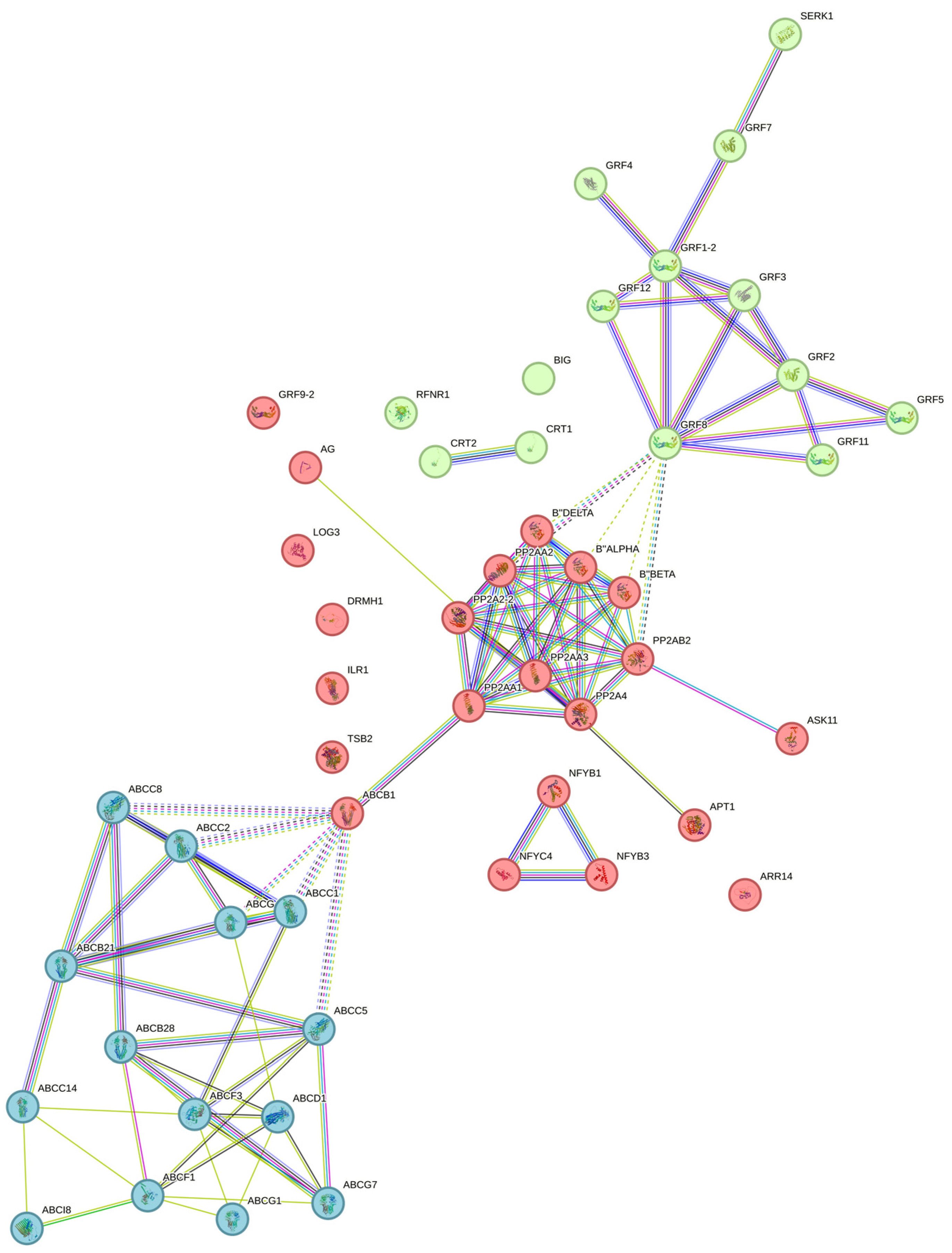
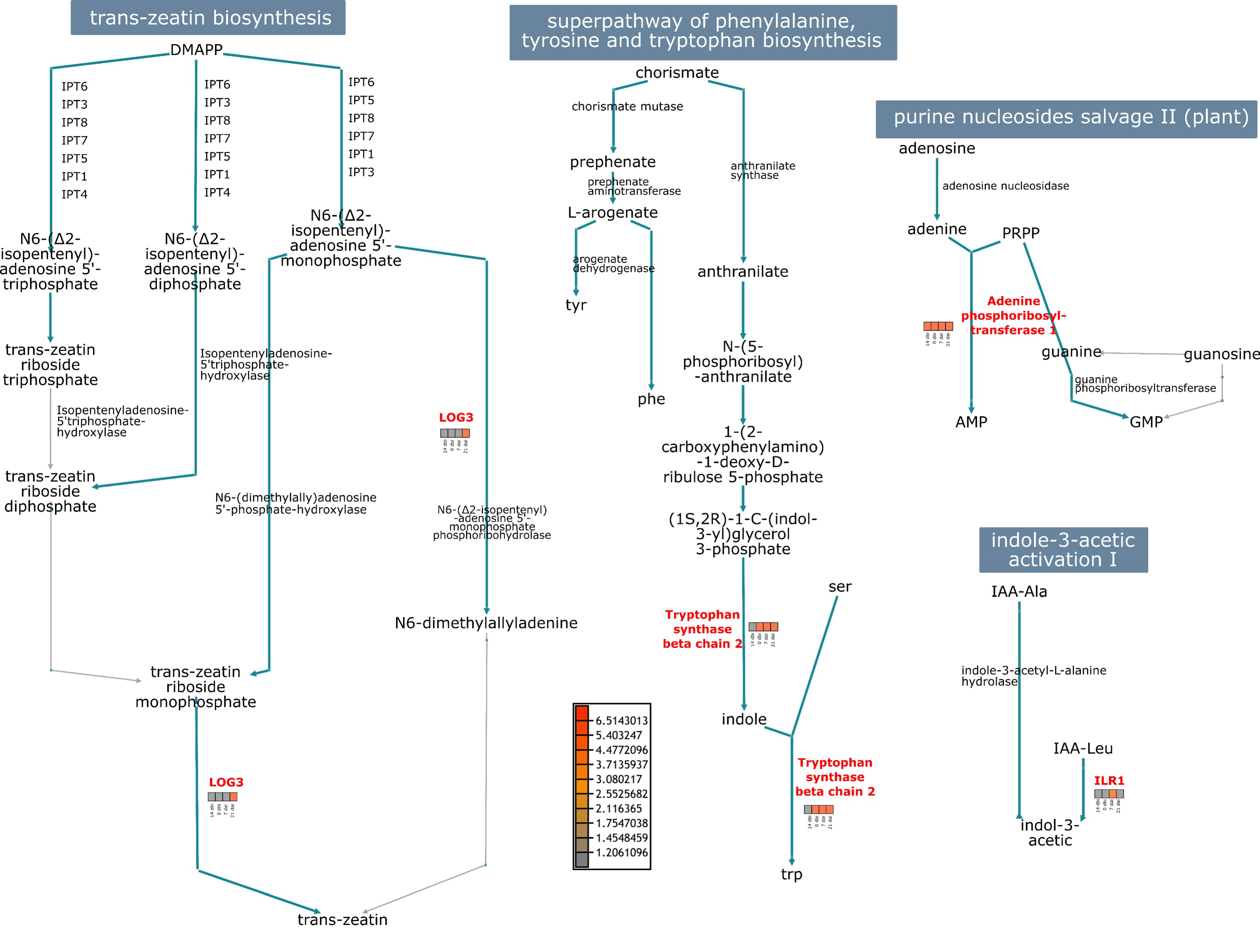
2. Results
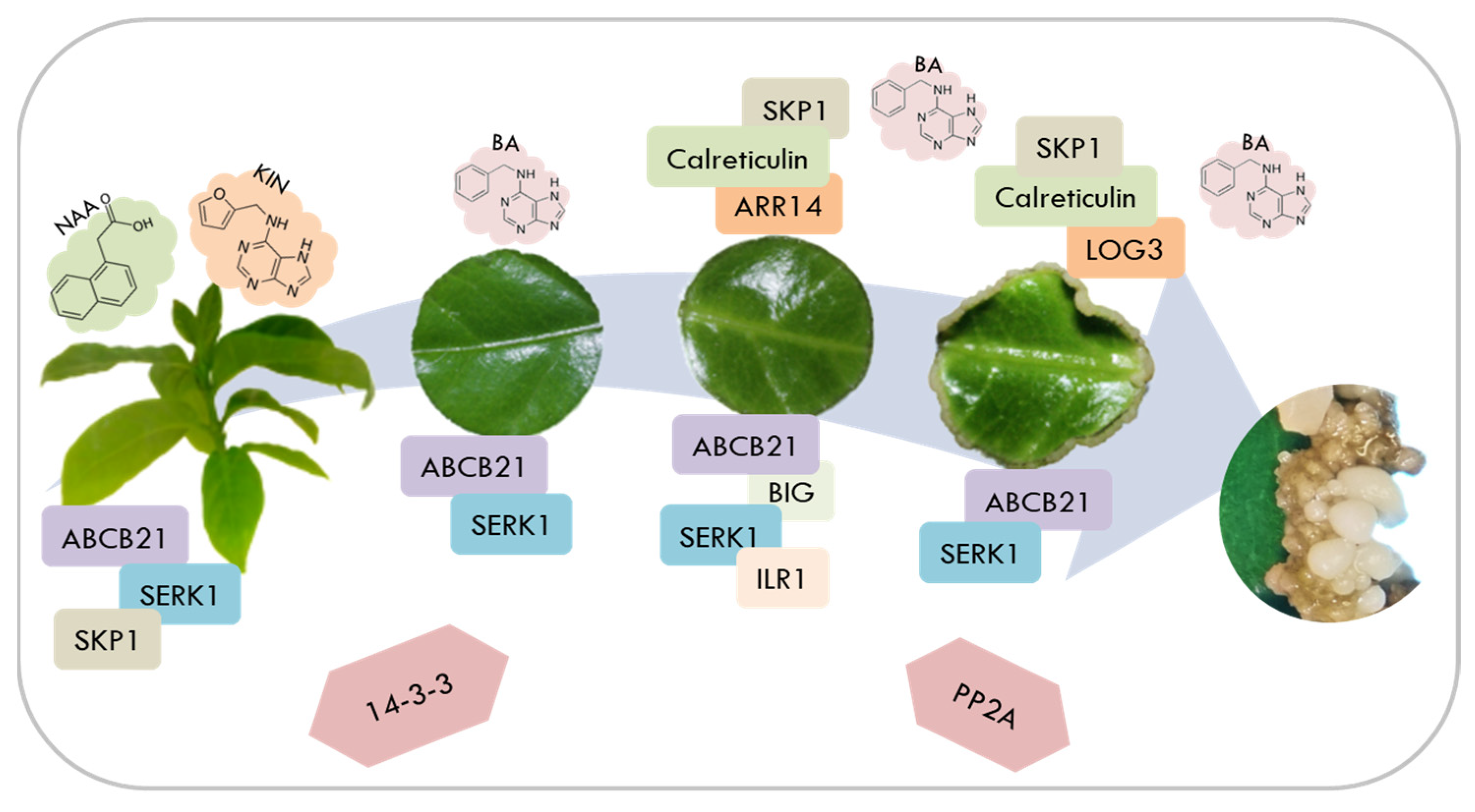
3. Discussion
4. Materials and Methods
4.1. Biological Material and Growth Conditions
4.2. Protein Extraction
4.3. Reduction, Alkylation, and Digestion
4.4. Nano LC/MS-MS Analysis
4.5. Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferreira, T.; Shuler, J.; Guimardes, R.; Farah, A. Introduction to coffee plant and genetics. In Coffee: Production, Quality and Chemistry; Farah, A., Ed.; The Royal Society of Chemistry: Croydon, UK, 2019; pp. 3–25. [Google Scholar]
- ICO Trade Statistics. ICO 2022. Available online: http://www.ico.org/trade_statistics.asp (accessed on 23 January 2022).
- Etienne, H. Somatic embryogenesis protocol: Coffee (Coffea arabica L. and C. canephora P.). In Protocol for Somatic Embryogenesis in Woody Plants; Jain, S., Gupta, P., Eds.; Springer: Dordrecht, The Netherlands, 2005; pp. 167–179. [Google Scholar]
- Méndez-Hernández, H.A.; Ledezma-Rodríguez, M.; Avilez-Montalvo, R.N.; Juárez-Gómez, Y.L.; Skeete, A.; Avilez-Montalvo, J.; De-la-Peña, C.; Loyola-Vargas, V.M. Signaling overview of plant somatic embryogenesis. Front. Plant Sci. 2019, 10, 77. [Google Scholar] [CrossRef]
- Nic-Can, G.I.; Loyola-Vargas, V.M. The role of the auxins during somatic embryogenesis. In Somatic Embryogenesis. Fundamental Aspects and Applications; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer: Handel, Switzerland, 2016; pp. 171–181. [Google Scholar]
- Santner, A.; Calderon-Villalobos, L.I.; Estelle, M. Plant hormones are versatile chemical regulators of plant growth. Nat. Chem. Biol. 2009, 5, 301–307. [Google Scholar] [CrossRef]
- Ljung, K. Auxin metabolism and homeostasis during plant development. Development 2013, 140, 943–950. [Google Scholar] [CrossRef]
- Márquez-López, R.E.; Quintana-Escobar, A.O.; Loyola-Vargas, V.M. Cytokinins, the Cinderella of plant growth regulators. Phytochem. Rev. 2019, 18, 1387–1408. [Google Scholar] [CrossRef]
- Enríquez-Valencia, A.J.; Vázquez-Flota, F.A.; Ku-Cauich, J.R.; Escobedo-GraciaMedrano, R.M. Differentially expressed genes during the transition from early to late development phases in somatic embryo of banana (Musa spp. AAB group, Silk subgroup) cv. Manzano. Plant Cell Tissue Organ Cult. 2019, 136, 289–302. [Google Scholar] [CrossRef]
- Góngora-Castillo, E.; Nic-Can, G.I.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M. Elaboration of transcriptome during the induction of somatic embryogenesis. In Plant Cell Culture Protocols; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer: New York, NY, USA, 2018; pp. 411–427. [Google Scholar]
- Chen, Y.; Xu, X.; Liu, Z.; Zhang, Z.; XuHan, X.; Lin, Y.; Lai, Z. Global scale transcriptome analysis reveals differentially expressed genes involve in early somatic embryogenesis in Dimocarpus longan Lour. BMC Genom. 2020, 21, 4. [Google Scholar] [CrossRef]
- Guo, H.; Guo, H.; Zhang, L.; Fan, Y.; Wu, J.; Tang, Z.; Zhang, Y.; Fan, Y.; Zeng, F. Dynamic transcriptome analysis reveals uncharacterized complex regulatory pathway underlying genotype-recalcitrant somatic embryogenesis transdifferentiation in cotton. Genes 2020, 11, 519. [Google Scholar] [CrossRef]
- Quintana-Escobar, A.O.; Nic-Can, G.I.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M.; Góngora-Castillo, E. Transcriptome analysis of the induction of somatic embryogenesis in Coffea canephora and the participation of arf and AUX/IAA genes. PeerJ 2019, 7, e7752. [Google Scholar] [CrossRef]
- Aguilar-Hernández, V.; Loyola-Vargas, V.M. Advanced proteomic approaches to elucidate somatic embryogenesis. Front. Plant Sci. 2018, 9, 1658. [Google Scholar] [CrossRef]
- Gulzar, B.; Mujib, A.; Rajam, M.V.; Frukh, A.; Zafar, N. Identification of somatic embryogenesis (SE) related proteins through label-free shotgun proteomic method and cellular role in Catharanthus roseus (L.) G. Don. Plant Cell Tissue Organ Cult. 2019, 137, 225–237. [Google Scholar] [CrossRef]
- Kumaravel, M.; Uma, S.; Backiyarani, S.; Saraswathi, M.S. Proteomic analysis of somatic embryo development in Musa spp. cv. Grand Naine (AAA). Sci. Rep. 2020, 10, 4501. [Google Scholar] [CrossRef]
- Feussner, I.; Polle, A. What the transcriptome does not tell—Proteomics and metabolomics are closer to the plants’ patho-phenotype. Curr. Opin. Plant Biol. 2015, 26, 26–31. [Google Scholar] [CrossRef]
- Tchorbadjieva, M. Advances in proteomics of somatic embryogenesis. In Somatic Embryogenesis in Ornamentals and Its Applications; Mujib, A., Ed.; Springer: New Delhi, India, 2016; pp. 67–90. [Google Scholar]
- Heringer, A.S.; Santa-Catarina, C.; Silveira, V. Insights from proteomic studies into plant somatic embryogenesis. Proteomics 2018, 18, 1700265. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, C.; Wang, Q.; Yang, Z.; Wang, Y.; Zhang, X.; Wu, Z.; Hou, Y.; Wu, J.; Li, F. iTRAQ protein profile differential analysis between somatic globular and cotyledonary embryos reveals stress, hormone, and respiration involved in increasing plantlet regeneration of Gossypium hirsutum L. J. Proteome Res. 2014, 14, 268–278. [Google Scholar] [CrossRef]
- Tonietto, Â.; Hiromi Sato, J.; Batista Teixeira, J.; de Souza, E.M.; Pedrosa, F.O.; Franco, O.L.; Metha, A. Proteomic analysis of developing somatic embryos of Coffea arabica. Plant Mol. Biol. Rep. 2012, 30, 1393–1399. [Google Scholar] [CrossRef]
- Campos, N.A.; Paiva, L.V.; Panis, B.; Carpentier, S.C. The proteome profile of embryogenic cell suspensions of Coffea arabica L. Proteomics 2016, 16, 1001–1005. [Google Scholar] [CrossRef]
- Mukul-López, H.G.; De-la-Peña, C.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M. Evaluation of the extracellular proteome profile during the somatic embryogenesis process of Coffea spp. J. Mex. Chem. Soc. 2012, 56, 72–79. [Google Scholar] [CrossRef]
- Iquebal, M.A.; Jaiswal, S.; Mukhopadhyay, C.S.; Sarkar, C.; Rai, A.; Kumar, D. Applications of bioinformatics in plant and agriculture. In PlantOmics: The Omics of Plant Science; Barh, D., Khan, M.S., Davies, E., Eds.; Springer: New Delhi, India, 2015; pp. 755–789. [Google Scholar]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Rhee, S.Y.; Dickerson, J.; Xu, D. Bioinformatics and its applications in plant biology. Annu. Rev. Plant Biol. 2006, 57, 335–360. [Google Scholar] [CrossRef]
- Nic-Can, G.I.; De-la-Peña, C. Epigenetic advances on somatic embryogenesis of agronomical and important crops. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications; Álvarez-Venegas, R., De-la-Peña, C., Casas-Mollano, J.A., Eds.; Springer: Cham, Switzerlands; Heidelberg, Germany; New York, NY, USA; Dordrecht, The Nertherlands; London, UK, 2014; pp. 91–109. [Google Scholar]
- Ayil-Gutiérrez, B.A.; Galaz-Ávalos, R.M.; Peña-Cabrera, E.; Loyola-Vargas, V.M. Dynamics of the concentration of IAA and some of its conjugates during the induction of somatic embryogenesis in Coffea canephora. Plant Signal. Behav. 2013, 8, e26998. [Google Scholar] [CrossRef]
- Avilez-Montalvo, J.; Quintana-Escobar, A.O.; Méndez-Hernández, H.A.; Uc-Chuc, M.Á.; Brito-Argáez, L.; Aguilar-Hernández, V.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M. Auxin-cytokinin cross talk in somatic embryogenesis of Coffea canephora. Plants 2022, 11, 2013. [Google Scholar] [CrossRef]
- Méndez-Hernández, H.A.; Quintana-Escobar, A.O.; Uc-Chuc, M.Á.; Loyola-Vargas, V.M. Genome-wide analysis, modeling, and identification of amino acid binding motifs suggest the involvement of GH3 genes during somatic embryogenesis of Coffea canephora. Plants 2021, 10, 2034. [Google Scholar] [CrossRef]
- Márquez-López, R.E.; Pérez-Hernández, C.A.; Kú-González, Á.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M. Localization and transport of indole-3-acetic acid during somatic embryogenesis in Coffea canephora. Protoplasma 2018, 255, 695–708. [Google Scholar] [CrossRef]
- Geisler, M.; Aryal, B.; di Donato, M.; Hao, P. A critical view on ABC transporters and their interacting partners in auxin transport. Plant Cell Physiol. 2017, 58, 1601–1614. [Google Scholar] [CrossRef]
- Xu, Y.X.; Liu, Y.; Chen, S.T.; Li, X.Q.; Xu, L.G.; Qi, Y.H.; Jiang, D.A.; Jin, S.H. The B subfamily of plant ATP binding cassette transporters and their roles in auxin transport. Biol. Plant. 2014, 58, 401–410. [Google Scholar] [CrossRef]
- Song, S.; Wang, Z.; Ren, Y.; Sun, H. Full-length transcriptome analysis of the ABCB, PIN/PIN-LIKES, and AUX/LAX families involved in somatic embryogenesis of Lilium pumilum DC. Fisch. Int. J. Mol. Sci. 2020, 21, 453. [Google Scholar] [CrossRef]
- Quintana-Escobar, A.O.; Galaz-Ávalos, R.M.; Elizalde-Contreras, J.M.; Reyes-Soria, F.A.; Aguilar-Hernández, V.; Ruíz-May, E.; Loyola-Vargas, V.M. Differences in the abundance of auxin homeostasis proteins suggest their central roles for in vitro tissue differentiation in Coffea arabica. Plants 2021, 10, 2607. [Google Scholar] [CrossRef]
- Su, L.; Xie, Y.; He, Z.; Zhang, J.; Tang, Y.; Zhou, X. Network response of two cherry tomato (Lycopersicon esculentum) cultivars to cadmium stress as revealed by transcriptome analysis. Ecotox. Environ. Safe 2021, 222, 112473. [Google Scholar] [CrossRef]
- Yu, R.; Jiang, Q.; Xv, C.; Li, L.; Bu, S.; Shi, G. Comparative proteomics analysis of peanut roots reveals differential mechanisms of cadmium detoxification and translocation between two cultivars differing in cadmium accumulation. BMC Plant Biol. 2019, 19, 137. [Google Scholar] [CrossRef]
- Faus, I.; Niñoles, R.; Kesari, V.; Gadea, J. The ABCF3 gene of Arabidopsis is functionally linked with GCN1 but not with GCN2 during stress and development. Plant Mol. Biol. Rep. 2021, 39, 663–672. [Google Scholar] [CrossRef]
- López-Bucio, J.; Hernández-Abreu, E.; Sánchez-Calderón, L.; Pérez-Torres, A.; Rampey, R.A.; Bartel, B.; Herrera-Estrella, L. An auxin transport independent pathway is involved in phosphate stress-induced root architectural alterations in Arabidopsis. Identification of BIG as a mediator of auxin in pericycle cell Activation. Plant Physiol. 2005, 137, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Kanyuka, K.; Praekelt, U.; Franklin, K.A.; Billingham, O.E.; Hooley, R.; Whitelam, G.C.; Halliday, K.J. Mutations in the huge Arabidopsis gene BIG affect a range of hormone and light responses. Plant J. 2003, 35, 57–70. [Google Scholar] [CrossRef]
- Zazimalová, E.; Krecek, P.; Skúpa, P.; Hoyerová, K.; Petrášek, J. Polar transport of the plant hormone auxin—the role of PIN-FORMED (PIN) proteins. Cell. Mol. Life Sci. 2007, 64, 1621–1637. [Google Scholar] [CrossRef]
- Blakeslee, J.J.; Peer, W.A.; Murphy, A.S. Auxin transport. Curr. Opin. Plant Biol. 2005, 8, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Sáez, R.; Mateo-Bonmatí, E.; Ljung, K. Auxin metabolism in plants. Cold Spring Harbor Perspect. Biol. 2021, 13, a039867. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Yuan, D.; Jin, F.; Zhang, Y.; Xu, J. Transcript profiling reveals complex auxin signalling pathway and transcription regulation involved in dedifferentiation and redifferentiation during somatic embryogenesis in cotton. BMC Plant Biol. 2012, 12, 110. [Google Scholar] [CrossRef]
- Saptari, R.T.; Susila, H. Data mining study of hormone biosynthesis gene expression reveals new aspects of somatic embryogenesis regulation. In Vitro Cell. Dev. Biol.-Plant 2018, 55, 139–152. [Google Scholar] [CrossRef]
- Kieber, J.J.; Schaller, G.E. Cytokinin signaling in plant development. Development 2018, 145, dev149344. [Google Scholar] [CrossRef]
- Allen, M.; Qin, W.; Moreau, F.; Moffatt, B. Adenine phosphoribosyltransferase isoforms of Arabidopsis and their potential contributions to adenine and cytokinin metabolism. Physiol. Plant. 2002, 115, 56–68. [Google Scholar] [CrossRef]
- Chen, C.M.; Melitz, D.K.; Clough, F.W. Metabolism of cytokinin: Phosphoribosylation of cytokinin bases by adenine phosphoribosyltransferase from wheat germ. Arch. Biochem. Biophys. 1982, 214, 634–641. [Google Scholar] [CrossRef]
- Ashihara, H.; Stasolla, C.; Loukanina, N.; Thorpe, T.A. Purine metabolism during white spruce somatic embryo development: Salvage of adenine, adenosine, and inosine. Plant Sci. 2001, 160, 647–657. [Google Scholar] [CrossRef]
- Jia, X.-Y.; He, L. −H.; Jing, R.−L.; Li, R.−Z. Calreticulin: Conserved protein and diverse functions in plants. Physiol. Plant. 2009, 136, 127–138. [Google Scholar] [CrossRef]
- Borisjuk, N.; Sitailo, L.; Adler, K.; Malysheva, L.; Tewes, A.; Borisjuk, L.; Manteuffel, R. Calreticulin expression in plant cells: Developmental regulation, tissue specificity and intracellular distribution. Planta 1998, 206, 504–514. [Google Scholar] [CrossRef]
- Libik, M.; Przywara, L. Immunolocalization of calreticulin in protoplasts and somatic embryos of Daucus carota L. grown in suspension culture. Acta Biol. Cracov. Ser. Bot. 2000, 42, 87–92. [Google Scholar]
- Steiner, N.; Santa-Catarina, C.; Guerra, M.; Cutri, L.; Carnier Dornelas, M.; Floh, E. A gymnosperm homolog of SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASE-1 (SERK1) is expressed during somatic embryogenesis. Plant Cell Tissue Organ Cult. 2012, 109, 41–50. [Google Scholar] [CrossRef]
- Pérez-Pascual, D.; Jiménez-Guillen, D.; Villanueva-Alonzo, H.; Souza-Perera, R.; Godoy-Hernández, G.; Zúñiga-Aguilar, J.J. Ectopic expression of the Coffea canephora SERK1 homologue induced differential transcription of genes involved in auxin metabolism and in the developmental control of embryogenesis. Physiol. Plant. 2018, 163, 530–551. [Google Scholar] [CrossRef] [PubMed]
- Rienties, I.M.; Vink, J.; Borst, J.W.; Russinova, E.; Vries, S.C. The Arabidopsis SERK1 protein interacts with the AAA-ATPase AtCDC48, the 14-3-3 protein GF14 and the PP2C phosphatase KAPP. Planta 2005, 221, 394–405. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, B.; Wang, X.; Zhang, Y.; Dong, M.; Zhang, J. iTRAQ-based comparative proteomic analysis of embryogenic and non-embryogenic tissues of Prince Rupprecht’s larch (Larix principis-rupprechtii Mayr). Plant Cell Tissue Organ Cult. 2015, 120, 655–669. [Google Scholar] [CrossRef]
- Zhang, L.f.; Li, W.F.; Xu, H.y.; Qi, L.w.; Han, S.y. Cloning and characterization of four differentially expressed cDNAs encoding NFYA homologs involved in responses to ABA during somatic embryogenesis in Japanese larch (Larix leptolepis). Plant Cell Tissue Organ Cult. 2014, 117, 293–304. [Google Scholar] [CrossRef]
- Yasuda, T.; Fujii, Y.; Yamaguchi, T. Embryogenic callus induction from Coffea arabica leaf explants by benzyladenine. Plant Cell Physiol. 1985, 26, 595–597. [Google Scholar] [CrossRef]
- Peterson, G.L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977, 83, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Valle, M.V.; Camacho—Vazquez, C.; Elizalde—Contreras, J.M.; Monribot—Villanueva, J.L.; Limón, A.M.V.; Bojórquez—Velázquez, E.; Zamora—Briseño, A.; Jorrín-Novo, J.V.; Ruiz—May, E. Comparing and integrating TMT-SPS-MS3 and label-free quantitative approaches for proteomics scrutiny in recalcitrant mango (Mangifera indica L.) peel tissue during postharvest period. Proteomics 2023, 2300239. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintana-Escobar, A.O.; Bojórquez-Velázquez, E.; Ruiz-May, E.; Loyola-Vargas, V.M. Proteomic Approach during the Induction of Somatic Embryogenesis in Coffea canephora. Plants 2023, 12, 4095. https://doi.org/10.3390/plants12244095
Quintana-Escobar AO, Bojórquez-Velázquez E, Ruiz-May E, Loyola-Vargas VM. Proteomic Approach during the Induction of Somatic Embryogenesis in Coffea canephora. Plants. 2023; 12(24):4095. https://doi.org/10.3390/plants12244095
Chicago/Turabian StyleQuintana-Escobar, Ana Odetth, Esaú Bojórquez-Velázquez, Eliel Ruiz-May, and Víctor Manuel Loyola-Vargas. 2023. "Proteomic Approach during the Induction of Somatic Embryogenesis in Coffea canephora" Plants 12, no. 24: 4095. https://doi.org/10.3390/plants12244095
APA StyleQuintana-Escobar, A. O., Bojórquez-Velázquez, E., Ruiz-May, E., & Loyola-Vargas, V. M. (2023). Proteomic Approach during the Induction of Somatic Embryogenesis in Coffea canephora. Plants, 12(24), 4095. https://doi.org/10.3390/plants12244095