
Identification of Plasma Biomarkers from Rheumatoid Arthritis Patients Using an Optimized Sequential Window Acquisition of All THeoretical Mass Spectra (SWATH) Proteomics Workflow

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Plasma Samples
2.2. Sample Fractionation and Spectral Library Generation
2.2.1. Sample Preparation
2.2.2. Size Exclusion Chromatography (SEC) Fractionation
2.2.3. Strong Anion Exchange (SAX) Fractionation
2.2.4. High-pH Reverse-Phase Fractionation
2.2.5. Top 14 High-Abundance Proteins Depleted for Peptide Spectral Library Building
2.2.6. Liquid Chromatography and Mass Spectrometry Analysis Using the DDA Method
2.2.7. Peptide Spectral Library Generation
2.2.8. In Silico Spectral Library Generation for Protein Isoform Analysis
2.3. Proteomics Analysis with SWATH-DIA Workflow
2.3.1. Sample Preparation
2.3.2. Liquid Chromatography and Mass Spectrometry Analysis for SWATH-DIA
2.3.3. Protein Identification and Relative Quantification
2.3.4. Data Analysis
3. Results
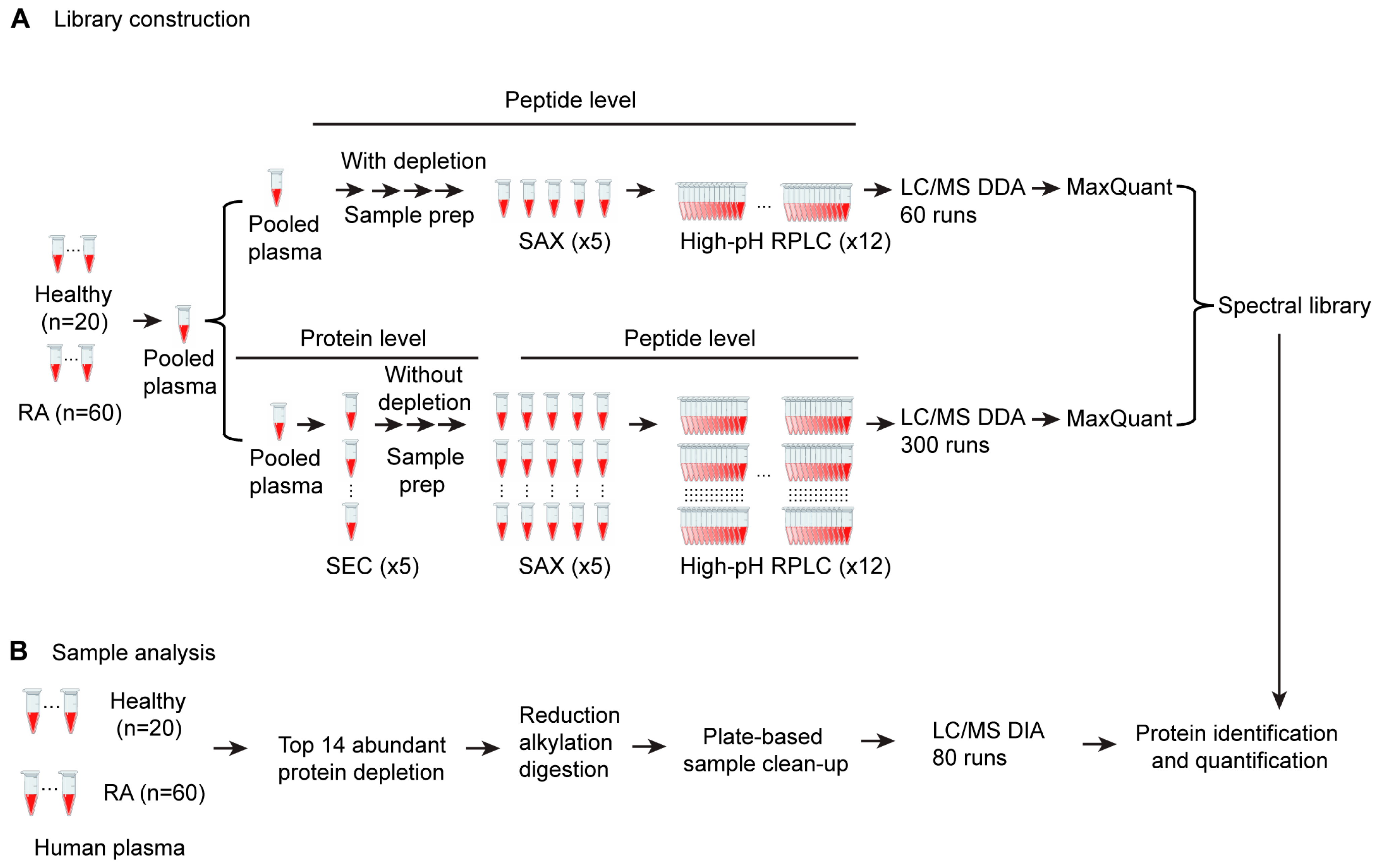
3.1. Development of a SWATH Proteomics Workflow for Large-Scale Plasma Sample Analysis
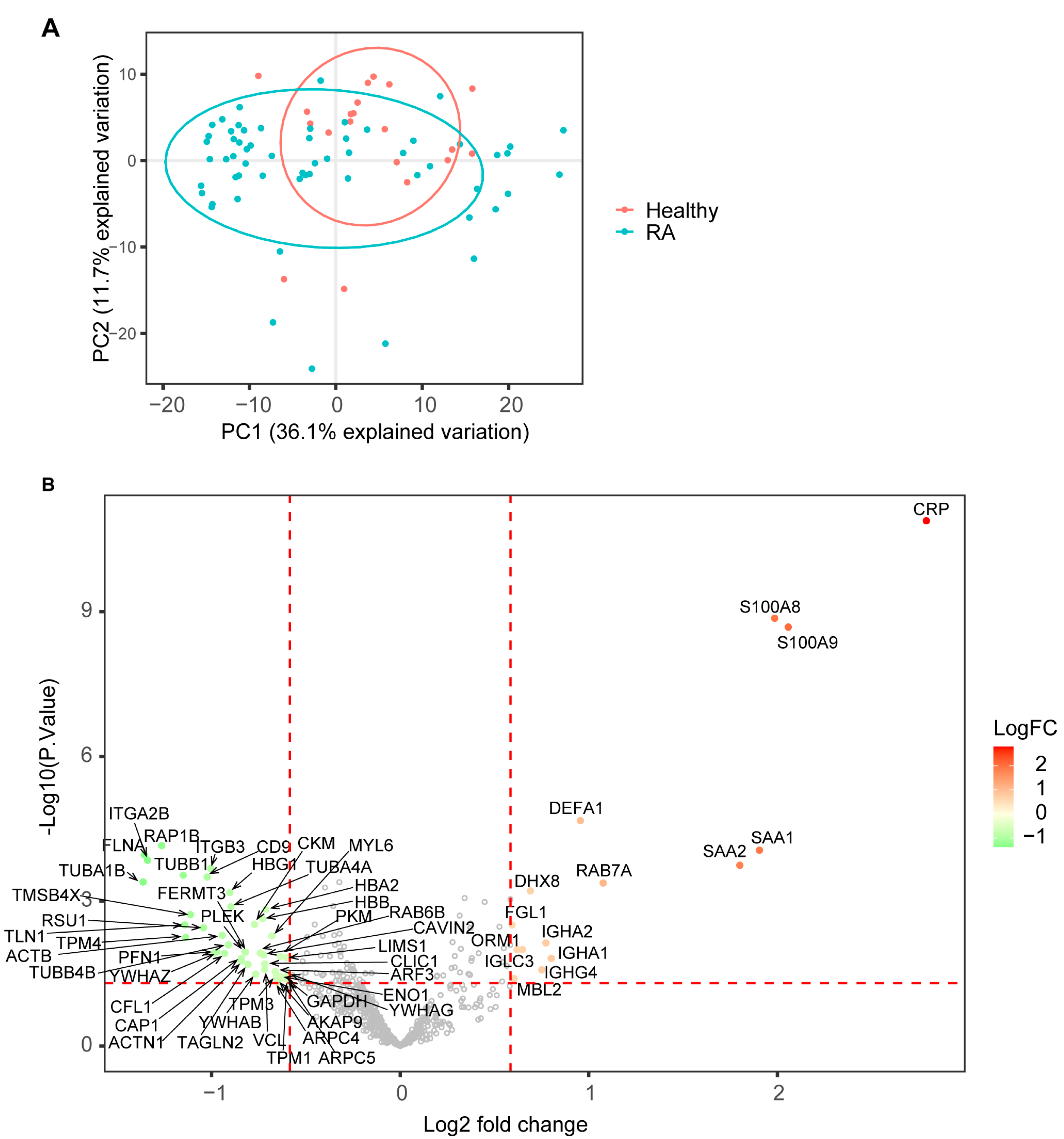
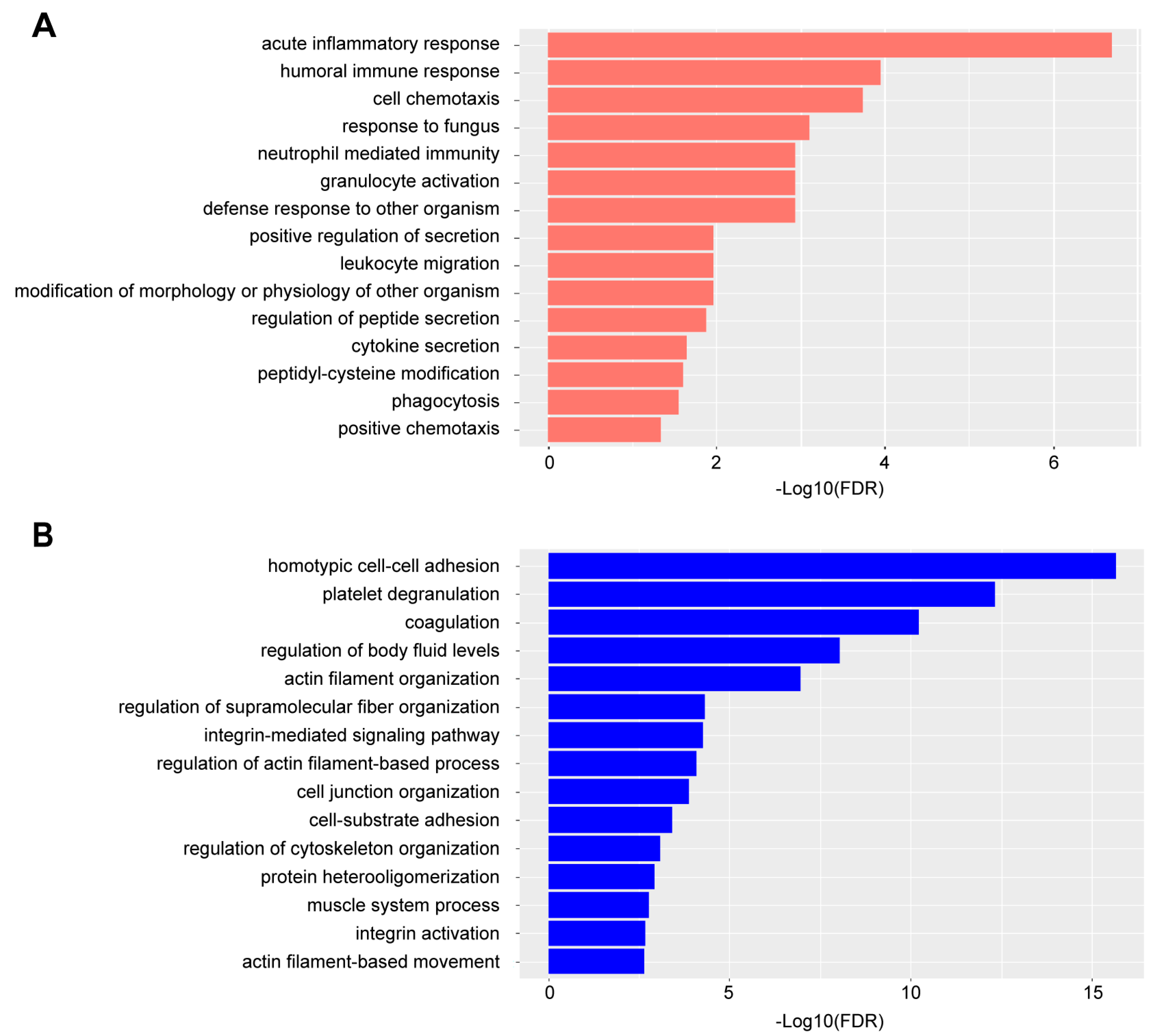
3.2. SWATH Proteomic Analysis Identified Differentially Expressed Proteins and Associated Biological Pathways in RA
3.3. Meta-Analysis to Compare This Study with Other RA Omics Studies
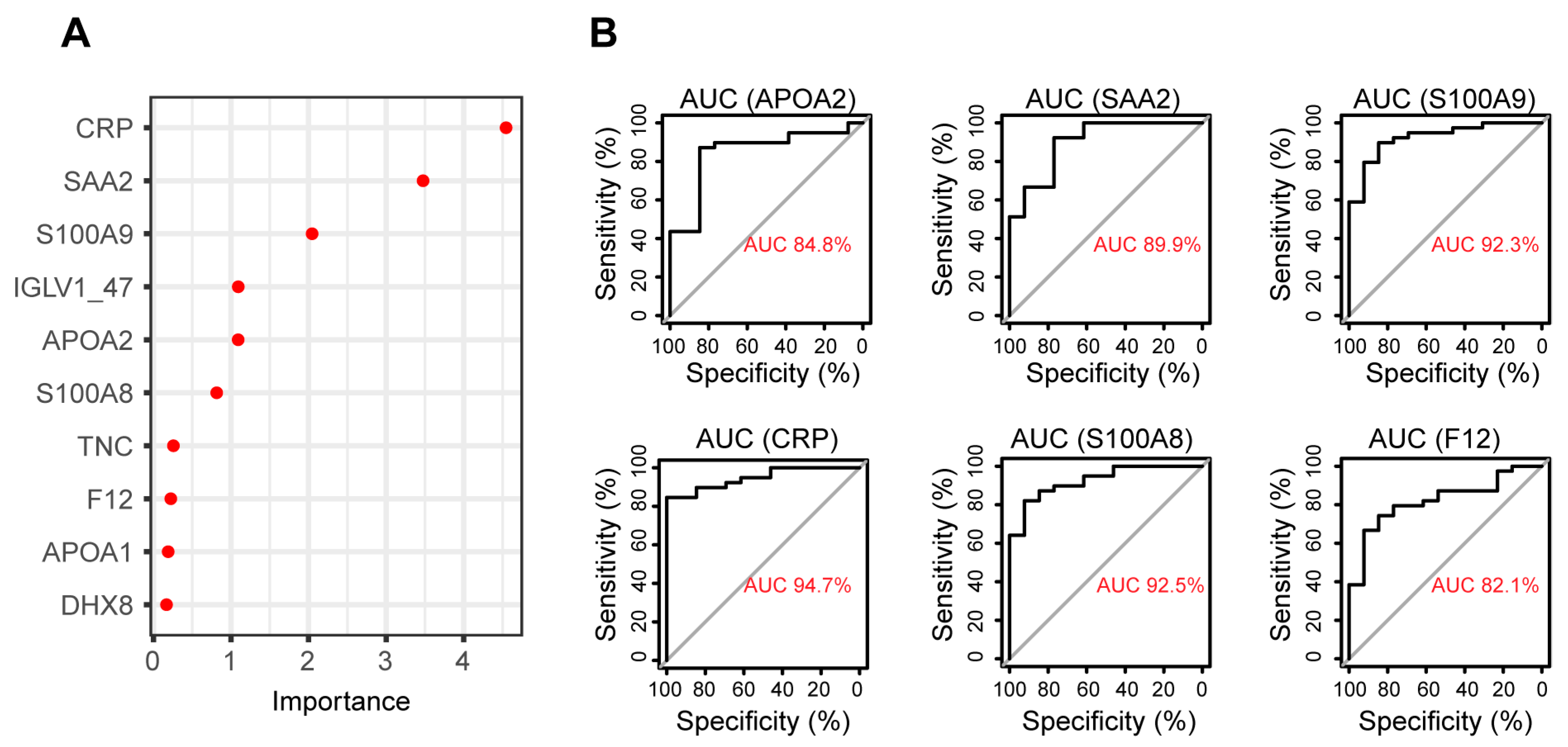
3.4. Biomarker Identification Using Random Forest to Discriminate between RA and Healthy Plasma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, Y.-J.; Chung, M.K.; Hwang, D.; Kim, W.-U. Proteomics in Rheumatoid Arthritis Research. Immune Netw. 2015, 15, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Salman, E.; Çetiner, S.; Boral, B.; Kibar, F.; Erken, E.; Ersözlü, E.D.; Badak, S.Ö.; Salman, R.B.; Sertdemir, Y.; Duran, A.Ç.; et al. Importance of 14-3-3eta, Anti-CarP, and Anti-Sa in the Diagnosis of Seronegative Rheumatoid Arthritis. Turk. J. Méd. Sci. 2019, 49, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Tenstad, H.B.; Nilsson, A.C.; Dellgren, C.D.; Lindegaard, H.M.; Rubin, K.H.; Lillevang, S.T. Use and Utility of Serologic Tests for Rheumatoid Arthritis in Primary Care. Dan. Méd. J. 2020, 67, A05190318b. [Google Scholar]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-Based Multiplexed Proteomic Technology for Biomarker Discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Dickens, E.R.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The Human Plasma Proteome History, Character, and Diagnostic Prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting Biomarker Discovery by Plasma Proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Bloomfield, N.; Yu, J.S.L.; White, M.; Kreidl, M.; Egger, A.-S.; Freiwald, A.; Ivosev, G.; Wasim, F.; et al. Ultra-Fast Proteomics with Scanning SWATH. Nat. Biotechnol. 2021, 39, 846–854. [Google Scholar] [CrossRef]
- Viode, A.; van Zalm, P.; Smolen, K.K.; Fatou, B.; Stevenson, D.; Jha, M.; Levy, O.; Steen, J.; Steen, H.; Network, O. behalf of the I. A Simple, Time- and Cost-Effective, High-Throughput Depletion Strategy for Deep Plasma Proteomics. Sci. Adv. 2023, 9, eadf9717. [Google Scholar] [CrossRef]
- Soni, R.K. High-Throughput Plasma Proteomic Profiling. Methods Mol. Biol. 2022, 2546, 411–420. [Google Scholar] [CrossRef]
- Blume, J.E.; Manning, W.C.; Troiano, G.; Hornburg, D.; Figa, M.; Hesterberg, L.; Platt, T.L.; Zhao, X.; Cuaresma, R.A.; Everley, P.A.; et al. Rapid, Deep and Precise Profiling of the Plasma Proteome with Multi-Nanoparticle Protein Corona. Nat. Commun. 2020, 11, 3662. [Google Scholar] [CrossRef] [PubMed]
- Ferdosi, S.; Tangeysh, B.; Brown, T.R.; Everley, P.A.; Figa, M.; McLean, M.; Elgierari, E.M.; Zhao, X.; Garcia, V.J.; Wang, T.; et al. Engineered Nanoparticles Enable Deep Proteomics Studies at Scale by Leveraging Tunable Nano–Bio Interactions. Proc. Natl. Acad. Sci. USA 2022, 119, e2106053119. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Pangan, A.L.; Song, I.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.J.; Li, Y.; Zhou, Y.; et al. Upadacitinib Versus Placebo or Adalimumab in Patients with Rheumatoid Arthritis and an Inadequate Response to Methotrexate: Results of a Phase III, Double-Blind, Randomized Controlled Trial. Arthritis Rheumatol. 2019, 71, 1788–1800. [Google Scholar] [CrossRef]
- Yang, F.; Shen, Y.; Camp, D.G.; Smith, R.D. High-PH Reversed-Phase Chromatography with Fraction Concatenation for 2D Proteomic Analysis. Expert Rev. Proteom. 2012, 9, 129–134. [Google Scholar] [CrossRef]
- Wang, H.; Sun, S.; Zhang, Y.; Chen, S.; Liu, P.; Liu, B. An Off-Line High PH Reversed-Phase Fractionation and Nano-Liquid Chromatography–Mass Spectrometry Method for Global Proteomic Profiling of Cell Lines. J. Chromatogr. B 2015, 974, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jin, L.; Hu, C.; Shen, S.; Qian, S.; Ma, M.; Zhu, X.; Li, F.; Wang, J.; Tian, Y.; et al. Ultra-High-Resolution IonStar Strategy Enhancing Accuracy and Precision of MS1-Based Proteomics and an Extensive Comparison with State-of-the-Art SWATH-MS in Large-Cohort Quantification. Anal. Chem. 2021, 93, 4884–4893. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Willforss, J.; Chawade, A.; Levander, F. NormalyzerDE: Online Tool for Improved Normalization of Omics Expression Data and High-Sensitivity Differential Expression Analysis. J. Proteome Res. 2019, 18, 732–740. [Google Scholar] [CrossRef]
- Stekhoven, D.J.; Bühlmann, P. MissForest—Non-Parametric Missing Value Imputation for Mixed-Type Data. Bioinformatics 2012, 28, 112–118. [Google Scholar] [CrossRef]
- Jin, L.; Bi, Y.; Hu, C.; Qu, J.; Shen, S.; Wang, X.; Tian, Y. A Comparative Study of Evaluating Missing Value Imputation Methods in Label-Free Proteomics. Sci. Rep. 2021, 11, 1760. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R Package Rsubread Is Easier, Faster, Cheaper and Better for Alignment and Quantification of RNA Sequencing Reads. Nucleic Acids Res. 2019, 47, gkz114. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. PROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Govaert, E.; Steendam, K.; Willems, S.; Vossaert, L.; Dhaenens, M.; Deforce, D. Comparison of Fractionation Proteomics for Local SWATH Library Building. Proteomics 2017, 17, 1700052. [Google Scholar] [CrossRef]
- Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a Tool for Biomarker Discovery: From Basic Research to Clinical Applications. Proteomics 2017, 17, 1600278. [Google Scholar] [CrossRef]
- Erre, G.L.; Paliogiannis, P.; Castagna, F.; Mangoni, A.A.; Carru, C.; Passiu, G.; Zinellu, A. Meta-analysis of Neutrophil-to-lymphocyte and Platelet-to-lymphocyte Ratio in Rheumatoid Arthritis. Eur. J. Clin. Investig. 2019, 49, e13037. [Google Scholar] [CrossRef]
- Jin, Z.; Cai, G.; Zhang, P.; Li, X.; Yao, S.; Zhuang, L.; Ren, M.; Wang, Q.; Yu, X. The Value of the Neutrophil-to-lymphocyte Ratio and Platelet-to-lymphocyte Ratio as Complementary Diagnostic Tools in the Diagnosis of Rheumatoid Arthritis: A Multicenter Retrospective Study. J. Clin. Lab. Anal. 2021, 35, e23569. [Google Scholar] [CrossRef]
- De Guadiana-Romualdo, L.G.; Rojas, C.R.; Morell-García, D.; Andaluz-Ojeda, D.; Mulero, M.D.R.; Rodríguez-Borja, E.; Ballesteros-Vizoso, A.; Calvo, M.D.; Albert-Botella, L.; Giráldez, A.P.; et al. Circulating Levels of Calprotectin, a Signature of Neutrophil Activation in Prediction of Severe Respiratory Failure in COVID-19 Patients: A Multicenter, Prospective Study (CalCov Study). Inflamm. Res. 2022, 71, 57–67. [Google Scholar] [CrossRef]
- Theilgaard-Mönch, K.; Jacobsen, L.C.; Nielsen, M.J.; Rasmussen, T.; Udby, L.; Gharib, M.; Arkwright, P.D.; Gombart, A.F.; Calafat, J.; Moestrup, S.K.; et al. Haptoglobin Is Synthesized during Granulocyte Differentiation, Stored in Specific Granules, and Released by Neutrophils in Response to Activation. Blood 2006, 108, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Kessel, C.; Koné-Paut, I.; Tellier, S.; Belot, A.; Masjosthusmann, K.; Wittkowski, H.; Fuehner, S.; Rossi-Semerano, L.; Dusser, P.; Marie, I.; et al. An Immunological Axis Involving Interleukin 1β and Leucine-Rich-A2-Glycoprotein Reflects Therapeutic Response of Children with Kawasaki Disease: Implications from the KAWAKINRA Trial. J. Clin. Immunol. 2022, 42, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil Elastase, Proteinase 3 and Cathepsin G: Physicochemical Properties, Activity and Physiopathological Functions. Biochimie 2008, 90, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Caccavo, D.; Garzia, P.; Sebastiani, G.D.; Ferri, G.M.; Galluzzo, S.; Vadacca, M.; Rigon, A.; Afeltra, A.; Amoroso, A. Expression of Lactoferrin on Neutrophil Granulocytes from Synovial Fluid and Peripheral Blood of Patients with Rheumatoid Arthritis. J. Rheumatol. 2003, 30, 220–224. [Google Scholar] [PubMed]
- Read, C.B.; Kuijper, J.L.; Hjorth, S.A.; Heipel, M.D.; Tang, X.; Fleetwood, A.J.; Dantzler, J.L.; Grell, S.N.; Kastrup, J.; Wang, C.; et al. Cutting Edge: Identification of Neutrophil PGLYRP1 as a Ligand for TREM-1. J. Immunol. 2015, 194, 1417–1421. [Google Scholar] [CrossRef]
- Lau, D.; Mollnau, H.; Eiserich, J.P.; Freeman, B.A.; Daiber, A.; Gehling, U.M.; Brümmer, J.; Rudolph, V.; Münzel, T.; Heitzer, T.; et al. Myeloperoxidase Mediates Neutrophil Activation by Association with CD11b/CD18 Integrins. Proc. Natl. Acad. Sci. USA 2005, 102, 431–436. [Google Scholar] [CrossRef]
- Sarr, D.; Oliveira, L.J.; Russ, B.N.; Owino, S.O.; Middii, J.D.; Mwalimu, S.; Ambasa, L.; Almutairi, F.; Vulule, J.; Rada, B.; et al. Myeloperoxidase and Other Markers of Neutrophil Activation Associate with Malaria and Malaria/HIV Coinfection in the Human Placenta. Front. Immunol. 2021, 12, 682668. [Google Scholar] [CrossRef]
- Scherlinger, M.; Richez, C.; Tsokos, G.C.; Boilard, E.; Blanco, P. The Role of Platelets in Immune-Mediated Inflammatory Diseases. Nat. Rev. Immunol. 2023, 23, 495–510. [Google Scholar] [CrossRef]
- Yamada, T.; Steinz, M.M.; Kenne, E.; Lanner, J.T. Muscle Weakness in Rheumatoid Arthritis: The Role of Ca2+ and Free Radical Signaling. eBioMedicine 2017, 23, 12–19. [Google Scholar] [CrossRef]
- Donovan, M.K.R.; Huang, Y.; Blume, J.E.; Wang, J.; Hornburg, D.; Ferdosi, S.; Mohtashemi, I.; Kim, S.; Ko, M.; Benz, R.W.; et al. Functionally Distinct BMP1 Isoforms Show an Opposite Pattern of Abundance in Plasma from Non-Small Cell Lung Cancer Subjects and Controls. PLoS ONE 2023, 18, e0282821. [Google Scholar] [CrossRef]
- Xu, L.; Zheng, H. The Isoform II of SRSF1: A Potential Biomarker in the Progression of Pediatric Acute Lymphoblastic Leukemia. Blood 2016, 128, 5275. [Google Scholar] [CrossRef]
- Ibáñez-Costa, A.; Perez-Sanchez, C.; Patiño-Trives, A.M.; Luque-Tevar, M.; Font, P.; de la Rosa, I.A.; Roman-Rodriguez, C.; Abalos-Aguilera, M.C.; Conde, C.; Gonzalez, A.; et al. Splicing Machinery Is Impaired in Rheumatoid Arthritis, Associated with Disease Activity and Modulated by Anti-TNF Therapy. Ann. Rheum. Dis. 2022, 81, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining Inflammatory Cell States in Rheumatoid Arthritis Joint Synovial Tissues by Integrating Single-Cell Transcriptomics and Mass Cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Dai, Z.; Xu, J.; Zhao, L.; Xu, Y.; Li, M.; Yu, J.; Zhang, L.; Deng, H.; Liu, L.; et al. Proteome Profiling Identifies Serum Biomarkers in Rheumatoid Arthritis. Front. Immunol. 2022, 13, 865425. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, Y.; Sun, X.; Li, Y.; Huang, C.; Deng, H.; Li, Z. Identification of Potential Serum Biomarkers for Rheumatoid Arthritis by High-Resolution Quantitative Proteomic Analysis. Inflammation 2014, 37, 1459–1467. [Google Scholar] [CrossRef]
- Liu, S.; Guo, Y.; Lu, L.; Lu, J.; Ke, M.; Xu, T.; Lu, Y.; Chen, W.; Wang, J.; Kong, D.; et al. Fibrinogen-Like Protein 1 Is a Novel Biomarker for Predicting Disease Activity and Prognosis of Rheumatoid Arthritis. Front. Immunol. 2020, 11, 579228. [Google Scholar] [CrossRef]
- Mun, S.; Lee, J.; Lim, M.-K.; Lee, Y.-R.; Ihm, C.; Lee, S.H.; Kang, H.-G. Development of a Novel Diagnostic Biomarker Set for Rheumatoid Arthritis Using a Proteomics Approach. BioMed Res. Int. 2018, 2018, 7490723. [Google Scholar] [CrossRef]
- Hayashi, J.; Kihara, M.; Kato, H.; Nishimura, T. A Proteomic Profile of Synoviocyte Lesions Microdissected from Formalin-Fixed Paraffin-Embedded Synovial Tissues of Rheumatoid Arthritis. Clin. Proteom. 2015, 12, 20. [Google Scholar] [CrossRef]
- Ren, X.; Geng, M.; Xu, K.; Lu, C.; Cheng, Y.; Kong, L.; Cai, Y.; Hou, W.; Lu, Y.; Aihaiti, Y.; et al. Quantitative Proteomic Analysis of Synovial Tissue Reveals That Upregulated OLFM4 Aggravates Inflammation in Rheumatoid Arthritis. J. Proteome Res. 2021, 20, 4746–4757. [Google Scholar] [CrossRef]
- Birkelund, S.; Bennike, T.B.; Kastaniegaard, K.; Lausen, M.; Poulsen, T.B.G.; Kragstrup, T.W.; Deleuran, B.W.; Christiansen, G.; Stensballe, A. Proteomic Analysis of Synovial Fluid from Rheumatic Arthritis and Spondyloarthritis Patients. Clin. Proteom. 2020, 17, 29. [Google Scholar] [CrossRef]
- Mateos, J.; Lourido, L.; Fernández-Puente, P.; Calamia, V.; Fernández-López, C.; Oreiro, N.; Ruiz-Romero, C.; Blanco, F.J. Differential Protein Profiling of Synovial Fluid from Rheumatoid Arthritis and Osteoarthritis Patients Using LC–MALDI TOF/TOF. J. Proteom. 2012, 75, 2869–2878. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Bhattacharjee, M.; Ahmad, S.; Nirujogi, R.S.; Renuse, S.; Subbannayya, Y.; Marimuthu, A.; Srikanth, S.M.; Raju, R.; Dhillon, M.; et al. Differential Proteomic Analysis of Synovial Fluid from Rheumatoid Arthritis and Osteoarthritis Patients. Clin. Proteom. 2014, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Rychkov, D.; Neely, J.; Oskotsky, T.; Yu, S.; Perlmutter, N.; Nititham, J.; Carvidi, A.; Krueger, M.; Gross, A.; Criswell, L.A.; et al. Cross-Tissue Transcriptomic Analysis Leveraging Machine Learning Approaches Identifies New Biomarkers for Rheumatoid Arthritis. Front. Immunol. 2021, 12, 638066. [Google Scholar] [CrossRef] [PubMed]
- Yim, A.Y.F.L.; Ferrero, E.; Maratou, K.; Lewis, H.D.; Royal, G.; Tough, D.F.; Larminie, C.; Mannens, M.M.A.M.; Henneman, P.; de Jonge, W.J.; et al. Novel Insights into Rheumatoid Arthritis Through Characterization of Concordant Changes in DNA Methylation and Gene Expression in Synovial Biopsies of Patients with Differing Numbers of Swollen Joints. Front. Immunol. 2021, 12, 651475. [Google Scholar] [CrossRef] [PubMed]
- Wittkowski, H.; Foell, D.; af Klint, E.; Rycke, L.D.; Keyser, F.D.; Frosch, M.; Ulfgren, A.-K.; Roth, J. Effects of Intra-Articular Corticosteroids and Anti-TNF Therapy on Neutrophil Activation in Rheumatoid Arthritis. Ann. Rheum. Dis. 2007, 66, 1020. [Google Scholar] [CrossRef]
- Mourão, A.F.; Canhão, H.; Sousa, E.; Cascão, R.; da Costa, J.B.; de Almeida, L.S.; Oliveira, M.E.; Gomes, M.M.; Queiroz, M.V.; Fonseca, J.E. From a Neutrophilic Synovial Tissue Infiltrate to a Challenging Case of Rheumatoid Arthritis. Acta Reum. Port. 2010, 35, 228–231. [Google Scholar]
- Wright, H.L.; Lyon, M.; Chapman, E.A.; Moots, R.J.; Edwards, S.W. Rheumatoid Arthritis Synovial Fluid Neutrophils Drive Inflammation Through Production of Chemokines, Reactive Oxygen Species, and Neutrophil Extracellular Traps. Front. Immunol. 2021, 11, 584116. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, G.; Lin, J.; Li, L.; Zeng, Z.; Chen, M.; Zhang, S. Serum Biomarkers for Inflammatory Bowel Disease. Front. Med. 2020, 7, 123. [Google Scholar] [CrossRef]
- Rice, S.J.; Belani, C.P. Optimizing Data-independent Acquisition (DIA) Spectral Library Workflows for Plasma Proteomics Studies. Proteomics 2022, 22, e2200125. [Google Scholar] [CrossRef]
- Fossati, A.; Richards, A.L.; Chen, K.-H.; Jaganath, D.; Cattamanchi, A.; Ernst, J.D.; Swaney, D.L. Toward Comprehensive Plasma Proteomics by Orthogonal Protease Digestion. J. Proteome Res. 2021, 20, 4031–4040. [Google Scholar] [CrossRef]
- Distler, U.; Łącki, M.K.; Schumann, S.; Wanninger, M.; Tenzer, S. Enhancing Sensitivity of Microflow-Based Bottom-Up Proteomics through Postcolumn Solvent Addition. Anal. Chem. 2019, 91, 7510–7515. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Mülleder, M.; Batruch, I.; Chelur, A.; Textoris-Taube, K.; Schwecke, T.; Hartl, J.; Causon, J.; Castro-Perez, J.; Demichev, V.; et al. High-Throughput Proteomics of Nanogram-Scale Samples with Zeno SWATH MS. eLife 2022, 11, e83947. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics Reveals a Remarkable Diversity of Lipids in Human Plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, X.; Shen, C.; Lin, Y.; Yang, P.; Qiao, L. In Silico Spectral Libraries by Deep Learning Facilitate Data-Independent Acquisition Proteomics. Nat. Commun. 2020, 11, 146. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Omenn, G.S.; Sun, Z.; Maes, M.; Pernemalm, M.; Palaniappan, K.K.; Letunica, N.; Vandenbrouck, Y.; Brun, V.; Tao, S.; et al. Advances and Utility of the Human Plasma Proteome. J. Proteome Res. 2021, 20, 5241–5263. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How Many Human Proteoforms Are There? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Palstrøm, N.B.; Matthiesen, R.; Rasmussen, L.M.; Beck, H.C. Recent Developments in Clinical Plasma Proteomics—Applied to Cardiovascular Research. Biomedicines 2022, 10, 162. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L.; Linial, M.; Goodlett, D.; Langridge-Smith, P.; Goo, Y.A.; Safford, G.; Bonilla, L.; Kruppa, G.; Zubarev, R.; et al. Proteoform: A Single Term Describing Protein Complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Paša-Tolić, L.; Tsybin, Y.O.; Kelleher, N.L.; Proteomics, T.C. for T.-D. The Human Proteoform Project: Defining the Human Proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef]
- Kim, H.K.; Pham, M.H.C.; Ko, K.S.; Rhee, B.D.; Han, J. Alternative Splicing Isoforms in Health and Disease. Pflügers Arch. Eur. J. Physiol. 2018, 470, 995–1016. [Google Scholar] [CrossRef]
- Wang, X.; Codreanu, S.G.; Wen, B.; Li, K.; Chambers, M.C.; Liebler, D.C.; Zhang, B. Detection of Proteome Diversity Resulted from Alternative Splicing Is Limited by Trypsin Cleavage Specificity*. Mol. Cell. Proteom. 2018, 17, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Gornik, O.; Lauc, G. Glycosylation of Serum Proteins in Inflammatory Diseases. Dis. Markers 2008, 25, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Cramer, D.A.T.; Franc, V.; Caval, T.; Heck, A.J.R. Charting the Proteoform Landscape of Serum Proteins in Individual Donors by High-Resolution Native Mass Spectrometry. Anal. Chem. 2022, 94, 12732–12741. [Google Scholar] [CrossRef]
- Bagdonaite, I.; Malaker, S.A.; Polasky, D.A.; Riley, N.M.; Schjoldager, K.; Vakhrushev, S.Y.; Halim, A.; Aoki-Kinoshita, K.F.; Nesvizhskii, A.I.; Bertozzi, C.R.; et al. Glycoproteomics. Nat. Rev. Methods Prim. 2022, 2, 48. [Google Scholar] [CrossRef]
- Melby, J.A.; Roberts, D.S.; Larson, E.J.; Brown, K.A.; Bayne, E.F.; Jin, S.; Ge, Y. Novel Strategies to Address the Challenges in Top-Down Proteomics. J. Am. Soc. Mass Spectrom. 2021, 32, 1278–1294. [Google Scholar] [CrossRef]
- Cheon, D.H.; Yang, E.G.; Lee, C.; Lee, J.E. Low-Molecular-Weight Plasma Proteome Analysis Using Top-Down Mass Spectrometry. Methods Mol. Biol. 2017, 1619, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Tiambeng, T.N.; Wu, Z.; Melby, J.A.; Ge, Y. Size Exclusion Chromatography Strategies and MASH Explorer for Large Proteoform Characterization. Methods Mol. Biol. 2022, 2500, 15–30. [Google Scholar] [CrossRef]
- Ntai, I.; Fornelli, L.; DeHart, C.J.; Hutton, J.E.; Doubleday, P.F.; LeDuc, R.D.; van Nispen, A.J.; Fellers, R.T.; Whiteley, G.; Boja, E.S.; et al. Precise Characterization of KRAS4b Proteoforms in Human Colorectal Cells and Tumors Reveals Mutation/Modification Crosstalk. Proc. Natl. Acad. Sci. USA 2018, 115, 4140–4145. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Tools to Cut the Sweet Layer-Cake That Is Glycoproteomics. Nat. Methods 2021, 18, 991–995. [Google Scholar] [CrossRef]
- Polasky, D.A.; Geiszler, D.J.; Yu, F.; Li, K.; Teo, G.C.; Nesvizhskii, A.I. MSFragger-Labile: A Flexible Method to Improve Labile PTM Analysis in Proteomics. Mol. Cell. Proteom. 2023, 22, 100538. [Google Scholar] [CrossRef]
- Roberts, D.S.; Mann, M.; Melby, J.A.; Larson, E.J.; Zhu, Y.; Brasier, A.R.; Jin, S.; Ge, Y. Structural OGlycoform Heterogeneity of the SARS-CoV-2 Spike Protein Receptor-Binding Domain Revealed by Top-Down Mass Spectrometry. J. Am. Chem. Soc. 2021, 143, 12014–12024. [Google Scholar] [CrossRef] [PubMed]
- Narzo, A.F.D.; Brodmerkel, C.; Telesco, S.E.; Argmann, C.; Peters, L.A.; Li, K.; Kidd, B.; Dudley, J.; Cho, J.; Schadt, E.E.; et al. High-Throughput Identification of the Plasma Proteomic Signature of Inflammatory Bowel Disease. J. Crohn’s Colitis 2019, 13, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Hu, S.; Spekhorst, L.M.; Zhernakova, D.V.; Vila, A.V.; Li, Y.; Voskuil, M.D.; van Berkel, L.A.; Folly, B.B.; Charrout, M.; et al. The Effect of Phenotype and Genotype on the Plasma Proteome in Patients with Inflammatory Bowel Disease. J. Crohn’s Colitis 2021, 16, jjab157. [Google Scholar] [CrossRef]
- Meuwis, M.-A.; Fillet, M.; Geurts, P.; de Seny, D.; Lutteri, L.; Chapelle, J.-P.; Bours, V.; Wehenkel, L.; Belaiche, J.; Malaise, M.; et al. Biomarker Discovery for Inflammatory Bowel Disease, Using Proteomic Serum Profiling. Biochem. Pharmacol. 2007, 73, 1422–1433. [Google Scholar] [CrossRef]
- Zhang, F.; Xu, C.; Ning, L.; Hu, F.; Shan, G.; Chen, H.; Yang, M.; Chen, W.; Yu, J.; Xu, G. Exploration of Serum Proteomic Profiling and Diagnostic Model That Differentiate Crohn’s Disease and Intestinal Tuberculosis. PLoS ONE 2016, 11, e0167109. [Google Scholar] [CrossRef]
- The Role of Fibronectin and Its Isoforms in the Pathogenesis and Progression of Rheumatoid Arthritis: A Review. Biointerface Res. Appl. Chem. 2022, 13, 341. [CrossRef]
- Yang, C.; Wang, C.; Zhou, J.; Liang, Q.; He, F.; Li, F.; Li, Y.; Chen, J.; Zhang, F.; Han, C.; et al. Fibronectin 1 Activates WNT/β-Catenin Signaling to Induce Osteogenic Differentiation via Integrin Β1 Interaction. Lab. Investig. 2020, 100, 1494–1502. [Google Scholar] [CrossRef]
- Katz, D.H.; Robbins, J.M.; Deng, S.; Tahir, U.A.; Bick, A.G.; Pampana, A.; Yu, Z.; Ngo, D.; Benson, M.D.; Chen, Z.-Z.; et al. Proteomic Profiling Platforms Head-to-Head: Leveraging Genetics and Clinical Traits to Compare Aptamer- and Antibody-Based Methods. Sci. Adv. 2022, 8, eabm5164. [Google Scholar] [CrossRef]
- Huang, T.; Wang, J.; Stukalov, A.; Donovan, M.; Ferdosi, S.; Williamson, L.; Just, S.; Astro, G.; Elgierari, E.; Benz, R.; et al. Functionalized Nanoparticles Enable Quantitative and Precise Large-Scale Unbiased, Deep Plasma Proteomics, WP 638, 2023 ASMS (Wednesday Poster, Poster ID: 314438). Available online: https://www.abstracts.asms.org/pages/dashboard.html#/conference/297/toc/297/details (accessed on 15 August 2023).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Serum/Plasma Proteomics (4) | Synovial Tissue Proteomics * (2) | Synovial Fluid Proteomics # (3) | Synovial Tissue Transcriptomics (2) |
|---|---|---|---|---|
| CRP | 3 | 1 | 0 | 0 |
| S100A9 | 0 | 2 | 1 | 1 |
| S100A8 | 0 | 2 | 2 | 1 |
| SAA1 | 4 | 0 | 0 | 0 |
| SAA2 | 2 | 0 | 0 | 0 |
| RAB7A | 1 | 0 | 0 | 0 |
| DEFA1 | 0 | 2 | 1 | 0 |
| IGHA1 | 0 | 0 | 1 | 0 |
| ORM1 | 1 | 1 | 0 | 0 |
| FGL1 | 1 | 0 | 0 | 0 |
| APCS | 3 | 0 | 0 | 0 |
| MMP9 | 0 | 1 | 1 | 1 |
| ORM2 | 1 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, L.; Wang, F.; Wang, X.; Harvey, B.P.; Bi, Y.; Hu, C.; Cui, B.; Darcy, A.T.; Maull, J.W.; Phillips, B.R.; et al. Identification of Plasma Biomarkers from Rheumatoid Arthritis Patients Using an Optimized Sequential Window Acquisition of All THeoretical Mass Spectra (SWATH) Proteomics Workflow. Proteomes 2023, 11, 32. https://doi.org/10.3390/proteomes11040032
Jin L, Wang F, Wang X, Harvey BP, Bi Y, Hu C, Cui B, Darcy AT, Maull JW, Phillips BR, et al. Identification of Plasma Biomarkers from Rheumatoid Arthritis Patients Using an Optimized Sequential Window Acquisition of All THeoretical Mass Spectra (SWATH) Proteomics Workflow. Proteomes. 2023; 11(4):32. https://doi.org/10.3390/proteomes11040032
Chicago/Turabian StyleJin, Liang, Fei Wang, Xue Wang, Bohdan P. Harvey, Yingtao Bi, Chenqi Hu, Baoliang Cui, Anhdao T. Darcy, John W. Maull, Ben R. Phillips, and et al. 2023. "Identification of Plasma Biomarkers from Rheumatoid Arthritis Patients Using an Optimized Sequential Window Acquisition of All THeoretical Mass Spectra (SWATH) Proteomics Workflow" Proteomes 11, no. 4: 32. https://doi.org/10.3390/proteomes11040032
APA StyleJin, L., Wang, F., Wang, X., Harvey, B. P., Bi, Y., Hu, C., Cui, B., Darcy, A. T., Maull, J. W., Phillips, B. R., Kim, Y., Jenkins, G. J., Sornasse, T. R., & Tian, Y. (2023). Identification of Plasma Biomarkers from Rheumatoid Arthritis Patients Using an Optimized Sequential Window Acquisition of All THeoretical Mass Spectra (SWATH) Proteomics Workflow. Proteomes, 11(4), 32. https://doi.org/10.3390/proteomes11040032