Complete Plastid Genome Sequencing of Eight Species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative Analyses and Phylogenetic Implications

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
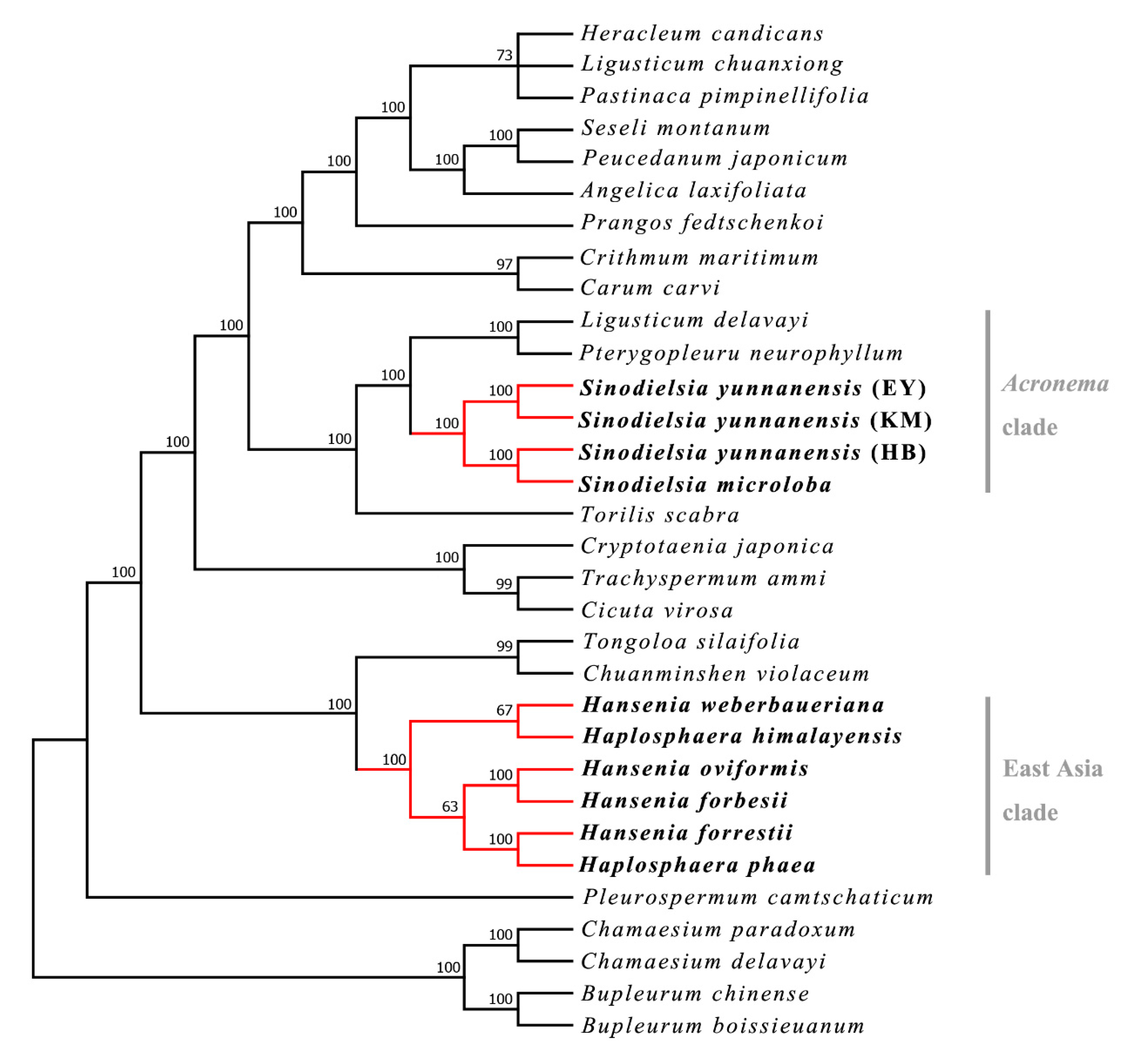
2.1. Phylogenetic Analysis
2.2. The Plastid Genomes of Hansenia, Haplosphaera and Sinodielsia Species
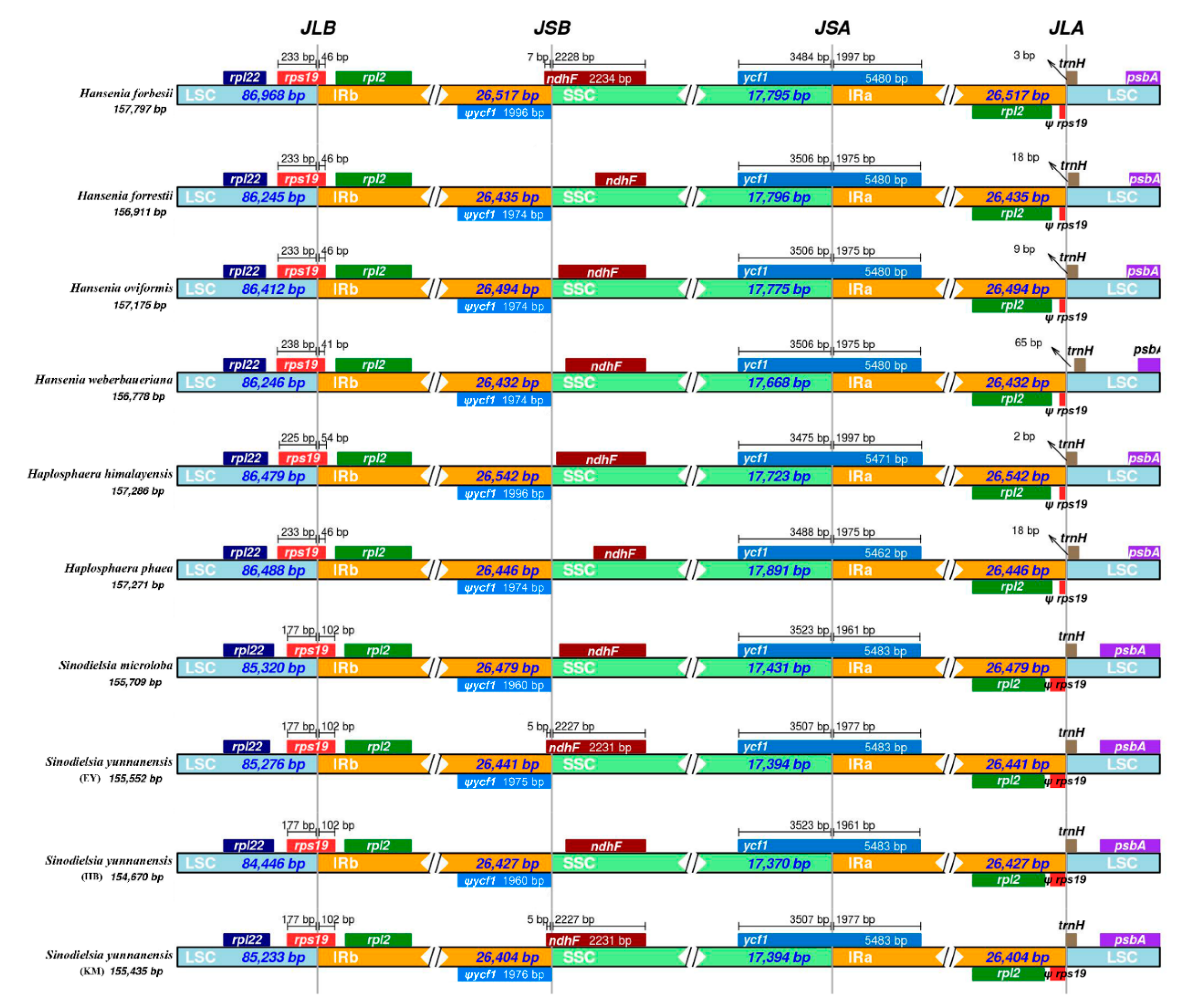
2.3. IR Boundaries and Simple Sequence Repeats (SSRs) Structure Analysis
2.4. Codon Usage Analysis
2.5. Nucleotide Diversity Analysis
3. Materials and Methods
3.1. Material, DNA Extraction and Complete Genome Sequencing
3.2. Genome Construction and Annotation
3.3. Phylogenetic Analysis
3.4. IRs Boundaries and SSR Analysis
3.5. Codon Usage Bias Analysis
3.6. Nucleotide Diversity Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pu, F.D.; Watson, M.F. Apiaceae. In Flora of China; Science Press: Beijing, China; Missouri Botanical garden Press: Saint Louis, MO, USA, 2005; Volume 14, pp. 1–205. [Google Scholar]
- Pimenov, M.G.; Kljuykov, E.V.; Ostroumova, T.A. Reduction of Notopterygium to Hansenia (Umbelliferae). Willdenowia 2008, 38, 155–172. [Google Scholar] [CrossRef]
- Jia, S.B.; Guo, X.L.; Zhou, S.D.; He, X.J. Hansenia pinnatiinvolucellata is conspecific with H. weberbaueriana (Apiaceae) based on morphology and molecular data. Phytotaxa 2019, 418, 203–210. [Google Scholar] [CrossRef]
- Pimenov, M.G.; Kljuykov, E.V. New nomenclatural combinations for Chinese Umbelliferae. Feddes Repert. 1999, 110, 481–491. [Google Scholar] [CrossRef]
- Pimenov, M.G. Updated checklist of Chinese Umbelliferea: Nomenclature, synonymy, typification, distribution. Turcz 2017, 20, 106–239. [Google Scholar]
- Valiejo-Roman, C.M.; Terentieva, E.I.; Samigullin, T.H.; Pimenov, M.G. nrDNA ITS sequences and affinities of Sino-Himalayan Apioideae (Umbelliferae). Taxon 2002, 51, 685–701. [Google Scholar]
- Valiejo-Roman, C.M.; Shneyer, V.S.; Samigullin, T.H.; Terentieva, E.I.; Pimenov, M.G. An attempt to clarify taxonomic relationships in “Verwandtschaftskreis der Gattung Ligusticum” (Umbelliferae-Apioideae) by molecular analysis. Plant Syst. Evol. 2006, 257, 25–43. [Google Scholar] [CrossRef]
- Zhou, J.; Peng, H.; Downie, S.R.; Liu, Z.W.; Gong, X. A molecular phylogeny of Chinese Apiaceae subfamily Apioideae inferred from nuclear ribosomal DNA internal transcribed spacer sequences. Taxon 2008, 57, 402–416. [Google Scholar]
- Zhou, J.; Gong, X.; Downie, S.R.; Peng, H. Towards a more robust molecular phylogeny of Chinese Apiaceae subfamily Apioideae: Additional evidence from nrDNA ITS and cpDNA intron (rpl16 and rps16) sequences. Mol. Phylogenetics Evol. 2009, 53, 56–68. [Google Scholar] [CrossRef]
- Wang, Z.X.; Downie, S.R.; Tan, J.B.; Liao, C.Y.; Yu, Y.; He, X.J. Molecular phylogenetics of Pimpinella and allied genera (Apiaceae), with emphasis on Chinese native species, inferred from nrDNA ITS and cpDNA intron sequence data. Nord. J. Bot. 2014, 32, 642–657. [Google Scholar] [CrossRef]
- Liu, Z.W.; Zhou, J.; Gao, Y.z.; Wei, J.; Downie, S.R. Molecular phylogenetics of Ligusticum (Apiaceae) based on nrDNA ITS sequences: Placement of the Chinese endemic species and a reduced circumscription of the genus. Int. J. Plant Sci. 2019. [Google Scholar]
- Downie, S.R.; Palmer, J.D. Use of Chloroplast DNA Rearrangements in Reconstructing Plant Phylogeny; Molecular Systematics of Plants; Soltis, P.S., Soltis, D.E., Doyle, J.J., Eds.; Springer: Boston, MA, USA, 1992. [Google Scholar]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Yu, X.L.; Tan, W.; Zhang, H.Y.; Gao, H.; Wang, W.X.; Tian, X.X. Complete chloroplast genomes of Ampelopsis humulifolia and Ampelopsis japonica: Molecular structure, comparative analysis, and phylogenetic analysis. Plants 2019, 8, 410. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Cui, L. Methods for Obtaining and Analyzing Whole Chloroplast Genome Sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [PubMed]
- Korpelainen, H. The evolutionary processes of mitochondrial and chloroplast genomes differ from those of nuclear genomes. Sci. Nat. 2004, 91, 505–518. [Google Scholar] [CrossRef]
- Huotari, T.; Korpelainen, H. Complete chloroplast genome sequence of Elodea canadensis and comparative analyses with other monocot plastid genomes. Gene 2012, 508, 96–105. [Google Scholar] [CrossRef]
- Jia, S.B.; Guo, X.L.; Xie, D.F.; Yu, Y.; Zhou, S.D.; He, X.J. The complete chloroplast genome of Haplosphaera phaea (Apiaceae). Mitochondrial Dna B 2019, 4, 1969–1970. [Google Scholar] [CrossRef] [Green Version]
- Gou, W.; Guo, X.L.; Yu, Y.; Zhou, S.D.; He, X.J. The complete chloroplast genome of Meeboldia yunnanensis (Apiaceae). Mitochondrial Dna B 2019, 4, 4176–4177. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Yu, Y.; Xie, D.F.; Peng, C.; Liu, Q.; Zhou, S.D.; He, X.J. A transcriptome-based study on the phylogeny and evolution for taxonomic controversial subfamily Apioideae (Apiaceae). Ann. Bot. 2020, 125, 937–953. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Ma, J.; Yang, B.X.; Li, R.Y.; Zhu, W.; Sun, L.L.; Tian, J.K.; Zhang, L. The complete chloroplast genome sequence of Taxus chinensis var. mairei (Taxaceae): Loss of an inverted repeat region and comparative analysis with related species. Gene 2014, 540, 201–209. [Google Scholar] [CrossRef]
- Guo, X.L.; Wang, C.B.; Wen, J.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Chamaesium (Apiaceae: Apioideae) inferred from ITS, cpDNA and morphological characters. Phytotaxa 2018, 376, 1–16. [Google Scholar] [CrossRef]
- Xiao, Q.Y.; Yu, Y.; Xie, D.F.; Guo, X.L.; He, X.J. Taxonomic revision of Angelica oncosepala and Heracleum yunnanense. Nord. J. Bot. 2018, 36, 1–10. [Google Scholar]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme Reconfiguration of Plastid Genomes in the Angiosperm Family Geraniaceae: Rearrangements, Repeats, and Codon Usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.J. Chloroplast genome structure and chloroplast genes. Plant. Physiol. Commun. 1985, 2, 65–71. (In Chinese) [Google Scholar]
- Li, J.; Xie, D.F.; Guo, X.L.; Zheng, Z.Y.; He, X.J.; Zhou, S.D. Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship. Plants 2020, 9, 543. [Google Scholar] [CrossRef]
- Guo, X.L.; Zheng, H.Y.; Price, M.; Zhou, S.D.; He, X.J. Phylogeny and Comparative Analysis of Chinese Chamaesium Species Revealed by the Complete Plastid Genome. Plants 2020, 9, 965. [Google Scholar] [CrossRef]
- Mustafina, F.U.; Yi, D.K.; Choi, K.; Shin, C.H.; Tojibaev, K.S.; Downie, S.R. A comparative analysis of complete plastid genomes from Prangos fedtschenkoi and Prangos lipskyi (Apiaceae). Ecol. Evol. 2018, 1–14. [Google Scholar] [CrossRef]
- Kang, L.; Xie, D.F.; Xiao, Q.Y.; Peng, C.; Yu, Y.; He, X.J. Sequencing and analyses on chloroplast genomes of Tetrataenium candicans and two allies give new insights on structural variants, DNA barcoding and phylogeny in Apiaceae subfamily Apioideae. PeerJ 2019, 7, e8063. [Google Scholar] [CrossRef] [Green Version]
- Peery, R.M. Understanding Angiosperm Genome Interactions and Evolution: Insights from Sacred Lotus (Nelumbo nucifera) and the Carrot Family (Apiaceae). Ph.D. Thesis, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 2015. [Google Scholar]
- Aii, J.; Kishima, Y.; Mikami, T.; Adachi, T. Expansion of the IR in the chloroplast genomes of buckwheat species is due to incorporation of an SSC sequence that could be mediated by an inversion. Curr. Genet. 1997, 31, 276–279. [Google Scholar] [CrossRef]
- Chung, H.J.; Jung, J.D.; Park, H.W.; Kim, J.H.; Cha, H.W. The complete chloroplast genome sequences of Solanum tuberosum and comparative analysis with Solanaceae species identified the presence of a 241-bp deletion in cultivated potato chloroplast DNA sequence. Plant Cell Rep. 2006, 25, 1369–1379. [Google Scholar] [CrossRef]
- Wang, W.B.; Yu, H.; Wang, J.H.; Lei, W.J.; Gao, J.H.; Qiu, X.P.; Wang, J.S. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaf, S.; Khan, A.L.; Khan, A.R.; Waqas, M.; Kang, S.M.; Khan, M.A.; Lee, S.M.; Lee, I.J. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant. Sci. 2016, 7, 447. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M. An evolutionary perspective on synopnymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar]
- Du, Y.; Bi, Y.; Chen, X.; Yang, F.; Xue, J.; Zhang, X. The complete chloroplast genome of Lilium cernuum: Genome structure and evolution. Conserv. Genet. Resour. 2016, 8, 375–378. [Google Scholar] [CrossRef]
- Grantham, R.; Gautier, C.; Gouy, M. Codon frequencies in 119 individual genes confirm consistent choices of degenerate bases according to genome type. Nucleic Acids Res. 1980, 8, 1893–1912. [Google Scholar] [CrossRef]
- Rao, Y.S.; Wu, G.Z.; Wang, Z.F.; Chai, X.W.; Nie, Q.H.; Zhang, X.Q. Mutation Bias is the Driving Force of Codon Usage in the genome. DNA Res. 2011, 18, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Rensing, S.A.; Fritzowsky, D.; Lang, D.; Reski, R. Protein encoding genes in an ancient plant: Analysis of codon usage, retained genes and splice sites in a moss, Physcomitrella patens. BMC Genom. 2005, 6, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, E.M.; Pouplana, L.R.D. Speeding with control: Codon usage, TRNAs, and ribosomes. Trends Genet. 2012, 28, 574–581. [Google Scholar] [CrossRef]
- Harrison, R.J.; Charlesworth, B. Biased Gene Conversion Affects Patterns of Codon Usage and Amino Acid Usage in the Saccharomyces sensu stricto Group of Yeasts. Mol. Biol. Evol. 2011, 28, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; Eisen, M.; Smith, D.; Singh, N.D.; Sackton, T.B.; Larracuente, A.M.; Bergman, C.M.; Gelbart, W.M.; Oliver, B.; Markow, T.A.; et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature 2007, 450, 203–218. [Google Scholar]
- Bulmer, M.G. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 4, 4. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. Irscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting est databases for the development and characterization of gene-derived ssr-markers in barley (hordeum vulgarel.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Peden, J.F. CodonW. Ph.D. Thesis, University of Nottingham, Nottinghamshire, UK, 1999. [Google Scholar]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Phylogenet. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Size (bp) | LSC Length (bp) | IR Length (bp) | SSC Length (bp) | Total Genes | Protein Coding Genes | tRNA Genes | rRNA Genes | Overall GC Content (%) |
|---|---|---|---|---|---|---|---|---|---|
| Hansenia forbesii | 157,797 | 86,968 | 26,517 | 17,795 | 131 | 85 | 37 | 8 | 37.6 |
| Hansenia forrestii | 156,911 | 86,245 | 26,435 | 17,796 | 131 | 85 | 37 | 8 | 37.6 |
| Hansenia oviformis | 157,175 | 86,412 | 26,494 | 17,775 | 131 | 85 | 37 | 8 | 37.6 |
| Hansenia weberbaueriana | 156,778 | 86,246 | 26,432 | 17,668 | 131 | 85 | 37 | 8 | 37.7 |
| Haplosphaera himalayensis | 157,286 | 86,479 | 26,542 | 17,723 | 131 | 85 | 37 | 8 | 37.6 |
| Haplosphaera phaea | 157,271 | 86,488 | 26,446 | 17,891 | 131 | 85 | 37 | 8 | 37.6 |
| Sinodielsia microloba | 155,709 | 85,320 | 26,479 | 17,431 | 131 | 85 | 37 | 8 | 37.5 |
| Sinodielsia yunnanensis (EY) | 155,552 | 85,276 | 26,441 | 17,394 | 131 | 85 | 37 | 8 | 37.6 |
| Sinodielsia yunnanensis (HB) | 154,670 | 84,446 | 26,427 | 17,370 | 131 | 85 | 37 | 8 | 37.6 |
| Sinodielsia yunnanensis (KM) | 155,435 | 85,233 | 26,404 | 17,394 | 131 | 85 | 37 | 8 | 37.5 |
| Category | Group of Genes | Name of Genes |
|---|---|---|
| Self-replication | transfer RNAs (tRNAs) | trnA-UGC *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC, trnH-GUG, trnI-CAU *, trnI-GAU *, trnK-UUU, trnL-CAA *, trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU *, trnP-UGG, trnQ-UUG, trnR-ACG *, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC *, trnV-UAC, trnW-CCA, trnY-GUA |
| ribosomal RNAs (rRNAs) | rrn4.5 *, rrna5 *, rrn16 *, rrn23 * | |
| RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | |
| Small subunit of ribosomal proteins (SSU) | rps2, rps3, rps4, rps7 *, rps8, rps11, rps12, rps14, rps15, rps16, rps18, rps19 * | |
| Large subunit of ribosomal proteins (LSU) | rpl2 *, rpl14, rpl16, rpl20, rpl22, rpl23 *, rpl32, rpl33, rpl36 | |
| Genes involved in photosynthesis | Subunits of NADH-dehydrogenase | ndhA, ndhB *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Subunits in photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Subunits in photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of cytochrome b/f complex | petA, petB, petD, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Other genes | Translational initiation factor | infA |
| Protease | clpP | |
| Maturase | matK | |
| Subunit of Acetyl-CoA-carboxylase | accD | |
| Envelope membrane protein | cemA | |
| C-type cytochrome synthesis gene | ccsA | |
| Conserved reading frames | Conserved open reading frames | ψrps19, ycf1 * (ycf1, ψycf1), ycf2 *, ycf3, ycf4, ψycf15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gou, W.; Jia, S.-B.; Price, M.; Guo, X.-L.; Zhou, S.-D.; He, X.-J. Complete Plastid Genome Sequencing of Eight Species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative Analyses and Phylogenetic Implications. Plants 2020, 9, 1523. https://doi.org/10.3390/plants9111523
Gou W, Jia S-B, Price M, Guo X-L, Zhou S-D, He X-J. Complete Plastid Genome Sequencing of Eight Species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative Analyses and Phylogenetic Implications. Plants. 2020; 9(11):1523. https://doi.org/10.3390/plants9111523
Chicago/Turabian StyleGou, Wei, Sheng-Bin Jia, Megan Price, Xian-Lin Guo, Song-Dong Zhou, and Xing-Jin He. 2020. "Complete Plastid Genome Sequencing of Eight Species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative Analyses and Phylogenetic Implications" Plants 9, no. 11: 1523. https://doi.org/10.3390/plants9111523
APA StyleGou, W., Jia, S. -B., Price, M., Guo, X. -L., Zhou, S. -D., & He, X. -J. (2020). Complete Plastid Genome Sequencing of Eight Species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative Analyses and Phylogenetic Implications. Plants, 9(11), 1523. https://doi.org/10.3390/plants9111523