Nanopore RNA Sequencing Revealed Long Non-Coding and LTR Retrotransposon-Related RNAs Expressed at Early Stages of Triticale SEED Development

 ,
, 
 ,
,  , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Material and DNA Isolation
2.2. Sample Collection and RNA Isolation
2.3. RT-PCR
2.4. Nanopore Direct RNA Sequencing and Transcript Assembly
2.5. Long Non-Coding RNA Prediction
2.6. Retrotransposon-Related Transcript Annotation
2.7. GAG Protein Analysis
2.8. Gene Ontology Enrichment
2.9. Expression Analysis
2.10. Extrachromosomal Circular DNA Isolation
2.11. Statistics and Visualization
3. Results
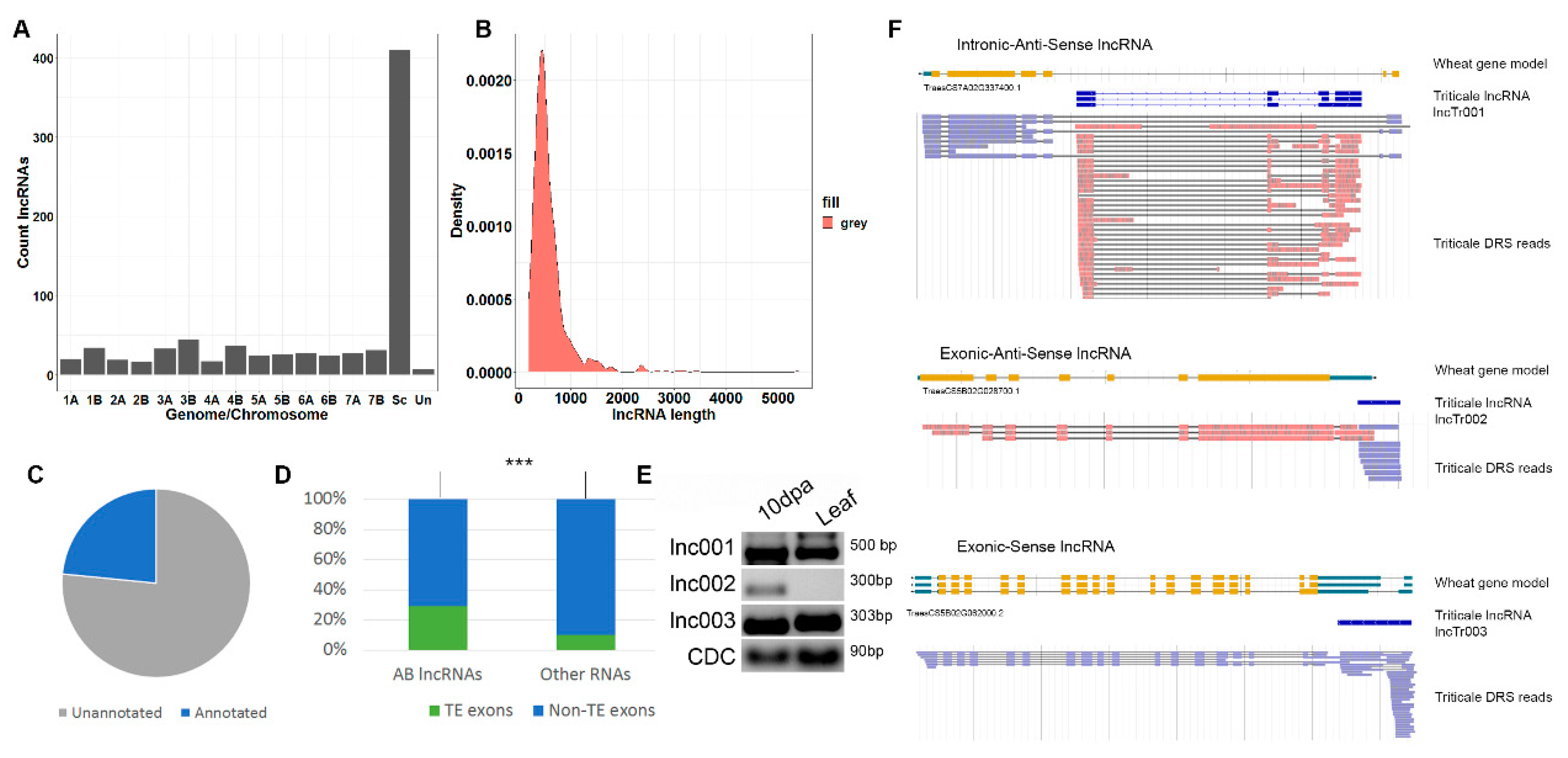
3.1. Direct Oxford Nanopore RNA Sequencing
3.2. Long Non-Coding RNA Prediction
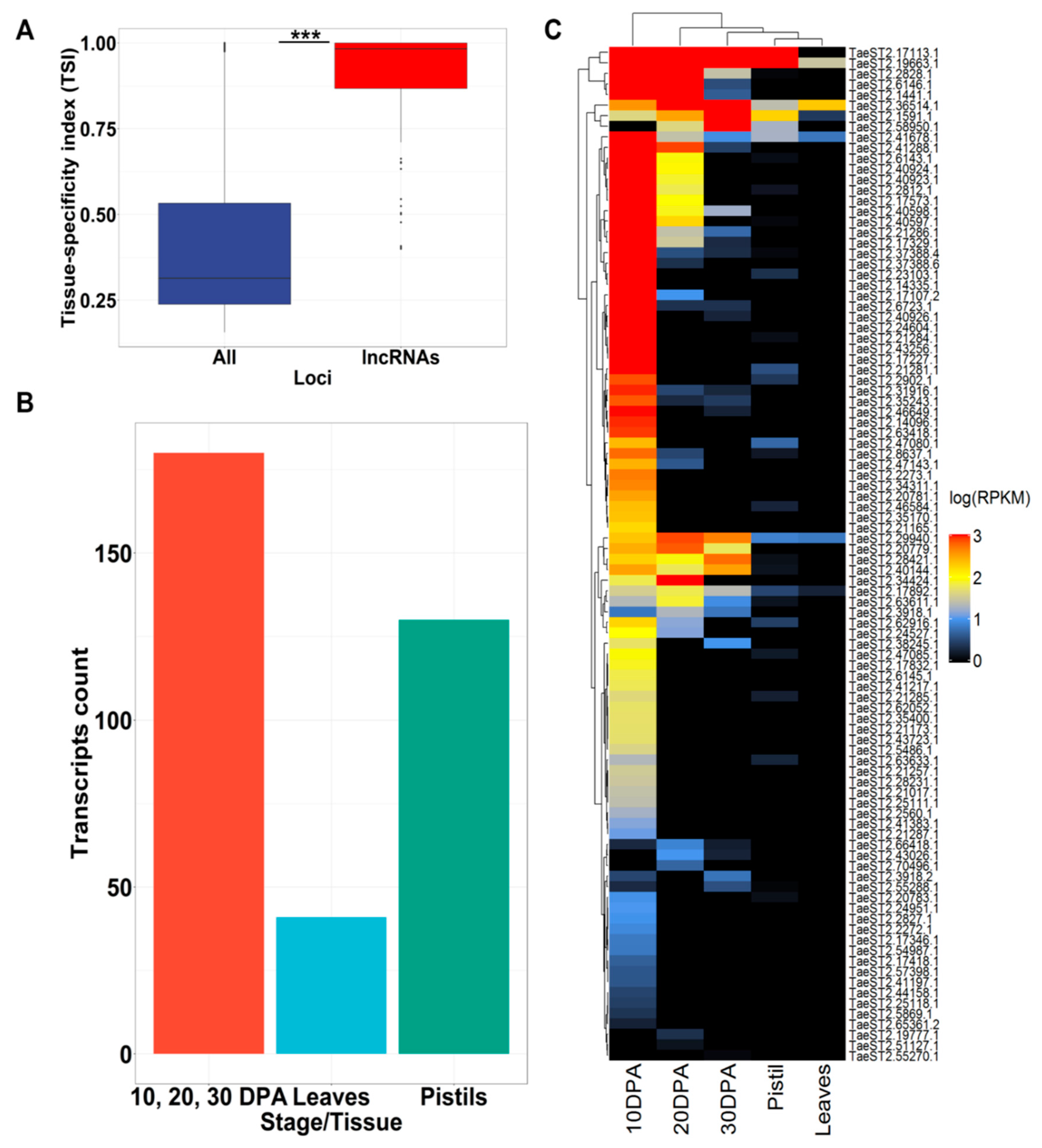
3.3. AB lncRNAs Are Prone to Tissue-Specific Expression
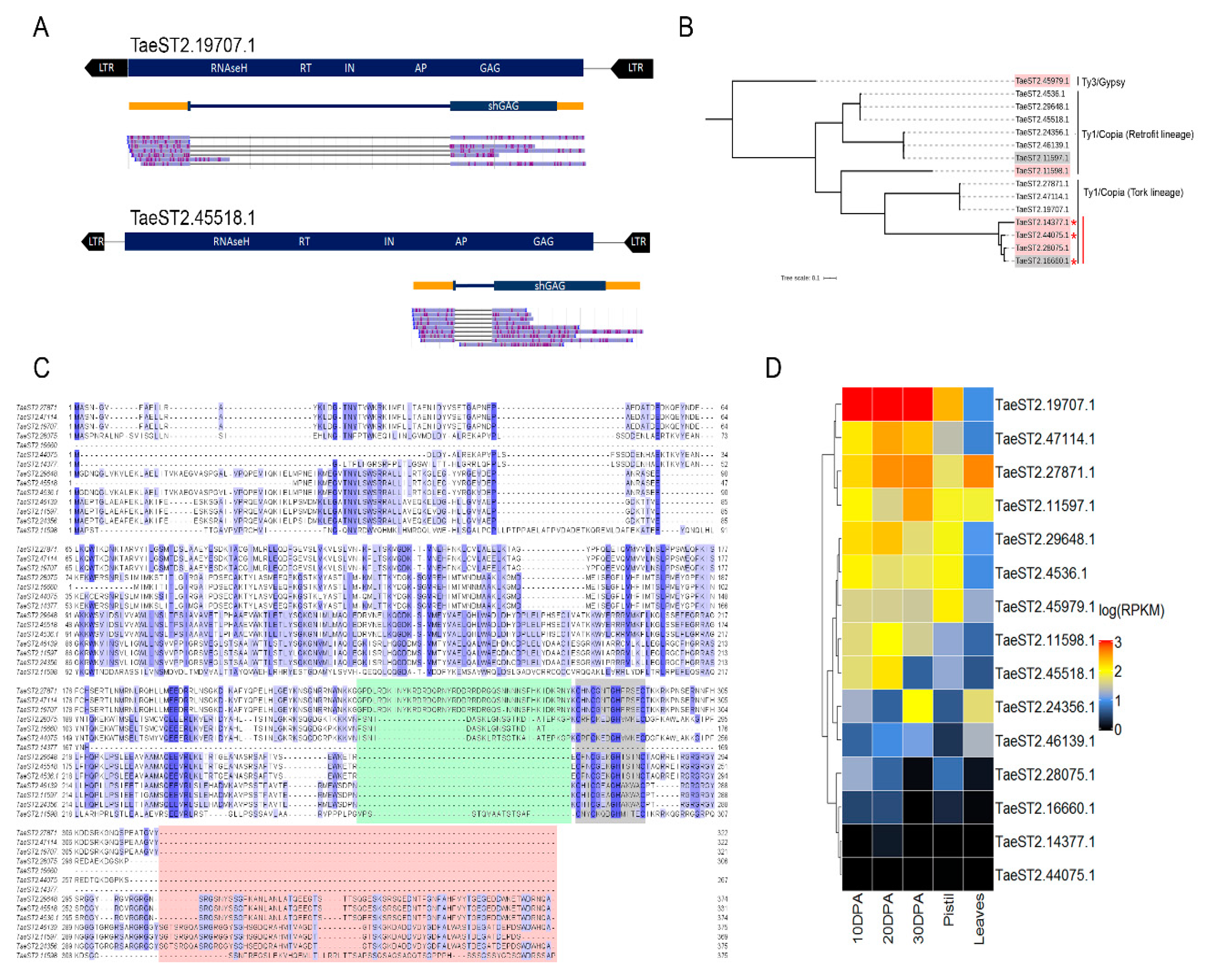
3.4. Retrotransposon-Related Transcripts Encoding GAG Proteins Are Expressed during Early Seed Development in Triticale
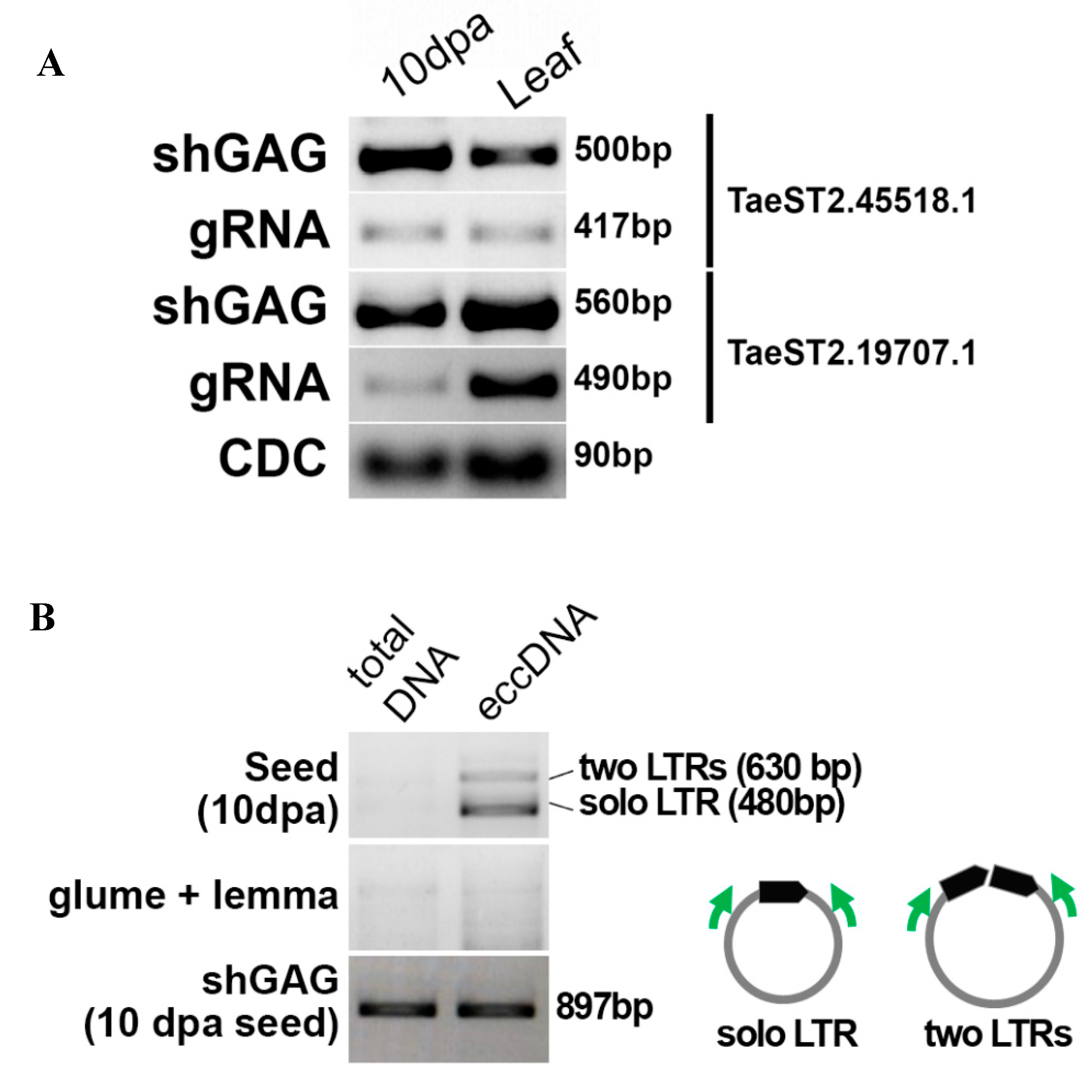
3.5. A Full-Length Ty1/Copia LTR Retrotransposon Is Active in Triticale Seeds
4. Discussion
4.1. A Set of Intergenic lncRNAs Detected by Nanopore Sequencing Is Expressed during the Early Stage of Triticale Seed Development
4.2. Transcripts Encoding Diverse GAG Proteins Are Expressed during the Early Stage of Seed Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability Statement
References
- Fesenko, I.; Kirov, I.; Kniazev, A.; Khazigaleeva, R.; Lazarev, V.; Kharlampieva, D.; Grafskaia, E.; Zgoda, V.; Butenko, I.; Arapidi, G.; et al. Distinct types of short open reading frames are translated in plant cells. Genome Res. 2019, 29, 1464–1477. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.I.; Alam, M.; Lightfoot, D.A.; Gurha, P.; Afzal, A.J. Classification and experimental identification of plant long non-coding RNAs. Genomics 2019, 111, 997–1005. [Google Scholar] [CrossRef]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. Rna Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef]
- Szcześniak, M.W.; Rosikiewicz, W.; Makałowska, I. CANTATAdb: A Collection of Plant Long Non-Coding RNAs. Plant Cell Physiol. 2015, 57, e8. [Google Scholar] [CrossRef] [Green Version]
- Paytuví Gallart, A.; Hermoso Pulido, A.; Anzar Martínez de Lagrán, I.; Sanseverino, W.; Aiese Cigliano, R. GREENC: A Wiki-based database of plant lncRNAs. Nucleic Acids Res. 2016, 44, D1161–D1166. [Google Scholar] [CrossRef]
- Xuan, H.; Zhang, L.; Liu, X.; Han, G.; Li, J.; Li, X.; Liu, A.; Liao, M.; Zhang, S.J.G. PLNlncRbase: A resource for experimentally identified lncRNAs in plants. Gene 2015, 573, 328–332. [Google Scholar] [CrossRef]
- Jin, J.; Lu, P.; Xu, Y.; Li, Z.; Yu, S.; Liu, J.; Wang, H.; Chua, N.-H.; Cao, P. PLncDB V2.0: A comprehensive encyclopedia of plant long noncoding RNAs. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Jha, U.C.; Nayyar, H.; Jha, R.; Khurshid, M.; Zhou, M.; Mantri, N.; Siddique, K.H.M. Long non-coding RNAs: Emerging players regulating plant abiotic stress response and adaptation. BMC Plant Biol. 2020, 20, 466. [Google Scholar] [CrossRef]
- Lucero, L.; Ferrero, L.; Fonouni-Farde, C.; Ariel, F. Functional classification of plant long noncoding RNAs: A transcript is known by the company it keeps. New Phytol. 2020. [Google Scholar] [CrossRef]
- Budak, H.; Kaya, S.B.; Cagirici, H.B. Long Non-coding RNA in Plants in the Era of Reference Sequences. Front. Plant Sci. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, A.F.; Koonin, E.V. Functional Long Non-coding RNAs Evolve from Junk Transcripts. Cell 2020. [Google Scholar] [CrossRef]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Tao, X.; Feng, S.; Wang, L.; Hong, H.; Ma, W.; Shang, G.; Guo, S.; He, Y.; Zhou, B.; et al. LncRNAs in polyploid cotton interspecific hybrids are derived from transposon neofunctionalization. Genome Biol. 2018, 19, 195. [Google Scholar] [CrossRef] [Green Version]
- De Quattro, C.; Pe, M.E.; Bertolini, E. Long noncoding RNAs in the model species Brachypodium distachyon. Sci. Rep. 2017, 7, 11252. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, L.; Marchetto, M.C.; Caron, M.; Chen, S.H.; Busche, S.; Kwan, T.; Pastinen, T.; Gage, F.H.; Bourque, G. Conserved expression of transposon-derived non-coding transcripts in primate stem cells. BMC Genom. 2017, 18, 214. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Hu, F.; Zhou, Y.; Wu, F.; Gaut, B.S. Maize transposable elements contribute to long non-coding RNAs that are regulatory hubs for abiotic stress response. BMC Genom. 2019, 20, 864. [Google Scholar] [CrossRef]
- Xue, L.; Wu, H.; Chen, Y.; Li, X.; Hou, J.; Lu, J.; Wei, S.; Dai, X.; Olson, M.S.; Liu, J.; et al. Evidences for a role of two Y-specific genes in sex determination in Populus deltoides. Nat. Commun. 2020, 11, 5893. [Google Scholar] [CrossRef]
- Cho, J.; Paszkowski, J. Regulation of rice root development by a retrotransposon acting as a microRNA sponge. eLife 2017, 6, e30038. [Google Scholar] [CrossRef] [Green Version]
- Warman, C.; Panda, K.; Vejlupkova, Z.; Hokin, S.; Unger-Wallace, E.; Cole, R.A.; Chettoor, A.M.; Jiang, D.; Vollbrecht, E.; Evans, M.M.S.; et al. High expression in maize pollen correlates with genetic contributions to pollen fitness as well as with coordinated transcription from neighboring transposable elements. PLoS Genet. 2020, 16, e1008462. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.N.; Stitzer, M.C.; Zhou, P.; Ross-Ibarra, J.; Hirsch, C.D.; Springer, N.M. Dynamic Patterns of Transcript Abundance of Transposable Element Families in Maize. G3 Genes|Genomes|Genet. 2019, 9, 3673. [Google Scholar] [CrossRef] [Green Version]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdžić, M.; Becker, J.D.; Feijó, J.A.; Martienssen, R.A. Epigenetic Reprogramming and Small RNA Silencing of Transposable Elements in Pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Slotkin, R.K. The case for not masking away repetitive DNA. Mob. DNA 2018, 9, 15. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Panda, K.; Slotkin, R.K. Long-Read cDNA Sequencing Enables a “Gene-Like” Transcript Annotation of Transposable Elements. Plant Cell 2020, 32, 2687–2698. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, M.; Wang, J.; Lei, M.; Li, C.; Zhao, D.; Huang, J.; Li, W.; Li, S.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019, 97, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Du, R.; Meng, X.; Zhao, W.; Kong, L.; Chen, J. Third-Generation Sequencing Indicated that LncRNA Could Regulate eIF2D to Enhance Protein Translation under Heat Stress in Populus simonii. Plant Mol. Biol. Rep. 2020. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, Y.; Pu, S.; Zhang, X.; Li, Z.; Chen, J. Third-generation sequencing found LncRNA associated with heat shock protein response to heat stress in Populus qiongdaoensis seedlings. BMC Genom. 2020, 21, 572. [Google Scholar] [CrossRef]
- Chao, Q.; Gao, Z.-F.; Zhang, D.; Zhao, B.-G.; Dong, F.-Q.; Fu, C.-X.; Liu, L.-J.; Wang, B.-C. The developmental dynamics of the Populus stem transcriptome. Plant Biotechnol. J. 2019, 17, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Yuan, J.; Li, S.; Jia, S.; Han, L.; Xu, L. Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Yu, T.; Cong, X.; Xu, F.; Lai, X.; Zhang, W.; Liao, Y.; Cheng, S. Integration analysis of PacBio SMRT- and Illumina RNA-seq reveals candidate genes and pathway involved in selenium metabolism in hyperaccumulator Cardamine violifolia. BMC Plant Biol. 2020, 20, 492. [Google Scholar] [CrossRef]
- Zhu, Z.; Chen, H.; Xie, K.; Liu, C.; Li, L.; Liu, L.; Han, X.; Jiao, C.; Wan, Z.; Sha, A. Characterization of Drought-Responsive Transcriptome During Seed Germination in Adzuki Bean (Vigna angularis L.) by PacBio SMRT and Illumina Sequencing. Front. Genet. 2020, 11, 996. [Google Scholar] [CrossRef]
- Rogers, S.O.; Quatrano, R.S. Morphological Staging of Wheat Caryopsis Development. Am. J. Bot. 1983, 70, 308–311. [Google Scholar] [CrossRef]
- Rangan, P.; Furtado, A.; Henry, R.J. The transcriptome of the developing grain: A resource for understanding seed development and the molecular control of the functional and nutritional properties of wheat. BMC Genom. 2017, 18, 766. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Poole, R.L.; Huttly, A.K.; Toscano-Underwood, C.; Feeney, K.; Welham, S.; Gooding, M.J.; Mills, C.; Edwards, K.J.; Shewry, P.R.; et al. Transcriptome analysis of grain development in hexaploid wheat. BMC Genom. 2008, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Pellny, T.K.; Lovegrove, A.; Freeman, J.; Tosi, P.; Love, C.G.; Knox, J.P.; Shewry, P.R.; Mitchell, R.A.C. Cell Walls of Developing Wheat Starchy Endosperm: Comparison of Composition and RNA-Seq Transcriptome. Plant Physiol. 2012, 158, 612–627. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Wu, Z.; Zhang, Y.; Guo, D.; Xu, Y.; Chen, W.; Zhou, H.; You, M.; Li, B. Transcriptome analysis of wheat grain using RNA-Seq. Front. Agric. Sci. Eng. 2014, 1, 214–222. [Google Scholar] [CrossRef]
- Pfeifer, M.; Kugler, K.G.; Sandve, S.R.; Zhan, B.; Rudi, H.; Hvidsten, T.R.; Mayer, K.F.X.; Olsen, O.-A. Genome interplay in the grain transcriptome of hexaploid bread wheat. Science 2014, 345, 1250091. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zhu, D.; Ma, C.; Cao, H.; Wang, Y.; Xu, Y.; Zhang, W.; Yan, Y. Transcriptome analysis reveals key differentially expressed genes involved in wheat grain development. Crop J. 2016, 4, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Gillies, S.A.; Futardo, A.; Henry, R.J. Gene expression in the developing aleurone and starchy endosperm of wheat. Plant Biotechnol. J. 2012, 10, 668–679. [Google Scholar] [CrossRef]
- Yamasaki, Y.; Gao, F.; Jordan, M.C.; Ayele, B.T. Seed maturation associated transcriptional programs and regulatory networks underlying genotypic difference in seed dormancy and size/weight in wheat (Triticum aestivum L.). BMC Plant Biol. 2017, 17, 154. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Li, G.; Chu, Z.; Ru, Z.; Jiang, X.; Wen, Z.; Zhang, G.; Wang, Y.; Zhang, Y.; Wei, W. Transcriptome analysis reveals important candidate genes involved in grain-size formation at the stage of grain enlargement in common wheat cultivar “Bainong 4199”. PLoS ONE 2019, 14, e0214149. [Google Scholar] [CrossRef]
- Madhawan, A.; Sharma, A.; Bhandawat, A.; Rahim, M.S.; Kumar, P.; Mishra, A.; Parveen, A.; Sharma, H.; Verma, S.K.; Roy, J. Identification and characterization of long non-coding RNAs regulating resistant starch biosynthesis in bread wheat (Triticum aestivum L.). Genomics 2020, 112, 3065–3074. [Google Scholar] [CrossRef]
- Rogers, S.O.; Bendich, A.J. Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol. Biol. 1985, 5, 69–76. [Google Scholar] [CrossRef]
- Paolacci, A.R.; Tanzarella, O.A.; Porceddu, E.; Ciaffi, M. Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol. Biol. 2009, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Ryan, P.R.; Dong, D.; Teuber, F.; Wendler, N.; Mühling, K.H.; Liu, J.; Xu, M.; Salvador Moreno, N.; You, J.; Maurer, H.-P.; et al. Assessing How the Aluminum-Resistance Traits in Wheat and Rye Transfer to Hexaploid and Octoploid Triticale. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- International Wheat Genome Sequencing, C. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Bauer, E.; Schmutzer, T.; Barilar, I.; Mascher, M.; Gundlach, H.; Martis, M.M.; Twardziok, S.O.; Hackauf, B.; Gordillo, A.; Wilde, P.; et al. Towards a whole-genome sequence for rye (Secale cereale L.). Plant J. Cell Mol. Biol. 2017, 89, 853–869. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Stromberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Han, S.; Liang, Y.; Ma, Q.; Xu, Y.; Zhang, Y.; Du, W.; Wang, C.; Li, Y. LncFinder: An integrated platform for long non-coding RNA identification utilizing sequence intrinsic composition, structural information and physicochemical property. Brief Bioinform. 2019, 20, 2009–2027. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef]
- Llorens, C.; Futami, R.; Covelli, L.; Dominguez-Escriba, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodriguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, D70–D74. [Google Scholar] [CrossRef] [Green Version]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mobile DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2019, 36, 2628–2629. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Lu, H.; Li, C.; Bhanbhro, N.; Cui, X.; Yang, C. Carbohydrate and plant hormone regulate the alkali stress response of hexaploid wheat (Triticum aestivum L.). Environ. Exp. Bot. 2020, 175, 104053. [Google Scholar] [CrossRef]
- Yang, Z.; Peng, Z.; Wei, S.; Liao, M.; Yu, Y.; Jang, Z. Pistillody mutant reveals key insights into stamen and pistil development in wheat (Triticum aestivum L.). BMC Genom. 2015, 16, 211. [Google Scholar] [CrossRef] [Green Version]
- Lanciano, S.; Carpentier, M.C.; Llauro, C.; Jobet, E.; Robakowska-Hyzorek, D.; Lasserre, E.; Ghesquiere, A.; Panaud, O.; Mirouze, M. Sequencing the extrachromosomal circular mobilome reveals retrotransposon activity in plants. PLoS Genet 2017, 13, e1006630. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Furtado, A.; Bundock, P.C.; Banks, P.M.; Fox, G.; Yin, X.; Henry, R.J. A novel highly differentially expressed gene in wheat endosperm associated with bread quality. Sci. Rep. 2015, 5, 10446. [Google Scholar] [CrossRef]
- Kirov, I.; Pirsikov, A.; Milyukova, N.; Dudnikov, M.; Kolenkov, M.; Gruzdev, I.; Siksin, S.; Khrustaleva, L.; Karlov, G.; Soloviev, A.J.A. Analysis of Wheat Bread-Making Gene (wbm) Evolution and Occurrence in Triticale Collection Reveal Origin via Interspecific Introgression into Chromosome 7AL. Agronomy 2019, 9, 854. [Google Scholar] [CrossRef] [Green Version]
- Unver, T.; Tombuloglu, H. Barley long non-coding RNAs (lncRNA) responsive to excess boron. Genomics 2020, 112, 1947–1955. [Google Scholar] [CrossRef]
- Wang, T.; Song, H.; Wei, Y.; Li, P.; Hu, N.; Liu, J.; Zhang, B.; Peng, R. High throughput deep sequencing elucidates the important role of lncRNAs in Foxtail millet response to herbicides. Genomics 2020, 112, 4463–4473. [Google Scholar] [CrossRef]
- Li, X.; Shahid, M.Q.; Wen, M.; Chen, S.; Yu, H.; Jiao, Y.; Lu, Z.; Li, Y.; Liu, X. Global identification and analysis revealed differentially expressed lncRNAs associated with meiosis and low fertility in autotetraploid rice. BMC Plant Biol. 2020, 20, 82. [Google Scholar] [CrossRef] [Green Version]
- Flórez-Zapata, N.M.V.; Reyes-Valdés, M.H.; Martínez, O. Long non-coding RNAs are major contributors to transcriptome changes in sunflower meiocytes with different recombination rates. BMC Genom. 2016, 17, 490. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Chapter Two—Association mapping in plants in the post-GWAS genomics era. In Advances in Genetics; Kumar, D., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 104, pp. 75–154. [Google Scholar]
- Oberlin, S.; Sarazin, A.; Chevalier, C.; Voinnet, O.; Marí-Ordóñez, A. A genome-wide transcriptome and translatome analysis of Arabidopsis transposons identifies a unique and conserved genome expression strategy for Ty1/Copia retroelements. Genome Res. 2017, 27, 1549–1562. [Google Scholar] [CrossRef] [Green Version]
- Chaparro, C.; Gayraud, T.; de Souza, R.F.; Domingues, D.S.; Akaffou, S.; Laforga Vanzela, A.L.; Kochko, A.; Rigoreau, M.; Crouzillat, D.; Hamon, S.; et al. Terminal-repeat retrotransposons with GAG domain in plant genomes: A new testimony on the complex world of transposable elements. Genome Biol. Evol. 2015, 7, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Campillos, M.; Doerks, T.; Shah, P.K.; Bork, P.J.T.i.G. Computational characterization of multiple Gag-like human proteins. Trends Genetics 2006, 22, 585–589. [Google Scholar] [CrossRef]
- Nefedova, L.N.; Kuzmin, I.V.; Makhnovskii, P.A.; Kim, A.I. Domesticated retroviral GAG gene in Drosophila: New functions for an old gene. Virology 2014, 450–451, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Ashley, J.; Cordy, B.; Lucia, D.; Fradkin, L.G.; Budnik, V.; Thomson, T. Retrovirus-like Gag Protein Arc1 Binds RNA and Traffics across Synaptic Boutons. Cell 2018, 172, 262–274.e11. [Google Scholar] [CrossRef] [Green Version]
- Makhnovskii, P.; Balakireva, Y.; Nefedova, L.; Lavrenov, A.; Kuzmin, I.; Kim, A.J.G. Domesticated gag Gene of Drosophila LTR Retrotransposons Is Involved in Response to Oxidative Stress. Genes 2020, 11, 396. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Mitchell, J.A.; Nishida, Y.; Hildreth, J.E.; Ariberre, J.A.; Gilbert, W.V.; Garfinkel, D.J. A trans-dominant form of Gag restricts Ty1 retrotransposition and mediates copy number control. J. Virol. 2015, 89, 3922–3938. [Google Scholar] [CrossRef] [Green Version]
- Tucker, J.M.; Larango, M.E.; Wachsmuth, L.P.; Kannan, N.; Garfinkel, D.J. The Ty1 Retrotransposon Restriction Factor p22 Targets Gag. PLoS Genet. 2015, 11, e1005571. [Google Scholar] [CrossRef] [Green Version]
- Kirov, I.; Omarov, M.; Merkulov, P.; Dudnikov, M.; Gvaramiya, S.; Kolganova, E.; Komakhin, R.; Karlov, G.; Soloviev, A. Genomic and Transcriptomic Survey Provides New Insight into the Organization and Transposition Activity of Highly Expressed LTR Retrotransposons of Sunflower (Helianthus annuus L.). Int. J. Mol. Sci. 2020, 21, 9331. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer Sequences | Amplicon Size |
|---|---|---|
| Lnc001 | lncTR001/F: AGGTTGCAAGTCTCTTGCTCTTGA lncTR001/R: TCATGCCCGCTAAGAATTACAGTGT | RNA/DNA = 500 bp/~1100 bp |
| Lnc002 | lncTR002/F: TGGGTTGTGACTTGTGATACGCA lncTR002/R: CGGTTAGGGCTGGGCTGAATG | RNA/DNA = 300 bp/300 bp |
| Lnc003 | lncTR003/F: ACAGTATGAAGCTAGCCGGCTTG lncTR003/R: TATCCTGTCGTCCTCTCGTCTCG | RNA/DNA = 303 bp/303 bp |
| CDC (the cell division control protein), Ta54227 [47] | CDC/F: GCCTGGTAGTCGCAGGAGAT CDC/R: ATGTCTGGCCTGTTGGTAGC | RNA/DNA = 76 bp/76 bp |
| gRNA TaeST2.19707.1 | gRNATae_19/F: ATTACACCCCCAAACCGCCAAAT gRNATae_19/R:TGGGGAATTTTCCACACCCACTT | RNA/DNA = 490 bp/490 bp |
| shGAG TaeST2.19707.1 | shGAGTae_19/F: TTGATTGCCGCCTGGTTATCACA shGAGTae_19/R: AGTGGGAATCGGAGGAACTGGAA | RNA/DNA = 560 bp/3200 bp |
| gRNA TaeST2.45518.1 | gRNATae_19/F: ATTACACCCCCAAACCGCCAAAT gRNATae_19/R: TGGGGAATTTTCCACACCCACTT | RNA/DNA = 417 bp/417 bp |
| shGAG TaeST2.45518.1 | shGAGTae_45/F: GCTTACTCTTGTCTACTCCACGCA shGAGTae_45/R: GGACTGGAGAAGCGAATGCATCT | RNA/DNA = 500 bp/897 bp |
| SRA Accession | Number of Reads | Development Stage/Organs | Reference |
|---|---|---|---|
| ERR392055 | 26,791,465 | 10 dpa/seed | [39] |
| ERR392076 | 29,714,230 | 20 dpa/seed | [39] |
| ERR392069 | 31,433,795 | 30 dpa/seed | [39] |
| SRR10522394 | 39,611,224 | Leaves | [70] |
| SRR1175868 | 64,825,850 | Pistils | [71] |
| Wheat Gene ID | Gene Expression, +/− | Reads per Million (RPM) | Gene Annotation | Genomic Coordinates |
|---|---|---|---|---|
| TraesCS4A02G418200 | + | 76 | GBSS/Starch synthase, chloroplastic/amyloplastic | 4A:688,097,145–688,100,962 |
| TraesCS4B02G029700 | + | 7 | (BGC1) Flo6/5′-AMP-activated protein kinase subunit beta-2 (PTST) | 4B:21,937,120–21,944,075 |
| TraesCS4A02G284000 | + | 3 | 4A:590,660,989–590,667,561 | |
| TraesCS7B02G139700 | + | 6 | ISA | 7B:175,999,323–176,007,332 |
| TraesCS7A02G251400 | + | 15 | 7A:235,460,629–235,468,417 | |
| TraesCS6A02G048900 | − | 0 | α/β-gliadins | 6A:24,921,651–24,922,607 |
| TraesCS6A02G049200 | − | 0 | 6A:25,203,493–25,204,413 | |
| TraesCS6A02G049100 | − | 0 | 6A:25,107,550–25,108,401 | |
| TraesCS6A02G049600 | − | 0 | 6A:25,472,841–25,473,704 | |
| TraesCS1A02G007400 | − | 0 | γ-gliadin-A3 | 1A:4,033,339–4,034,196 |
| TraesCS7A02G531903 | − | 0 | wbm | 7A:710,471,331–710,471,679 |
| TraesCS1A02G317311 | − | 0 | HMW Glu-1Ax | 1A:508,723,999–508,726,319 |
| TraesCS1B02G329711 | − | 0 | HMW Glu-1Bx | 1B:555,765,127–555,766,152 |
| RTE Group | ||||
|---|---|---|---|---|
| Encoded proteins | Full-length RTEs (all domains are detectable) | Non-complete RTEs (one or more canonical protein domains are not detectable) | No associated RTEs identified | Total number of RTE-RNAs |
| GAG | 8 | 5 | 2 | 15 |
| Other RTE proteins (AP, RT, RNAse H) | 1 | 1 | 3 | 5 |
| Total number of RTE-RNAs | 9 | 6 | 5 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirov, I.; Dudnikov, M.; Merkulov, P.; Shingaliev, A.; Omarov, M.; Kolganova, E.; Sigaeva, A.; Karlov, G.; Soloviev, A. Nanopore RNA Sequencing Revealed Long Non-Coding and LTR Retrotransposon-Related RNAs Expressed at Early Stages of Triticale SEED Development. Plants 2020, 9, 1794. https://doi.org/10.3390/plants9121794
Kirov I, Dudnikov M, Merkulov P, Shingaliev A, Omarov M, Kolganova E, Sigaeva A, Karlov G, Soloviev A. Nanopore RNA Sequencing Revealed Long Non-Coding and LTR Retrotransposon-Related RNAs Expressed at Early Stages of Triticale SEED Development. Plants. 2020; 9(12):1794. https://doi.org/10.3390/plants9121794
Chicago/Turabian StyleKirov, Ilya, Maxim Dudnikov, Pavel Merkulov, Andrey Shingaliev, Murad Omarov, Elizaveta Kolganova, Alexandra Sigaeva, Gennady Karlov, and Alexander Soloviev. 2020. "Nanopore RNA Sequencing Revealed Long Non-Coding and LTR Retrotransposon-Related RNAs Expressed at Early Stages of Triticale SEED Development" Plants 9, no. 12: 1794. https://doi.org/10.3390/plants9121794
APA StyleKirov, I., Dudnikov, M., Merkulov, P., Shingaliev, A., Omarov, M., Kolganova, E., Sigaeva, A., Karlov, G., & Soloviev, A. (2020). Nanopore RNA Sequencing Revealed Long Non-Coding and LTR Retrotransposon-Related RNAs Expressed at Early Stages of Triticale SEED Development. Plants, 9(12), 1794. https://doi.org/10.3390/plants9121794