

Optimization of a Protocol for Protein Extraction from Calcified Aortic Valves for Proteomics Applications: Development of a Standard Operating Procedure

 , , , ,
, , , ,  ,
, 
Abstract
:1. Introduction
2. Material and Methods
2.1. Sample Collection
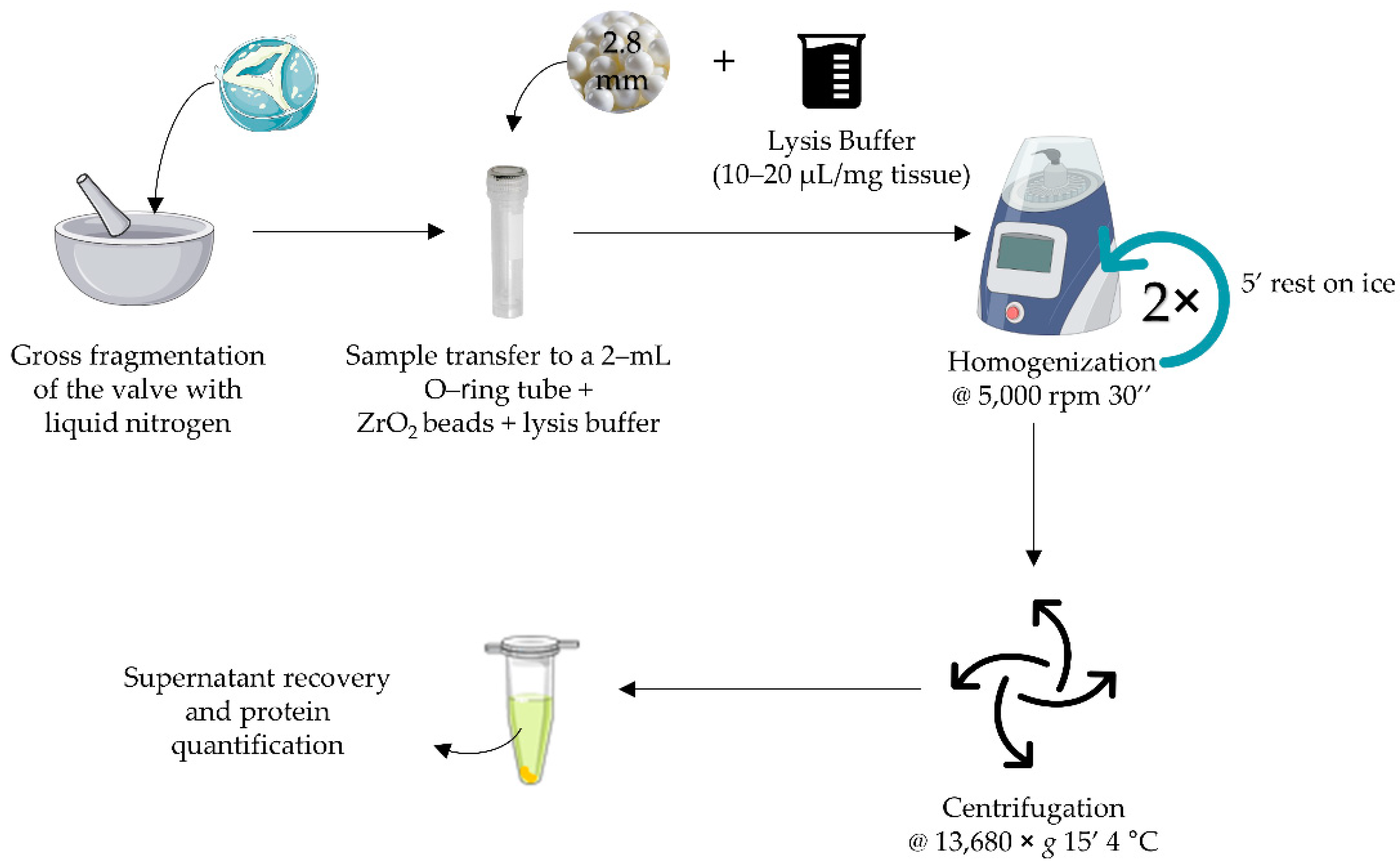
2.2. Tissue Homogenization and Protein Extraction Optimization
2.3. Protein Quantification
2.4. SDS-PAGE
2.5. Proteomics Analysis
2.5.1. Sample Preparation
2.5.2. LC-MS Methodology
2.5.3. Data Analysis—Ion-Library Construction (DDA)
2.5.4. Data Analysis—Relative Quantification of Proteins by SWATH-MS
2.5.5. Bioinformatics Analyses
2.6. Statistical Analysis
3. Results and Discussion
3.1. The Bead-Beating System Outperforms the Traditional Liquid Nitrogen Grinding Followed by Mechanical Disruption with a Potter–Elvehjem Pestle
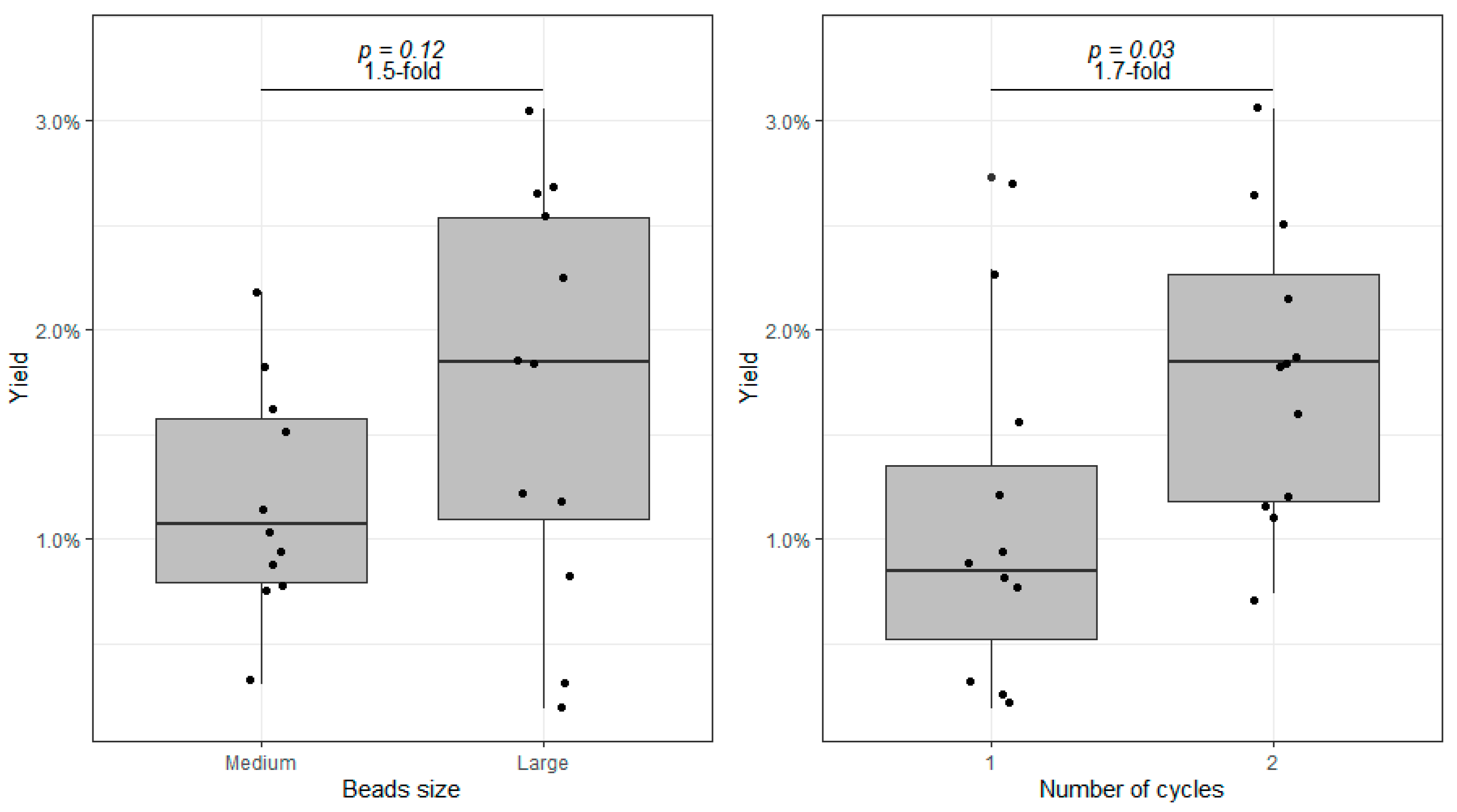
3.2. The Effect of Increasing Beads Size and a Second Homogenization Cycle
3.3. The Effect of Tube Capacity, Further Homogenization Cycles, and Increasing Lysis Buffer Volume to Tissue Mass Ratios
3.4. The Effect of Lysis Buffer Chemistry
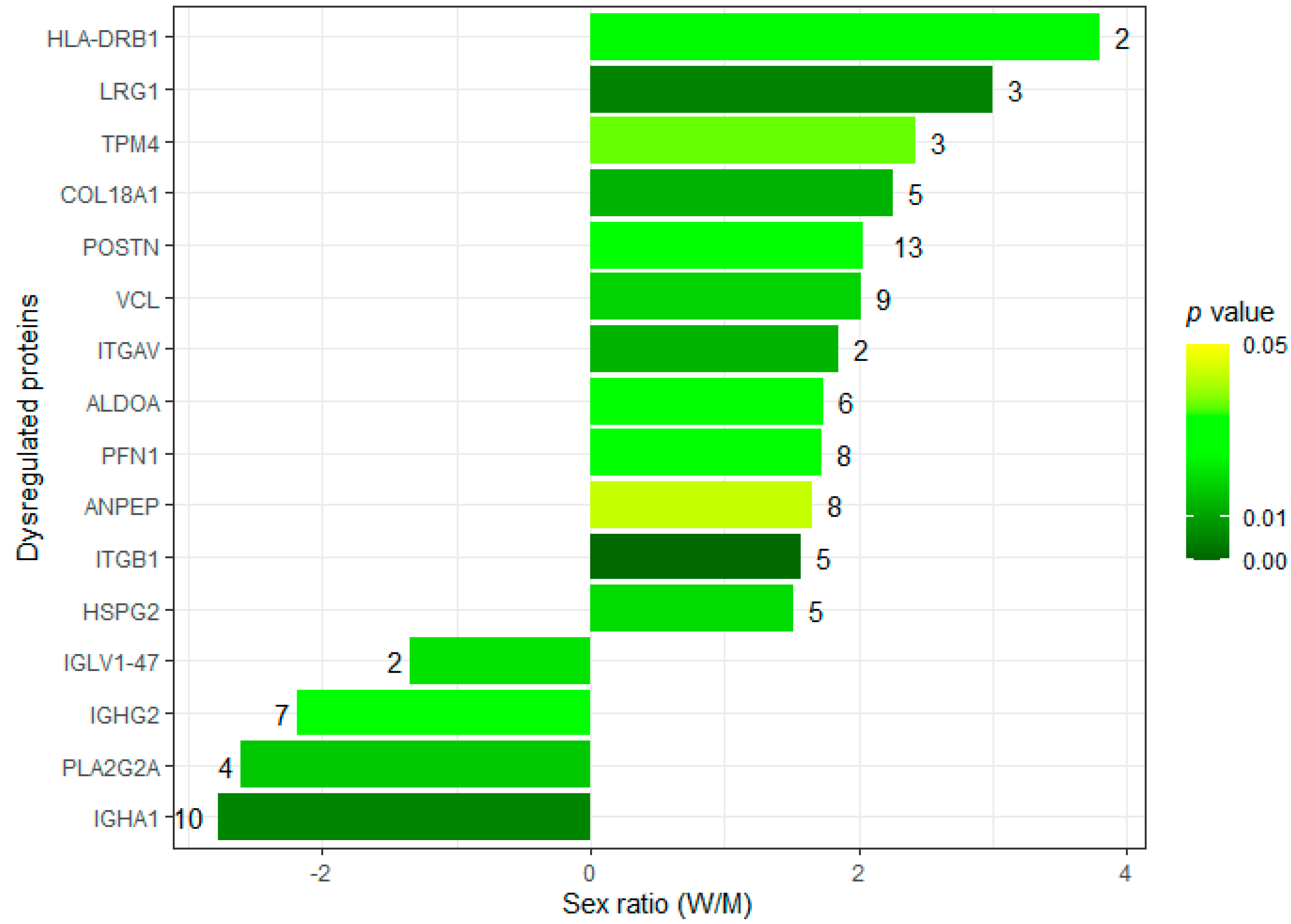
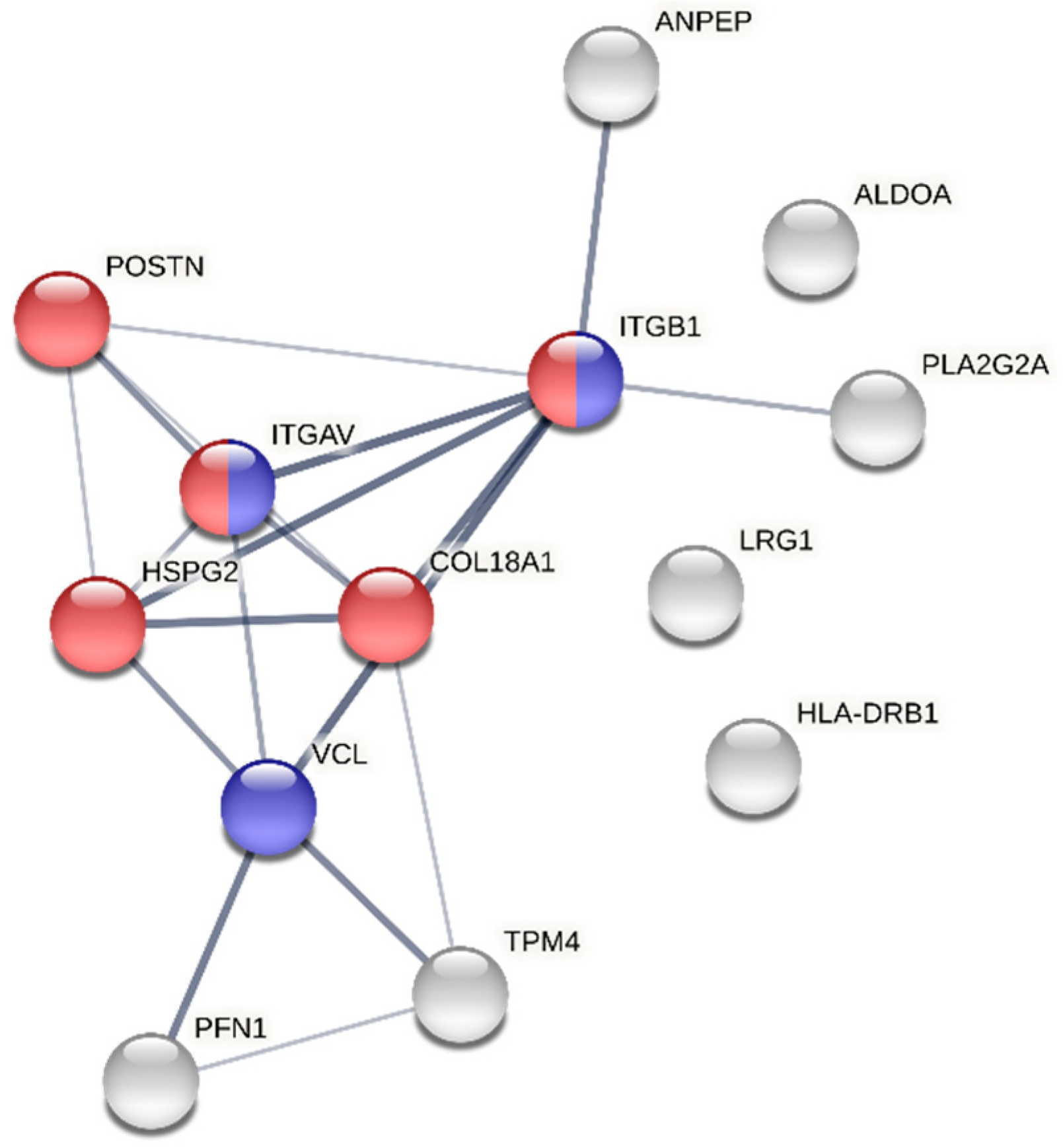
3.5. Exploring the Sexual Dimorphism in Aortic Valve Stenosis through Proteomics: A Proof-of-Concept Demonstration of the Usefulness of the SOP for Aortic Valve Homogenization and Protein Extraction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iung, B.; Delgado, V.; Rosenhek, R.; Price, S.; Prendergast, B.; Wendler, O.; de Bonis, M.; Tribouilloy, C.; Evangelista, A.; Bogachev-Prokophiev, A.; et al. Contemporary Presentation and Management of Valvular Heart Disease. Circulation 2019, 140, 1156–1169. [Google Scholar] [CrossRef] [PubMed]
- Rossebø, A.B.; Pedersen, T.R.; Boman, K.; Brudi, P.; Chambers, J.B.; Egstrup, K.; Gerdts, E.; Gohlke-Bärwolf, C.; Holme, I.; Kesäniemi, Y.A.; et al. Intensive Lipid Lowering with Simvastatin and Ezetimibe in Aortic Stenosis. N. Engl. J. Med. 2008, 359, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J. Effect of Lipid Lowering with Rosuvastatin on Progression of Aortic Stenosis. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A. A Randomized Trial of Intensive Lipid-Lowering Therapy in Calcific Aortic Stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef]
- Martín-Rojas, T.; Gil-Dones, F.; Lopez-Almodovar, L.F.; Padial, L.R.; Vivanco, F.; Barderas, M.G. Proteomic Profile of Human Aortic Stenosis: Insights into the Degenerative Process. J. Proteome Res. 2012, 11, 1537–1550. [Google Scholar] [CrossRef]
- Schlotter, F.; Halu, A.; Goto, S.; Blaser, M.C.; Body, S.C.; Lee, L.H.; Higashi, H.; DeLaughter, D.M.; Hutcheson, J.D.; Vyas, P.; et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation 2018, 138, 377–393. [Google Scholar] [CrossRef]
- Gil-Dones, F.; Martin-Rojas, T.; Lopez-Almodovar, L.F.; de la Cuesta, F.; Darde, V.M.; Alvarez-Llamas, G.; Juarez-Tosina, R.; Barroso, G.; Vivanco, F.; Padial, L.R.; et al. Valvular Aortic Stenosis: A Proteomic Insight. Clin. Med. Insights Cardiol. 2010, 4, 1–7. [Google Scholar] [CrossRef]
- Suzuki, H.; Chikada, M.; Yokoyama, M.K.; Kurokawa, M.S.; Ando, T.; Furukawa, H.; Arito, M.; Miyairi, T.; Kato, T. Aberrant Glycosylation of Lumican in Aortic Valve Stenosis Revealed by Proteomic Analysis. Int. Heart J. 2016, 57, 104–111. [Google Scholar] [CrossRef]
- Han, R.I.; Hu, C.W.; Loose, D.S.; Yang, L.; Li, L.; Connell, J.P.; Reardon, M.J.; Lawrie, G.M.; Qutub, A.A.; Morrisett, J.D.; et al. Differential Proteome Profile, Biological Pathways, and Network Relationships of Osteogenic Proteins in Calcified Human Aortic Valves. Heart Vessel. 2022, 37, 347–358. [Google Scholar] [CrossRef]
- Weisell, J.; Ohukainen, P.; Näpänkangas, J.; Ohlmeier, S.; Bergmann, U.; Peltonen, T.; Taskinen, P.; Ruskoaho, H.; Rysä, J. Heat Shock Protein 90 Is Downregulated in Calcific Aortic Valve Disease. BMC Cardiovasc. Disord. 2019, 19, 306. [Google Scholar] [CrossRef] [Green Version]
- Martin-Rojas, T.; Mourino-Alvarez, L.; Alonso-Orgaz, S.; Rosello-Lleti, E.; Calvo, E.; Lopez-Almodovar, L.F.; Rivera, M.; Padial, L.R.; Lopez, J.A.; de la Cuesta, F.; et al. ITRAQ Proteomic Analysis of Extracellular Matrix Remodeling in Aortic Valve Disease. Sci. Rep. 2015, 5, 17290. [Google Scholar] [CrossRef]
- Mourino-Alvarez, L.; Iloro, I.; de la Cuesta, F.; Azkargorta, M.; Sastre-Oliva, T.; Escobes, I.; Lopez-Almodovar, L.F.; Sanchez, P.L.; Urreta, H.; Fernandez-Aviles, F.; et al. MALDI-Imaging Mass Spectrometry: A Step Forward in the Anatomopathological Characterization of Stenotic Aortic Valve Tissue. Sci. Rep. 2016, 6, 27106. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.-I.; Satoh, K.; Maniwa, T.; Araki, A.; Maruyama, R.; Oda, T. Noticeable Decreased Expression of Tenascin-X in Calcific Aortic Valves. Connect. Tissue Res. 2012, 53, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Martin-Rojas, T.; Mourino-Alvarez, L.; Gil-Dones, F.; de la Cuesta, F.; Rosello-Lleti, E.; Laborde, C.M.; Rivera, M.; Lopez-Almodovar, L.F.; Lopez, J.A.; Akerstrom, F.; et al. A Clinical Perspective on the Utility of Alpha 1 Antichymotrypsin for the Early Diagnosis of Calcific Aortic Stenosis. Clin Proteom. 2017, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Llamas, G.; Martín-Rojas, T.; de la Cuesta, F.; Calvo, E.; Gil-Dones, F.; Dardé, V.M.; Lopez-Almodovar, L.F.; Padial, L.R.; Lopez, J.-A.; Vivanco, F.; et al. Modification of the Secretion Pattern of Proteases, Inflammatory Mediators, and Extracellular Matrix Proteins by Human Aortic Valve Is Key in Severe Aortic Stenosis. Mol. Cell. Proteom. 2013, 12, 2426–2439. [Google Scholar] [CrossRef]
- Mourino-Alvarez, L.; Baldan-Martin, M.; Gonzalez-Calero, L.; Martinez-Laborde, C.; Sastre-Oliva, T.; Moreno-Luna, R.; Lopez-Almodovar, L.F.; Sanchez, P.L.; Fernandez-Aviles, F.; Vivanco, F.; et al. Patients with Calcific Aortic Stenosis Exhibit Systemic Molecular Evidence of Ischemia, Enhanced Coagulation, Oxidative Stress and Impaired Cholesterol Transport. Int. J. Cardiol. 2016, 225, 99–106. [Google Scholar] [CrossRef]
- Bouchareb, R.; Guauque-Olarte, S.; Snider, J.; Zaminski, D.; Anyanwu, A.; Stelzer, P.; Lebeche, D. Proteomic Architecture of Valvular Extracellular Matrix: FNDC1 and MXRA5 Are New Biomarkers of Aortic Stenosis. JACC Basic Transl. Sci. 2021, 6, 25–39. [Google Scholar] [CrossRef]
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best Practices and Considerations for Mass-Spectrometry-Based Protein Biomarker Discovery and Validation. Nat. Protoc. 2021, 16, 3737–3760. [Google Scholar] [CrossRef]
- Mischak, H.; Apweiler, R.; Banks, R.E.; Conaway, M.; Coon, J.; Dominiczak, A.; Ehrich, J.H.H.; Fliser, D.; Girolami, M.; Hermjakob, H.; et al. Clinical Proteomics: A Need to Define the Field and to Begin to Set Adequate Standards. Proteom.—Clin. Appl. 2007, 1, 148–156. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Anjo, S.I.; Santa, C.; Manadas, B. Short GeLC-SWATH: A Fast and Reliable Quantitative Approach for Proteomic Screenings. Proteomics 2015, 15, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Louis, S.; Nancy, C.; François, D.; Patrick, M.; Christian, C.; Sylvain, T.; Yohan, B.; Siamak, M.; Sylvain, P.; Philippe, J.; et al. Sex-Related Discordance Between Aortic Valve Calcification and Hemodynamic Severity of Aortic Stenosis. Circ. Res. 2017, 120, 681–691. [Google Scholar] [CrossRef]
- Vahanian, A.; Beyersdorf, F.; Praz, F.; Milojevic, M.; Baldus, S.; Bauersachs, J.; Capodanno, D.; Conradi, L.; de Bonis, M.; de Paulis, R.; et al. 2021 ESC/EACTS Guidelines for the Management of Valvular Heart Disease: Developed by the Task Force for the Management of Valvular Heart Disease of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2022, 43, 561–632. [Google Scholar] [CrossRef] [PubMed]
- Mahmut, A.; Boulanger, M.-C.; el Husseini, D.; Fournier, D.; Bouchareb, R.; Després, J.-P.; Pibarot, P.; Bossé, Y.; Mathieu, P. Elevated Expression of Lipoprotein-Associated Phospholipase A2 in Calcific Aortic Valve Disease: Implications for Valve Mineralization. J. Am. Coll. Cardiol. 2014, 63, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Kardys, I.; Oei, H.-H.S.; Hofman, A.; Oudkerk, M.; Witteman, J.C.M. Lipoprotein-Associated Phospholipase A2 and Coronary Calcification: The Rotterdam Coronary Calcification Study. Atherosclerosis 2007, 191, 377–383. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Lysis Buffer | Protein Yield (mg) | p-Value 1 | Protein Yield (%) | p-Value 1 |
| RIPA | 0.55 ± 0.08 | 0.88 | 1.8 ± 0.2 | 0.74 |
| Urea | 0.57 ± 0.17 | 1.9 ± 0.6 | ||
| Second Lysis Buffer | Protein Yield (mg) | p-Value 1 | Protein Yield (%) | p-Value 1 |
| Urea | 0.06 ± 0.05 | 0.09 | 0.2 ± 0.2 | 0.07 |
| RIPA | 0.14 ± 0.02 | 0.5 ± 0.1 |
| Men (n = 3) | Women (n = 3) | |
|---|---|---|
| Age, years | 70 ± 8.2 | 72 ± 8.1 |
| Diabetes (type 2), n | 1 | 2 |
| Smoking (active or past history), n | 1 | 0 |
| Arterial hypertension, n | 3 | 3 |
| Dislipidemia, n | 3 | 3 |
| Body Mass Index, kg/m2 | 29.1 ± 4.9 | 32.0 ± 4.5 |
| Indexed aortic valve area, cm2/m2 | 0.5 ± 0.07 | 0.4 ± 0.05 |
| Mean transvalvular pressure gradient, mmHg | 54 ± 11 | 53 ± 16 |
| Indexed left ventricle mass, g/m2 | 147 ± 48 | 132 ± 28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trindade, F.; Ferreira, A.F.; Saraiva, F.; Martins, D.; Mendes, V.M.; Sousa, C.; Gavina, C.; Leite-Moreira, A.; Manadas, B.; Falcão-Pires, I.; et al. Optimization of a Protocol for Protein Extraction from Calcified Aortic Valves for Proteomics Applications: Development of a Standard Operating Procedure. Proteomes 2022, 10, 30. https://doi.org/10.3390/proteomes10030030
Trindade F, Ferreira AF, Saraiva F, Martins D, Mendes VM, Sousa C, Gavina C, Leite-Moreira A, Manadas B, Falcão-Pires I, et al. Optimization of a Protocol for Protein Extraction from Calcified Aortic Valves for Proteomics Applications: Development of a Standard Operating Procedure. Proteomes. 2022; 10(3):30. https://doi.org/10.3390/proteomes10030030
Chicago/Turabian StyleTrindade, Fábio, Ana F. Ferreira, Francisca Saraiva, Diana Martins, Vera M. Mendes, Carla Sousa, Cristina Gavina, Adelino Leite-Moreira, Bruno Manadas, Inês Falcão-Pires, and et al. 2022. "Optimization of a Protocol for Protein Extraction from Calcified Aortic Valves for Proteomics Applications: Development of a Standard Operating Procedure" Proteomes 10, no. 3: 30. https://doi.org/10.3390/proteomes10030030
APA StyleTrindade, F., Ferreira, A. F., Saraiva, F., Martins, D., Mendes, V. M., Sousa, C., Gavina, C., Leite-Moreira, A., Manadas, B., Falcão-Pires, I., & Vitorino, R. (2022). Optimization of a Protocol for Protein Extraction from Calcified Aortic Valves for Proteomics Applications: Development of a Standard Operating Procedure. Proteomes, 10(3), 30. https://doi.org/10.3390/proteomes10030030