
Artificial Intelligence: A Promising Tool in Exploring the Phytomicrobiome in Managing Disease and Promoting Plant Health


Abstract
:1. Introduction
2. Major AI Models
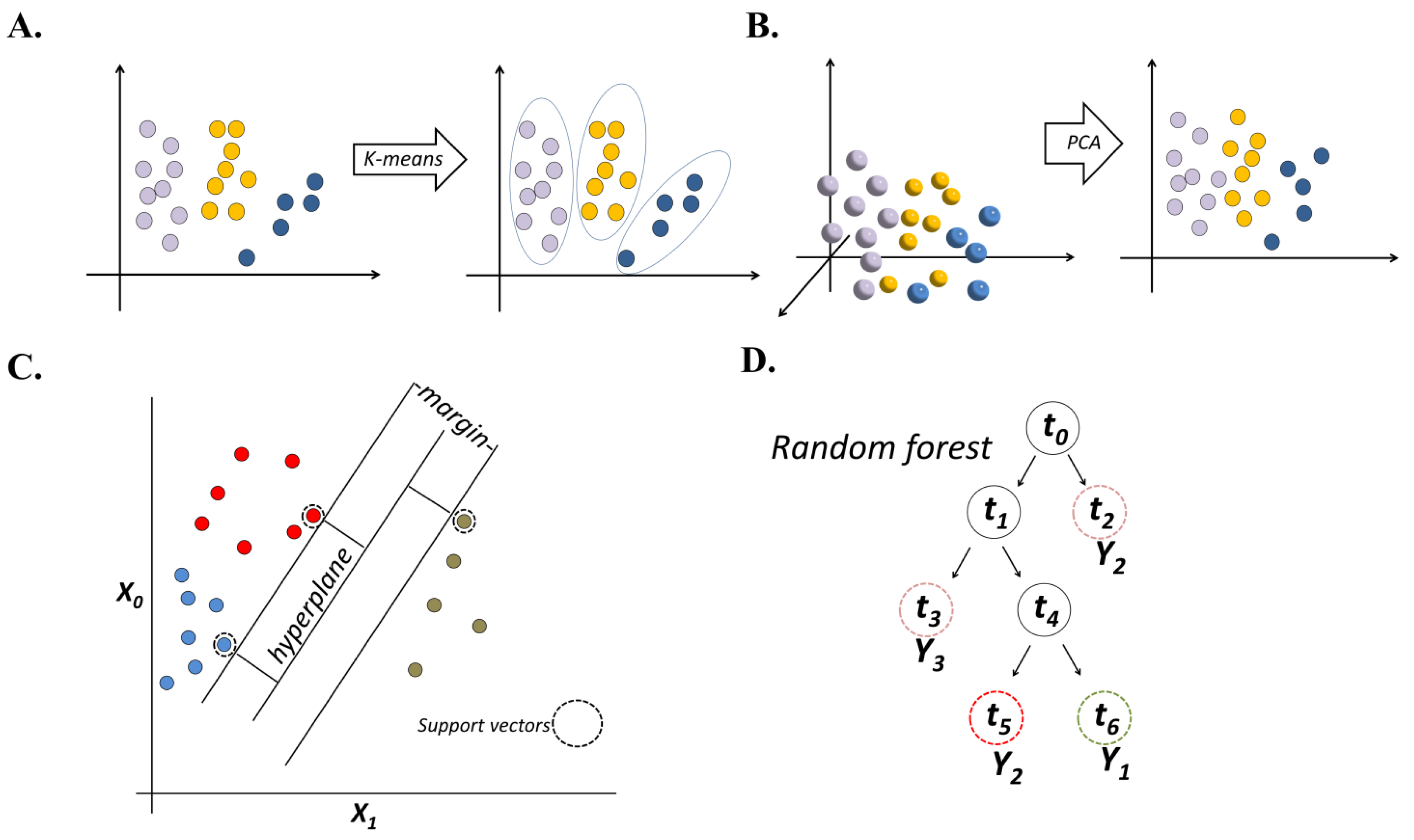
2.1. Unsupervised ML
2.2. Supervised ML
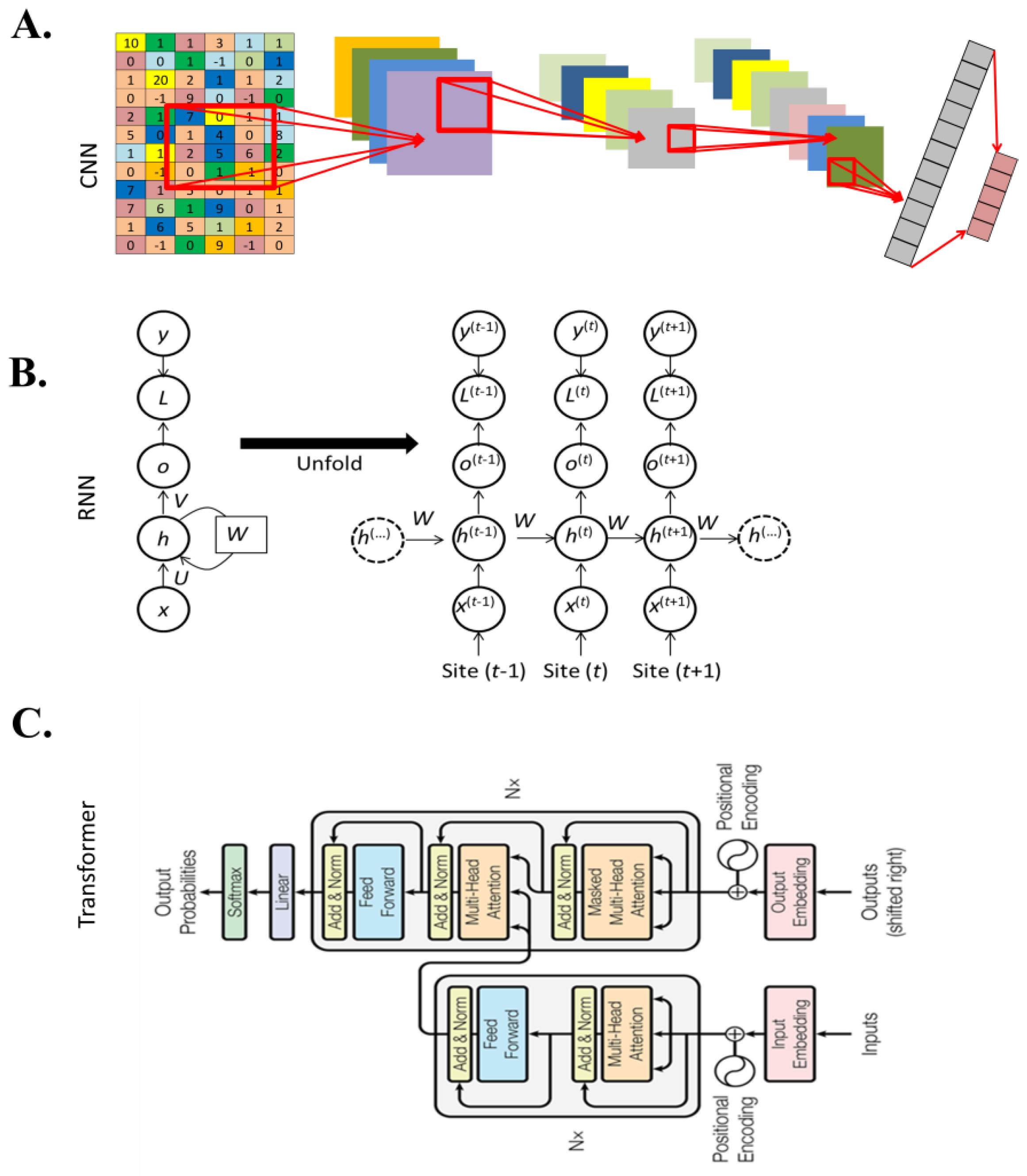
2.3. Major Deep Learning Architectures
3. AI Applications in Taxonomic Annotation, Gene Function Annotation, Associating Plant Traits, and Designing Synthetic Microbe Communities

{kind=link}
{kind=link}
| Reference | Research Type | Research Priority | Raw Sequences | AI Method |
|---|---|---|---|---|
| [56] | Taxonomic analysis | Assignment of raw sequences to the origin of the genome without knowing the reference genome. | Microbiome datasets from several different habitats from GMGCv1 (Global Microbial Gene Catalog), including the human gut, non-human guts, and environmental habitats (ocean and soil). | Semi-supervised learning |
| [57] | Taxonomic analysis | Annotation of viral components in mixed metagenomes containing both viral and host contigs. | Sequences subsampled from prokaryotes and viral genome sequences at several contig lengths: 500, 1000, 3000, 5000, and 10,000 bp. | Unspecified machine learning |
| [58] | Taxonomic analysis | Taxonomic identification of microbial eukaryotes from integral components of natural microbial communities. | Raw sequence reads of microbial samples mainly originating from groundwater. | SVM |
| [59] | Taxonomic analysis | Classification of microbes into species and genera, and the estimation of abundance for human gut microbiomes. | 2505 representative genomes of human gut microbe species. | LSTM; self-attention |
| [60] | Taxonomic analysis | Identification of eukaryotic sequences in metagenomic datasets. | Datasets from NCBI and the Joint Genome Institute, including 8220 genomic sequences representing Eukarya (4381) (nuclear (73), plastid (2260) and mitochondrial genomes (2048)), Bacteria (1860), and Archaea (1979). | ANN |
| [61] | Taxonomic analysis | Identification of phage sequences without a reference genome. | Metagenomic sequences from NCBI. | LSTM |
| [62] | Functional annotation | Prediction of antibacterial or antifungal activity based on features of known natural product biosynthetic gene clusters. | Biosynthetic gene clusters that were available from the Minimum Information about a Biosynthetic Gene Custer database (version 1.4). | SVM; RF |
| [63] | Functional annotation | Functional annotation and classification of the complete (genomic proteins) and partial (metagenomic ORFs) protein sequences. | Protein sequences and associated information of orthologous groups of genes (from eggNOGv3.0). | RF |
| [64] | Functional annotation | Identification of biosynthetic gene clusters in bacterial genomes, and improved identification precision and ability to identify novel functional gene classes. | Open reading frames in 3376 reference bacterial genomes. | BiLSTM |
| [65] | Functional annotation | Identification of transcription activator-like effector that causes bacterial leaf streak of rice. | Promoter sequences, defined as the 1000 bases upstream of the start codon, for the approximately 56,000 rice genes annotated in the MSU Rice Genome Annotation Project Release 7. | Naive Bayes and logistic regression classifiers |
| [66] | Functional annotation | Identification of promoters in atypical microbial hosts. | Promoter sequences from the Geobacillus 7544 core coding sequence. | RF, ANN, and partial least squares regression (PLS) |
| [67] | Functional annotation | Annotation of Lactococcus genes with molecular functions needed for biological nitrogen fixation in Sierra Mixe maize, including mucilage carbohydrate catabolism, glycan-mediated host adhesion, iron/siderophore utilization, and oxidation/reduction control. | Whole genome sequences. | RF |
| [68] | Functional annotation | Identification of bacterial sequence functions that are associated with the growth of the plant Brassica rapa in different soil microbial treatments and at different stages of plant development. | 16S rRNA amplicon variants. | Generalized linear and Bayesian multilevel modeling |
| [69] | Functional annotation | Annotation of DNA sequences of crop pathogens for functions in nutrient acquisition, avoidance of host defenses, regulation of symbiosis, symbiosis, and movement in the environment of another organism. | 16S rRNA amplicons. | SVM; RF |
| [70] | Functional annotation | Classification of non-ribosomal peptides from soil-associated microbes with a high tolerance to sequence modification. | The DNA sequences of microbial datasets from Xenorhabdus and Photorhabdus families (XPF), Staphylococcus (SkinStaph), soil-dwelling Actinobacteria (SoilActi), and a collection of soil-associated bacteria within Bacillus, Pseudomonas, Buttiauxella, and Rahnella genera generated under the Tiny Earth antibiotic discovery project (TinyEarth). | SVM |
| [71] | Functional annotation | Annotation of non-ribosomal peptides. | Nucleotide sequences including complete and draft genome assemblies. | SVM |
| [72] | Functional annotation | Identification of potential sources of novel antibiotic resistance genes (ARGs). | ARG genes were obtained from three major databases: CARD, ARDB, and UNIPROT. | ANN |
| [73] | Functional annotation | Gene prediction using metagenomics fragments. | Sequencing reads from Orphelia and MGC metagenomic dataset. | CNN |
| [74] | Association with host traits | Rice traits (dried biomass, tissue nitrogen concentrations, and net photosynthetic rate) were associated with bacterial microbiota, including those in the seed, root endosphere, and rhizosphere. | 16S rRNA amplicons. | RF |
| [75] | Association with host traits | Classification of fungi into lifestyle classes (pathogen, saprobe, or others). | The whole genome of 101 Dothideomycetes. | SVM |
| [16] | Association with host traits | Association of crop productivity with bulk soil microbiome composition and several nitrogen utility-related taxa. | Shotgun sequences for bulk soil samples. | RF |
| [76] | Association with host traits | Association of rhizosphere microflora and root exudate profiles to cucumber resistance to Fusarium wilt disease. | 16S rRNA amplicons. | PCA; RF |
| [77] | Association with host traits | Association of a microbiome profile with its original location. | Microbiome datasets (16S rRNA amplicon and shotgun sequences) from Boston urban and blinded samples from eight cities. | RF |
| [78] | Association with host traits | Association of root microbiomes with rice traits, including sulfur oxidation and reduction, biofilm production, nitrogen fixation, denitrification, and phosphorus metabolism. | 16S rRNA amplicons. | RF |
| [79] | Association with host traits | Association of the relative OTUs abundance with rice age, and identification of OTUs in the rhizosphere and endosphere compartments that discriminate rice age. | 16S rRNA of 1510 samples from root spatial compartments in field-grown rice (Oryza sativa) throughout three consecutive growing seasons, as well as two geographic sites. | PCoA, RF |
| [80] | Association with host traits | Association of root microbiota with rice developmental stages. | 16S rRNA amplicons. | RF |
| [81] | Association with host traits | Association of root microbiota with different Panax species. | Amplicon sequencing for 405 multi-niche samples of three Holarctic distinct Panax species. | RF |
| [82] | Association with host traits | Revealing worldwide soil microbial community patterns by merging independent taxonomy-based data sets. | 16S rRNA amplicons. | RF |
| [83] | Association with host traits | Deciphering the functional relationship between soil-specific microbes and ecosystem properties. | 16S rRNA amplicons. | Neural network; RF |
| [84] | Designing SynComs | Development of a novel approach to design microbe communities and to predict plant response to phosphate starvation. | 16S rRNA amplicons | ANN |
3.1. AI Applications in Taxonomic Analysis
3.2. AI Applications in Functional Annotation
3.3. AI Application in Plant–Microbiota Association Analysis
3.4. AI Design of Synthetic Communities
4. AI Applications in Plant Genomic Prediction against Pathogens, Phenotyping, Plant–Microbiome Interactions, and Disease Forecasting
4.1. AI Applications in Genomic Prediction against Pathogens
4.2. High-Throughput Plant Disease Phenotyping and In-Field Plant Disease Forecasting
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Glossary
| AI | artificial intelligence |
| ML | machine learning |
| DL | deep learning |
| NLP | natural language processing |
| PCA | principal component analysis |
| PCs | principal components |
| PCoA | principle coordinate analysis |
| OTUs | operational taxonomic units |
| SVM | support vector machine |
| RF | random forest |
| ANN | artificial neural network |
| CNNs | convolutional neural networks |
| RNNs | recurrent neural networks |
| SNPs | single nucleotide polymorphisms |
| LSTM | long short-term memory |
| ITS | internal transcribed spacer |
| RDP | ribosome database project |
| NB | Naive Bayesian |
| ARGs | antibiotic resistance genes |
| ACC | 1- aminocyclopropane-1-carboxylic acid |
| PGPRs | plant growth promoting rhizobacteria |
| SynComs | synthetic microbial communities |
| MAS | marker-assisted selection |
| GP | genomic prediction |
| GBLUP | genomic best linear unbiased prediction |
| LMM | linear mixed model |
| RKHS | reproducing kernel Hilbert spaces |
| GTB | Gradient Tree Boosting |
| FHB | Fusarium head blight |
| DON | deoxynivalenol |
| CYMMIT | International Maize and Wheat Improvement Center |
| MS | mass spectrometry |
| NMR | nuclear magnetic resonance |
| STRNN | spatial–temporal recurrent neural network |
References
- Müller, D.B.; Vogel, C.; Bai, Y.; Vorholt, J.A. The plant microbiota: Systems-level insights and perspectives. Annu. Rev. Genet. 2016, 50, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 2018, 175, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Song, Y.; Yang, K.; Gu, Y.; Wei, Z.; Kowalchuk, G.A.; Xu, Y.; Jousset, A.; Shen, Q.; Geisen, S. Rhizosphere protists are key determinants of plant health. Microbiome 2020, 8, 27. [Google Scholar] [CrossRef]
- Yang, J.; Kloepper, J.W.; Ryu, C.M. Rhizosphere bacteria help plants tolerate abiotic stress. Trends Plant Sci. 2009, 14, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kim, A.; Rosenbluh, J.; Horn, H.; Greenfeld, L.; An, D.; Zimmer, A.; Liberzon, A.; Bistline, J.; Natoli, T. GeNets: A unified web platform for network-based genomic analyses. Nat. Methods 2018, 15, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Pennock, D.; McKenzie, N.; Montanarella, L. Status of the World’s Soil Resources; Technical Summary; FAO: Rome, Italy, 2015. [Google Scholar]
- Wei, Z.; Gu, Y. Initial soil microbiome composition and functioning predetermine future plant health. Sci. Adv. 2019, 5, eaaw0759. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef]
- Campbell, C.L.; Noe, J.P. The spatial analysis of soilborne pathogens and root diseases. Annu. Rev. Phytopathol. 1985, 23, 129–148. [Google Scholar] [CrossRef]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Kwak, M.J.; Kong, H.G.; Choi, K.; Kwon, S.K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; De Bruijn, I.; Dekkers, E.; van Der Voort, M.; Schneider, J.H.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Huang, J.F.; Hu, J.; Gu, Y.A.; Yang, C.L.; Mei, X.L.; Shen, Q.R.; Xu, Y.C.; Friman, V.P. Altering transplantation time to avoid periods of high temperature can efficiently reduce bacterial wilt disease incidence with tomato. PLoS ONE 2015, 10, e0139313. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Hu, J.; Yin, S.; Xu, Y.; Jousset, A.; Shen, Q.; Friman, V.P. Ralstonia solanacearum pathogen disrupts bacterial rhizosphere microbiome during an invasion. Soil Biol. Biochem. 2018, 118, 8–17. [Google Scholar] [CrossRef]
- Gu, Y.; Dong, K.; Geisen, S.; Yang, W.; Yan, Y.; Gu, D.; Liu, N.; Borisjuk, N.; Luo, Y.; Friman, V.P. The effect of microbial inoculant origin on the rhizosphere bacterial community composition and plant growth-promotion. Plant Soil 2020, 452, 105–117. [Google Scholar] [CrossRef]
- Chang, H.X.; Haudenshield, J.S.; Bowen, C.R.; Hartman, G.L. Metagenome-wide association study and machine learning prediction of bulk soil microbiome and crop productivity. Front. Microbiol. 2017, 8, 519. [Google Scholar] [CrossRef]
- Hermans, S.M.; Buckley, H.L.; Case, B.S.; Curran-Cournane, F.; Taylor, M.; Lear, G. Using soil bacterial communities to predict physico-chemical variables and soil quality. Microbiome 2020, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Panch, T.; Szolovits, P.; Atun, R. Artificial intelligence, machine learning and health systems. J. Glob. Health 2018, 8, 020303. [Google Scholar] [CrossRef]
- Lund, B.D.; Wang, T. Chatting about ChatGPT: How may AI and GPT impact academia and libraries? Libr. Hi Tech News, 2023; epub ahead of print. [Google Scholar] [CrossRef]
- Cahan, P.; Treutlein, B. A conversation with ChatGPT on the role of computational systems biology in stem cell research. Stem Cell Rep. 2023, 18, 1–2. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Namkung, J. Machine learning methods for microbiome studies. J. Microbiol. 2020, 58, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.; Tenenbaum, J. Global versus local methods in nonlinear dimensionality reduction. Proc. Adv. Neural Inf. Process. Syst. 2002, 15, 705–712. [Google Scholar]
- Hotelling, H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 417. [Google Scholar] [CrossRef]
- Wakita, Y.; Shimomura, Y.; Kitada, Y.; Yamamoto, H.; Ohashi, Y.; Matsumoto, M. Taxonomic classification for microbiome analysis, which correlates well with the metabolite milieu of the gut. BMC Microbiol. 2018, 18, 188. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, D.; Fang, Z.; Jie, Z.; Qiu, X.; Zhang, C.; Chen, Y.; Ji, L. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS ONE 2013, 8, e71108. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, R.B.; Techtmann, S.M. Machine learning applications in microbial ecology: Human microbiome studies, and environmental monitoring. Comput. Struct. Biotechnol. J. 2021, 19, 1092–1107. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Wilhelm, R.C.; van Es, H.M.; Buckley, D.H. Predicting measures of soil health using the microbiome and supervised machine learning. Soil Biol. Biochem. 2022, 164, 108472. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Calle, M.L.; Urrea, V.; Boulesteix, A.L.; Malats, N. AUC-RF: A new strategy for genomic profiling with random forest. Hum. Hered. 2011, 72, 121–132. [Google Scholar] [CrossRef]
- Probst, P.; Bischl, B.; Boulesteix, A.L. Tunability: Importance of hyperparameters of machine learning algorithms. arXiv 2018, arXiv:1802.09596. [Google Scholar]
- Lantz, B. Machine Learning with R: Expert Techniques for Predictive Modeling; Packt Publishing Ltd.: Birmingham, UK, 2019. [Google Scholar]
- Fiannaca, A.; La Paglia, L.; La Rosa, M.; Bosco, L.; Renda, G.; Rizzo, R.; Gaglio, S.; Urso, A. Deep learning models for bacteria taxonomic classification of metagenomic data. BMC Bioinform. 2018, 19, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, S.; Song, Y.S. Deep learning for population genetic inference. PLoS Comput. Biol. 2016, 12, e1004845. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A.; Bengio, Y. Deep Learning; MIT Press: Cambridge, MA, USA, 2016; Volume 1. [Google Scholar]
- Kamilaris, A.; Prenafeta-Boldú, F.X. A review of the use of convolutional neural networks in agriculture. J. Agric. Sci. 2018, 156, 312–322. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.M.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. arXiv 2017, arXiv:abs/1706.03762. [Google Scholar]
- Bengio, Y.; Simard, P.; Frasconi, P. Learning long-term dependencies with gradient descent is difficult. IEEE Trans Neural Netw. 1994, 5, 157–166. [Google Scholar] [CrossRef]
- Xiao, Q.; Li, W.; Chen, P.; Wang, B. Prediction of crop pests and diseases in cotton by long short term memory network. In Proceedings of the 2nd International Conference on Intelligent Computing and Control Systems (ICICCS), Madurai, India, 14–15 June 2018; Springer: Berlin/Heidelberg, Germany, 2018; pp. 11–16. [Google Scholar]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Ryberg, M.; Abarenkov, K.; Sjökvist, E.; Kristiansson, E. The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol. Lett. 2009, 296, 97–101. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33 (Suppl. S2), W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Panda, K.C.; Sagar, A.; Al-Ansari, N.; Duan, H.F.; Paramaguru, P.K.; Vishwakarma, D.K.; Kumar, A.; Kumar, D.; Kashyap, P. Novel Genetic Algorithm (GA) based hybrid machine learning-pedotransfer Function (ML-PTF) for prediction of spatial pattern of saturated hydraulic conductivity. Eng. Appl. Comput. Fluid Mech. 2022, 16, 1082–1099. [Google Scholar] [CrossRef]
- Urtecho, G.; Insigne, K.D.; Tripp, A.D.; Brinck, M.; Lubock, N.B.; Kim, H.; Chan, T.; Kosuri, S. Genome-wide functional characterization of Escherichia coli promoters and regulatory elements responsible for their function. bioRxiv 2020. bioRxiv:2020.01.04.894907. [Google Scholar]
- Calle, M.L. Statistical analysis of metagenomics data. Genom. Inform. 2019, 17, e6. [Google Scholar] [CrossRef]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine learning meta-analysis of large metagenomic datasets: Tools and biological insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Zhu, C.; Zhao, X.M.; Coelho, L.P. A deep siamese neural network improves metagenome-assembled genomes in microbiome datasets across different environments. Nat. Commun. 2022, 13, 2326. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- West, P.T.; Probst, A.J.; Grigoriev, I.V.; Thomas, B.C.; Banfield, J.F. Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res. 2018, 28, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Bible, P.W.; Liu, Y.; Zou, B.; Wei, L. DeepMicrobes: Taxonomic classification for metagenomics with deep learning. NAR Genom. Bioinform. 2020, 2, lqaa009. [Google Scholar] [CrossRef]
- Karlicki, M.; Antonowicz, S.; Karnkowska, A. Tiara. deep learning-based classification system for eukaryotic sequences. Bioinformatics 2022, 38, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Auslander, N.; Gussow, A.B.; Benler, S.; Wolf, Y.I.; Koonin, E.V. Seeker: Alignment-free identification of bacteriophage genomes by deep learning. Nucleic Acids Res. 2020, 48, e121. [Google Scholar] [CrossRef]
- Walker, A.S.; Clardy, J. A machine learning bioinformatics method to predict biological activity from biosynthetic gene clusters. J. Chem. Inf. Model. 2021, 61, 2560–2571. [Google Scholar] [CrossRef]
- Sharma, A.K.; Gupta, A.; Kumar, S.; Dhakan, D.B.; Sharma, V.K. Woods: A fast and accurate functional annotator and classifier of genomic and metagenomic sequences. Genomics 2015, 106, 1–6. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Prihoda, D.; Palicka, A.; Soukup, J.; Klempir, O.; Rampula, L.; Durcak, J.; Wurst, M.; Kotowski, J.; Chang, D. A deep learning genome-mining strategy for biosynthetic gene cluster prediction. Nucleic Acids Res. 2019, 47, e110. [Google Scholar] [CrossRef]
- Cernadas, R.A.; Doyle, E.L.; Niño-Liu, D.O.; Wilkins, K.E.; Bancroft, T.; Wang, L.; Schmidt, C.L.; Caldo, R.; Yang, B.; White, F.F. Code-assisted discovery of TAL effector targets in bacterial leaf streak of rice reveals contrast with bacterial blight and a novel susceptibility gene. PLoS Pathog. 2014, 10, e1003972. [Google Scholar] [CrossRef] [PubMed]
- Gilman, J.; Singleton, C.; Tennant, R.K.; James, P.; Howard, T.P.; Lux, T.; Parker, D.A.; Love, J. Rapid, heuristic discovery and design of promoter collections in non-model microbes for industrial applications. ACS Synth. Biol. 2019, 8, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Higdon, S.M.; Huang, B.C.; Bennett, A.B.; Weimer, B.C. Identification of nitrogen fixation genes in Lactococcus isolated from maize using population genomics and machine learning. Microorganisms 2020, 8, 2043. [Google Scholar] [CrossRef] [PubMed]
- Klasek, S.A.; Brock, M.T.; Calder, W.J.; Morrison, H.G.; Weinig, C.; Maïgnien, L. Spatiotemporal heterogeneity and intragenus variability in rhizobacterial associations with Brassica rapa growth. Msystems 2022, 7, e00060-22. [Google Scholar] [CrossRef]
- Ma, B.; Charkowski, A.O.; Glasner, J.D.; Perna, N.T. Identification of host-microbe interaction factors in the genomes of soft rot-associated pathogens Dickeya dadantii 3937 and Pectobacterium carotovorum WPP14 with supervised machine learning. BMC Genom. 2014, 15, 508. [Google Scholar] [CrossRef]
- Behsaz, B.; Bode, E.; Gurevich, A.; Shi, Y.N.; Grundmann, F.; Acharya, D.; Caraballo-Rodríguez, A.M.; Bouslimani, A.; Panitchpakdi, M.; Linck, A. Integrating genomics and metabolomics for scalable non-ribosomal peptide discovery. Nat. Commun. 2021, 12, 3225. [Google Scholar] [CrossRef]
- Kunyavskaya, O.; Tagirdzhanov, A.M.; Caraballo-Rodríguez, A.M.; Nothias, L.F.; Dorrestein, P.C.; Korobeynikov, A.; Mohimani, H.; Gurevich, A. Nerpa: A tool for discovering biosynthetic gene clusters of bacterial nonribosomal peptides. Metabolites 2021, 11, 693. [Google Scholar] [CrossRef]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018, 6, 23. [Google Scholar] [CrossRef]
- Al-Ajlan, A.; El Allali, A. CNN-MGP: Convolutional neural networks for metagenomics gene prediction. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 628–635. [Google Scholar] [CrossRef]
- Guo, J.; Ling, N.; Li, Y.; Li, K.; Ning, H.; Shen, Q.; Guo, S.; Vandenkoornhuyse, P. Seed-borne, endospheric and rhizospheric core microbiota as predictors of plant functional traits across rice cultivars are dominated by deterministic processes. New Phytol. 2021, 230, 2047–2060. [Google Scholar] [CrossRef]
- Haridas, S.; Albert, R.; Binder, M.; Bloem, J.; LaButti, K.; Salamov, A.; Andreopoulos, B.; Baker, S.; Barry, K.; Bills, G. 101 Dothideomycetes genomes: A test case for predicting lifestyles and emergence of pathogens. Stud. Mycol. 2020, 95, 5–169. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Yuan, J.; He, X.; Lin, Y.; Huang, Q.; Shen, Q. Enrichment of beneficial cucumber rhizosphere microbes mediated by organic acid secretion. Hortic. Res. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.Y.; Tyler, A.D. Systematic evaluation of supervised machine learning for sample origin prediction using metagenomic sequencing data. Biol. Direct. 2020, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kumar, M.; Chakdar, H.; Pandiyan, K.; Kumar, S.C.; Zeyad, M.T.; Singh, B.N.; Ravikiran, K.; Mahto, A.; Srivastava, A.K. Influence of host genotype in establishing root associated microbiome of indica rice cultivars for plant growth promotion. Front. Microbiol. 2022, 13, 1033158. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.A.; Santos-Medellín, C.M.; Liechty, Z.S.; Nguyen, B.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, N.; Liu, Y.X.; Zhang, X.; Hu, B.; Qin, Y.; Xu, H.; Wang, H.; Guo, X.; Qian, J. Root microbiota shift in rice correlates with resident time in the field and developmental stage. Sci. China Life Sci. 2018, 61, 613–621. [Google Scholar] [CrossRef]
- Zhang, G.; Wei, F.; Chen, Z.; Wang, Y.; Jiao, S.; Yang, J.; Chen, Y.; Liu, C.; Huang, Z.; Dong, L. Evidence for saponin diversity–mycobiome links and conservatism of plant–fungi interaction patterns across Holarctic disjunct Panax species. Sci. Total Environ. 2022, 830, 154583. [Google Scholar] [CrossRef]
- Ramirez, K.S.; Knight, C.G.; De Hollander, M.; Brearley, F.Q.; Constantinides, B.; Cotton, A.; Creer, S.; Crowther, T.W.; Davison, J.; Delgado-Baquerizo, M. Detecting macroecological patterns in bacterial communities across independent studies of global soils. Nat. Microbiol. 2018, 3, 189–196. [Google Scholar] [CrossRef]
- Thompson, J.; Johansen, R.; Dunbar, J.; Munsky, B. Machine learning to predict microbial community functions: An analysis of dissolved organic carbon from litter decomposition. PLoS ONE 2019, 14, e0215502. [Google Scholar] [CrossRef]
- Herrera Paredes, S.; Gao, T.; Law, T.F.; Finkel, O.M.; Mucyn, T.; Teixeira, P.; Salas González, I.; Feltcher, M.E.; Powers, M.J.; Shank, E.A.; et al. Design of synthetic bacterial communities for predictable plant phenotypes. PLoS Biol. 2018, 16, e2003962. [Google Scholar] [CrossRef]
- McHardy, A.C.; Martín, H.G.; Tsirigos, A.; Hugenholtz, P.; Rigoutsos, I. Accurate phylogenetic classification of variable-length DNA fragments. Nat. Methods 2007, 4, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Patil, K.R.; Roune, L.; McHardy, A.C. The PhyloPythiaS web server for taxonomic assignment of metagenome sequences. PLoS ONE 2012, 7, e38581. [Google Scholar] [CrossRef] [PubMed]
- Vervier, K.; Mahé, P.; Tournoud, M.; Veyrieras, J.B.; Vert, J.P. Large-scale machine learning for metagenomics sequence classification. Bioinformatics 2016, 32, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Ounit, R.; Lonardi, S. Higher classification sensitivity of short metagenomic reads with CLARK-S. Bioinformatics 2016, 32, 3823–3825. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Menegaux, R.; Vert, J. P: Continuous embeddings of DNA sequencing reads and application to metagenomics. Comput. Biol. 2019, 26, 509–518. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, H.; Ye, Y. RAPSearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 2012, 28, 125–126. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Nathan, C. Resisting antimicrobial resistance. Nat. Rev. Microbiol. 2020, 18, 259–260. [Google Scholar] [CrossRef]
- Zaman, S.B.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A review on antibiotic resistance: Alarm bells are ringing. Cureus 2017, 9, e1403. [Google Scholar] [CrossRef] [PubMed]
- Wassan, J.T.; Wang, H.; Browne, F.; Zheng, H. Phy-PMRFI: Phylogeny-aware prediction of metagenomic functions using random forest feature importance. IEEE Trans. Nanobioscience 2019, 18, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Khodabandelou, G.; Routhier, E.; Mozziconacci, J. Genome functional annotation across species using deep convolutional neural networks. bioRxiv 2019. bioRxiv:330308. [Google Scholar]
- Galperin, M.Y.; Kristensen, D.M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Microbial genome analysis: The COG approach. Brief. Bioinform. 2019, 20, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.A.; Chai, B.; Wang, Q.; Sun, Y.; Brown, C.T.; Tiedje, J.M.; Cole, J.R. FunGene: The functional gene pipeline and repository. Front. Microbiol. 2013, 4, 291. [Google Scholar] [CrossRef]
- Wilke, A.; Bischof, J.; Harrison, T.; Brettin, T.; D’Souza, M.; Gerlach, W.; Matthews, H.; Paczian, T.; Wilkening, J.; Glass, E.M. A RESTful API for accessing microbial community data for MG-RAST. PLoS Comput. Biol. 2015, 11, e1004008. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Dreher, K.; Fulcher, C.A.; Subhraveti, P.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012, 40, D742–D753. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Wei, Y.; Hu, W.; Liu, G.; Zeng, H.; Shi, H. Microbiome-wide association studies reveal correlations between the structure and metabolism of the rhizosphere microbiome and disease resistance in cassava. Plant Biotechnol. J. 2021, 19, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Wang, Y.; Huang, Y.; Xu, J.; Zhang, P.; Wang, N.; Liu, X.; Chu, H.; Liu, G.; Jiang, H. Taxonomic structure and functional association of foxtail millet root microbiome. Gigascience 2017, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wagg, C.; Schlaeppi, K.; Banerjee, S.; Kuramae, E.E.; van der Heijden, M.G. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 2019, 10, 4841. [Google Scholar] [CrossRef]
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. USA 2018, 115, 7368–7373. [Google Scholar] [CrossRef]
- Blaustein, R.A.; Lorca, G.L.; Meyer, J.L.; Gonzalez, C.F.; Teplitski, M. Defining the core citrus leaf-and root-associated microbiota: Factors associated with community structure and implications for managing huanglongbing (citrus greening) disease. Appl. Environ. Microbiol. 2017, 83, e00210-17. [Google Scholar] [CrossRef]
- Santos-Medellín, C.; Edwards, J.; Liechty, Z.; Nguyen, B.; Sundaresan, V. Drought stress results in a compartment-specific restructuring of the rice root-associated microbiomes. mBio 2017, 8, e00764-17. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Riera, N.; Jin, T.; Li, J.; Wang, N. Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 2017, 5, 97. [Google Scholar] [CrossRef]
- Ali, S.; Tyagi, A.; Park, S.; Mir, R.A.; Mushtaq, M.; Bhat, B.; Mahmoudi, H.; Bae, H. Deciphering the plant microbiome to improve drought tolerance: Mechanisms and perspectives. Environ. Exp. Bot. 2022, 201, 104933. [Google Scholar] [CrossRef]
- Niu, B.; Paulson, J.N.; Zheng, X.; Kolter, R. Simplified and representative bacterial community of maize roots. Proc. Natl. Acad. Sci. USA 2017, 114, E2450–E2459. [Google Scholar] [CrossRef] [PubMed]
- Parlevliet, J.E. Durability of resistance against fungal, bacterial and viral pathogens: Present situation. Euphytica 2002, 124, 147–156. [Google Scholar] [CrossRef]
- Bernardo, R.; Yu, J. Prospects for genomewide selection for quantitative traits in maize. Crop Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef]
- Heffner, E.L.; Lorenz, A.J.; Jannink, J.L.; Sorrells, M.E. Plant breeding with genomic selection: Gain per unit time and cost. Crop Sci. 2010, 50, 1681–1690. [Google Scholar] [CrossRef]
- Bernardo, R. A model for marker-assisted selection among single crosses with multiple genetic markers. Theor. Appl. Genet. 1998, 97, 473–478. [Google Scholar] [CrossRef]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.J.; Goddard, M.E. Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Pérez, P.; de Los Campos, G.; Crossa, J.; Gianola, D. Genomic-enabled prediction based on molecular markers and pedigree using the Bayesian linear regression package in R. Plant Genome 2010, 3, 106. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Monir, M.M.; Zhu, J. Dominance and epistasis interactions revealed as important variants for leaf traits of maize NAM population. Front. Plant Sci. 2018, 9, 627. [Google Scholar] [CrossRef]
- Holland, J.B. Genetic architecture of complex traits in plants. Curr. Opin. Plant Biol. 2007, 10, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kasnavi, S.A.; Afshar, M.A.; Shariati, M.M.; Kashan, N.E.J.; Honarvar, M. Performance evaluation of support vector machine (SVM)-based predictors in genomic selection. Indian J. Anim. Sci. 2017, 87, 1226–1231. [Google Scholar] [CrossRef]
- Long, N.; Gianola, D.; Rosa, G.J.; Weigel, K.A. Application of support vector regression to genome-assisted prediction of quantitative traits. Theor. Appl. Genet. 2011, 123, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- De Los Campos, G.; Gianola, D.; Rosa, G.J.; Weigel, K.A.; Crossa, J. Semi-parametric genomic-enabled prediction of genetic values using reproducing kernel Hilbert spaces methods. Genet. Res. 2010, 92, 295–308. [Google Scholar] [CrossRef]
- Gianola, D.; Fernando, R.L.; Stella, A. Genomic-assisted prediction of genetic value with semiparametric procedures. Genetics 2006, 173, 1761–1776. [Google Scholar] [CrossRef] [PubMed]
- González-Recio, O.; Forni, S. Genome-wide prediction of discrete traits using Bayesian regressions and machine learning. Genet. Sel. 2011, 43, 7. [Google Scholar] [CrossRef]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redona, E.; Atlin, G.; Jannink, J.L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar]
- Drummond, S.T.; Sudduth, K.A.; Joshi, A.; Birrell, S.J.; Kitchen, N.R. Statistical and neural methods for site–specific yield prediction. Trans. ASAE 2003, 46, 5. [Google Scholar] [CrossRef]
- Gianola, D.; Okut, H.; Weigel, K.A.; Rosa, G.J. Predicting complex quantitative traits with Bayesian neural networks: A case study with Jersey cows and wheat. BMC Genet. 2011, 12, 87. [Google Scholar] [CrossRef]
- González-Recio, O.; Rosa, G.J.; Gianola, D. Machine learning methods and predictive ability metrics for genome-wide prediction of complex traits. Livest. Sci. 2014, 166, 217–231. [Google Scholar] [CrossRef]
- Leung, M.K.; Delong, A.; Alipanahi, B.; Frey, B.J. Machine learning in genomic medicine: A review of computational problems and data sets. Proc. IEEE 2015, 104, 176–197. [Google Scholar] [CrossRef]
- Glória, L.S.; Cruz, C.D.; Vieira, R.A.M.; de Resende, M.D.V.; Lopes, P.S.; de Siqueira, O.H.D.; e Silva, F.F. Accessing marker effects and heritability estimates from genome prediction by Bayesian regularized neural networks. Livest. Sci. 2016, 191, 91–96. [Google Scholar] [CrossRef]
- Romagnoni, A.; Jégou, S.; Van Steen, K.; Wainrib, G.; Hugot, J.P. Comparative performances of machine learning methods for classifying Crohn Disease patients using genome-wide genotyping data. Sci. Rep. 2019, 9, 10351. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Balvert, M.; van der Spek, R.A.; Dutilh, B.E.; Bohté, S.; Veldink, J.; Schönhuth, A. Using the structure of genome data in the design of deep neural networks for predicting amyotrophic lateral sclerosis from genotype. Bioinformatics 2019, 35, i538–i547. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, N.F.; Orhobor, O.I.; King, R.D. An evaluation of machine-learning for predicting phenotype: Studies in yeast, rice, and wheat. Mach. Learn. 2020, 109, 251–277. [Google Scholar] [CrossRef]
- Ranganathan, S.; Nakai, K.; Schonbach, C. Encyclopedia of Bioinformatics and Computational Biology: ABC of Bioinformatics; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Ma, W.; Qiu, Z.; Song, J.; Cheng, Q.; Ma, C. DeepGS: Predicting phenotypes from genotypes using deep learning. bioRxiv 2017. bioRxiv:241414. [Google Scholar]
- Khaki, S.; Wang, L. Crop yield prediction using deep neural networks. Front. Plant Sci. 2019, 10, 621. [Google Scholar] [CrossRef]
- Jubair, S.; Tucker, J.R.; Henderson, N.; Hiebert, C.W.; Badea, A.; Domaratzki, M.; Fernando, W. GPTransformer: A transformer-based deep learning method for predicting Fusarium related traits in barley. Front. Plant Sci. 2021, 12, 2984. [Google Scholar] [CrossRef]
- Montesinos-López, O.A.; Montesinos-López, A.; Crossa, J.; Gianola, D.; Hernández-Suárez, C.M.; Martín-Vallejo, J. Multi-trait, multi-environment deep learning modeling for genomic-enabled prediction of plant traits. G3 Genes Genomes Genet. 2018, 8, 3829–3840. [Google Scholar] [CrossRef]
- Montesinos-López, O.A.; Montesinos-López, A.; Pérez-Rodríguez, P.; Barrón-López, J.A.; Martini, J.W.; Fajardo-Flores, S.B.; Gaytan-Lugo, L.S.; Santana-Mancilla, P.C.; Crossa, J. A review of deep learning applications for genomic selection. BMC Genom. 2021, 22, 19. [Google Scholar] [CrossRef]
- Pook, T.; Freudenthal, J.; Korte, A.; Simianer, H. Using local convolutional neural networks for genomic prediction. Front. Genet. 2020, 11, 561497. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Enciso, M.; Zingaretti, L.M. A guide on deep learning for complex trait genomic prediction. Genes 2019, 10, 553. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Qiu, Z.; Song, J.; Li, J.; Cheng, Q.; Zhai, J.; Ma, C. A deep convolutional neural network approach for predicting phenotypes from genotypes. Planta 2018, 248, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, C.; Mora, F.; Contreras-Soto, R.; Ahmar, S.; Chen, J.T.; do Amaral Júnior, A.T.; Scapim, C.A. Genome-wide prediction of complex traits in two outcrossing plant species through deep learning and Bayesian regularized neural network. Front. Plant Sci. 2020, 11, 1734. [Google Scholar] [CrossRef] [PubMed]
- Zingaretti, L.M.; Gezan, S.A.; Ferrão, L.F.V.; Osorio, L.F.; Monfort, A.; Muñoz, P.R.; Whitaker, V.M.; Pérez-Enciso, M. Exploring deep learning for complex trait genomic prediction in polyploid outcrossing species. Front. Plant Sci. 2020, 11, 25. [Google Scholar] [CrossRef]
- Jeong, S.; Kim, J.Y.; Kim, N. GMStool: GWAS-based marker selection tool for genomic prediction from genomic data. Sci. Rep. 2020, 10, 19653. [Google Scholar] [CrossRef]
- Yang, W.; Feng, H.; Zhang, X.; Zhang, J.; Doonan, J.H.; Batchelor, W.D.; Xiong, L.; Yan, J. Crop phenomics and high-throughput phenotyping: Past decades, current challenges, and future perspectives. Mol. Plant. 2020, 13, 187–214. [Google Scholar] [CrossRef]
- Fahlgren, N.; Gehan, M.A.; Baxter, I. Lights, camera, action: High-throughput plant phenotyping is ready for a close-up. Curr. Opin. Plant Biol. 2015, 24, 93–99. [Google Scholar] [CrossRef]
- Khamparia, A.; Saini, G.; Gupta, D.; Khanna, A.; Tiwari, S.; de Albuquerque, V.H.C. Seasonal crops disease prediction and classification using deep convolutional encoder network. Circuits Syst. Signal Process. 2020, 39, 818–836. [Google Scholar] [CrossRef]
- Anagnostis, A.; Asiminari, G.; Papageorgiou, E.; Bochtis, D. A convolutional neural networks based method for anthracnose infected walnut tree leaves identification. Appl. Sci. 2020, 10, 469. [Google Scholar] [CrossRef]
- Agarwal, M.; Sinha, A.; Gupta, S.K.; Mishra, D.; Mishra, R. Potato crop disease classification using convolutional neural network. In Proceedings of the 2nd International Conference on Smart IOT Systems: Innovations in Computing 2019 (SSIC 2019), Manipal, India, 18–20 January 2019; Springer: Singapore, 2020; pp. 391–400. [Google Scholar]
- Picon, A.; Alvarez-Gila, A.; Seitz, M.; Ortiz-Barredo, A.; Echazarra, J.; Johannes, A. Deep convolutional neural networks for mobile capture device-based crop disease classification in the wild. Comput. Electron. Agric. 2019, 161, 280–290. [Google Scholar] [CrossRef]
- Sibiya, M.; Sumbwanyambe, M. A Computational procedure for the recognition and classification of maize leaf diseases out of healthy leaves using convolutional neural networks. Agric. Eng. J. 2019, 1, 119–131. [Google Scholar] [CrossRef]
- Lin, K.; Gong, L.; Huang, Y.; Liu, C.; Pan, J. Deep learning-based segmentation and quantification of cucumber powdery mildew using convolutional neural network. Front. Plant Sci. 2019, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, S.; Kamsu-Foguem, B.; Kamissoko, D.; Traore, D. Deep neural networks with transfer learning in millet crop images. Comput. Ind. 2019, 108, 115–120. [Google Scholar] [CrossRef]
- Bergsträsser, S.; Fanourakis, D.; Schmittgen, S.; Cendrero-Mateo, M.P.; Jansen, M.; Scharr, H.; Rascher, U. HyperART: Non-invasive quantification of leaf traits using hyperspectral absorption-reflectance-transmittance imaging. Plant Methods 2015, 11, 1. [Google Scholar] [CrossRef]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant Metabolomics: An indispensable system biology tool for plant science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef]
- Wani, J.A.; Sharma, S.; Muzamil, M.; Ahmed, S.; Sharma, S.; Singh, S. Machine learning and deep learning based computational techniques in automatic agricultural diseases detection: Methodologies, applications, and challenges. Arch. Comput. Methods Eng. 2022, 29, 641–677. [Google Scholar] [CrossRef]
- Hasan, R.I.; Yusuf, S.M.; Alzubaidi, L. Review of the state of the art of deep learning for plant diseases: A broad analysis and discussion. Plants 2020, 9, 1302. [Google Scholar] [CrossRef]
- Neupane, K.; Baysal-Gurel, F. Automatic identification and monitoring of plant diseases using unmanned aerial vehicles: A review. Remote Sens. 2021, 13, 3841. [Google Scholar] [CrossRef]
- Fenu, G.; Malloci, F.M. Forecasting plant and crop disease: An explorative study on current algorithms. Big Data Cogn. Comput. 2021, 5, 2. [Google Scholar] [CrossRef]
- Pryzant, R.; Ermon, S.; Lobell, D. Monitoring Ethiopian wheat fungus with satellite imagery and deep feature learning. In Proceedings of the 2017 IEEE Conference on Computer Vision and Pattern Recognition Workshops (CVPRW), Honolulu, HI, USA, 21–26 July 2017; pp. 1524–1532. [Google Scholar]
- Xu, W.; Wang, Q.; Chen, R. Spatio-temporal prediction of crop disease severity for agricultural emergency management based on recurrent neural networks. GeoInformatica 2018, 22, 363–381. [Google Scholar] [CrossRef]
- Fernando, W.D.; Zhang, X.; Selin, C.; Zou, Z.; Liban, S.H.; McLaren, D.L.; Kubinec, A.; Parks, P.S.; Rashid, M.H.; Padmathilake, K.R.E. A six–year investigation of the dynamics of avirulence allele profiles, blackleg incidence, and mating type alleles of Leptosphaeria maculans populations associated with canola crops in Manitoba, Canada. Plant Dis. 2018, 102, 790–798. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Walkowiak, S.; Fernando, W.G.D. Artificial Intelligence: A Promising Tool in Exploring the Phytomicrobiome in Managing Disease and Promoting Plant Health. Plants 2023, 12, 1852. https://doi.org/10.3390/plants12091852
Zhao L, Walkowiak S, Fernando WGD. Artificial Intelligence: A Promising Tool in Exploring the Phytomicrobiome in Managing Disease and Promoting Plant Health. Plants. 2023; 12(9):1852. https://doi.org/10.3390/plants12091852
Chicago/Turabian StyleZhao, Liang, Sean Walkowiak, and Wannakuwattewaduge Gerard Dilantha Fernando. 2023. "Artificial Intelligence: A Promising Tool in Exploring the Phytomicrobiome in Managing Disease and Promoting Plant Health" Plants 12, no. 9: 1852. https://doi.org/10.3390/plants12091852
APA StyleZhao, L., Walkowiak, S., & Fernando, W. G. D. (2023). Artificial Intelligence: A Promising Tool in Exploring the Phytomicrobiome in Managing Disease and Promoting Plant Health. Plants, 12(9), 1852. https://doi.org/10.3390/plants12091852