

Observations from the Proteomics Bench



Abstract
:1. Introduction
2. Results and Discussion—Challenges in Proteomics
2.1. Pre-Analytical Issues
2.1.1. Example: Serum Quality
2.1.2. Example: Tissue Origin
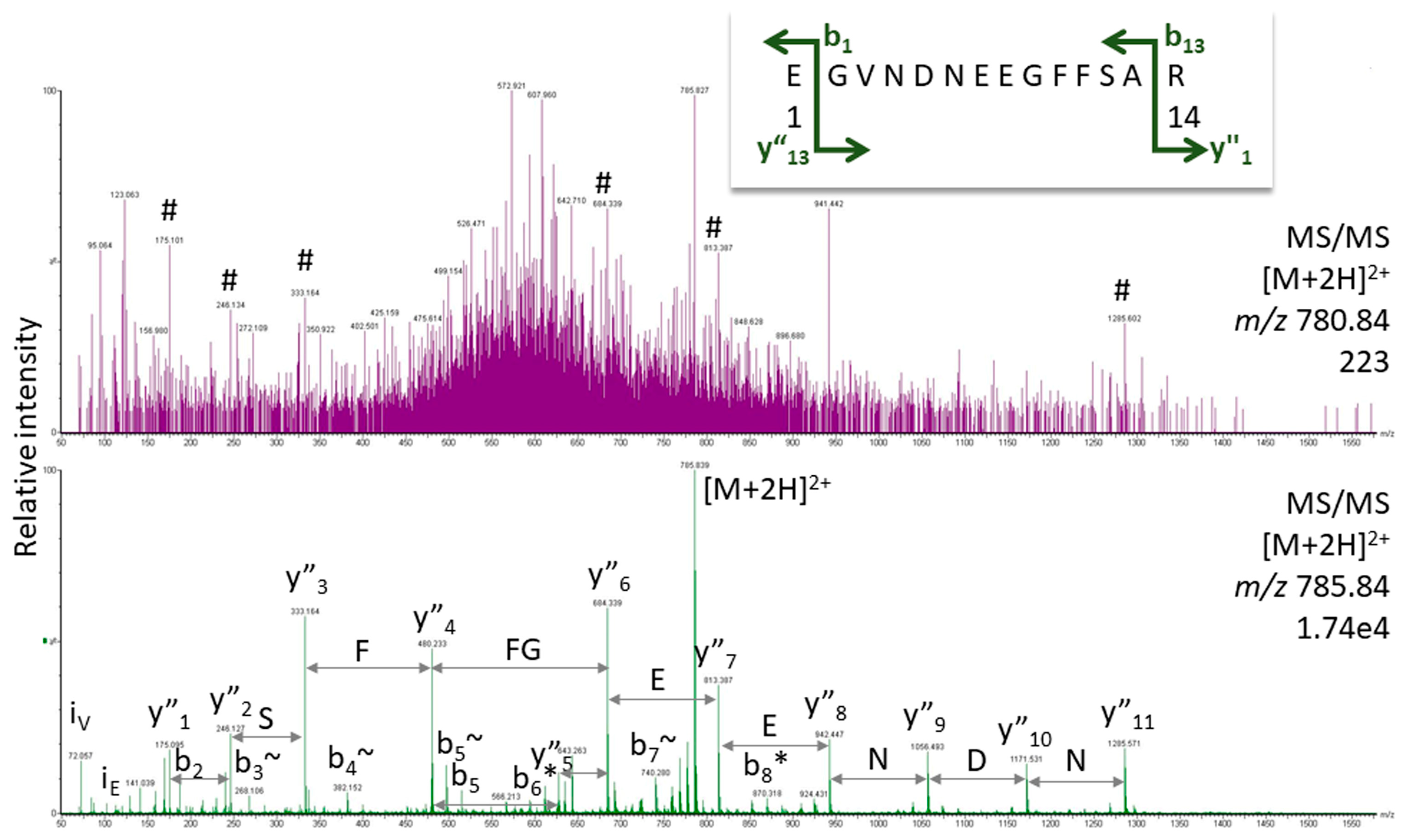
2.2. Peptide Sequencing
2.3. Protein Identification
2.4. Modifications
2.5. Bioinformatics/Data Analysis Issues
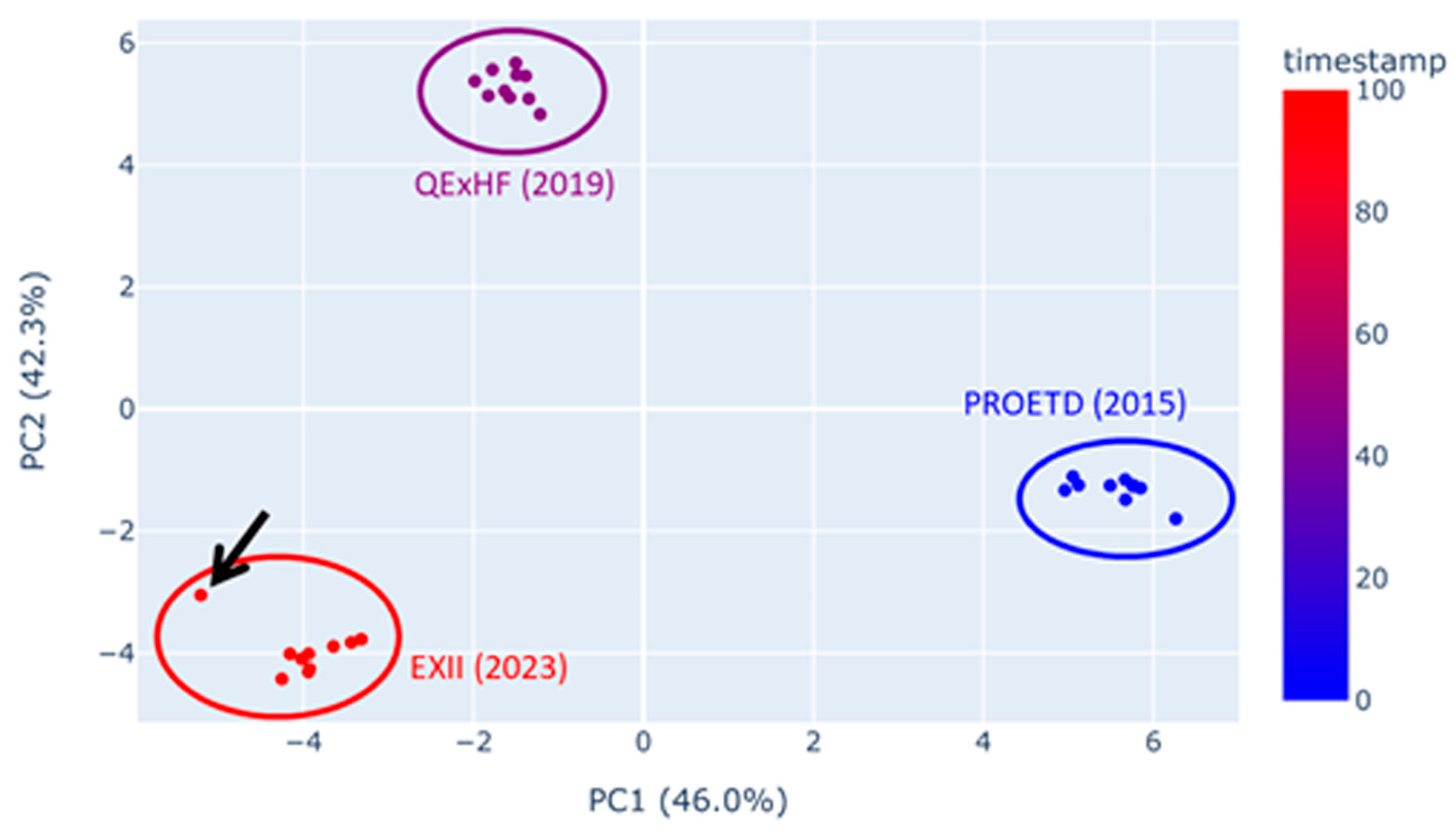
2.5.1. Quality Assessment/Experiment Reproducibility
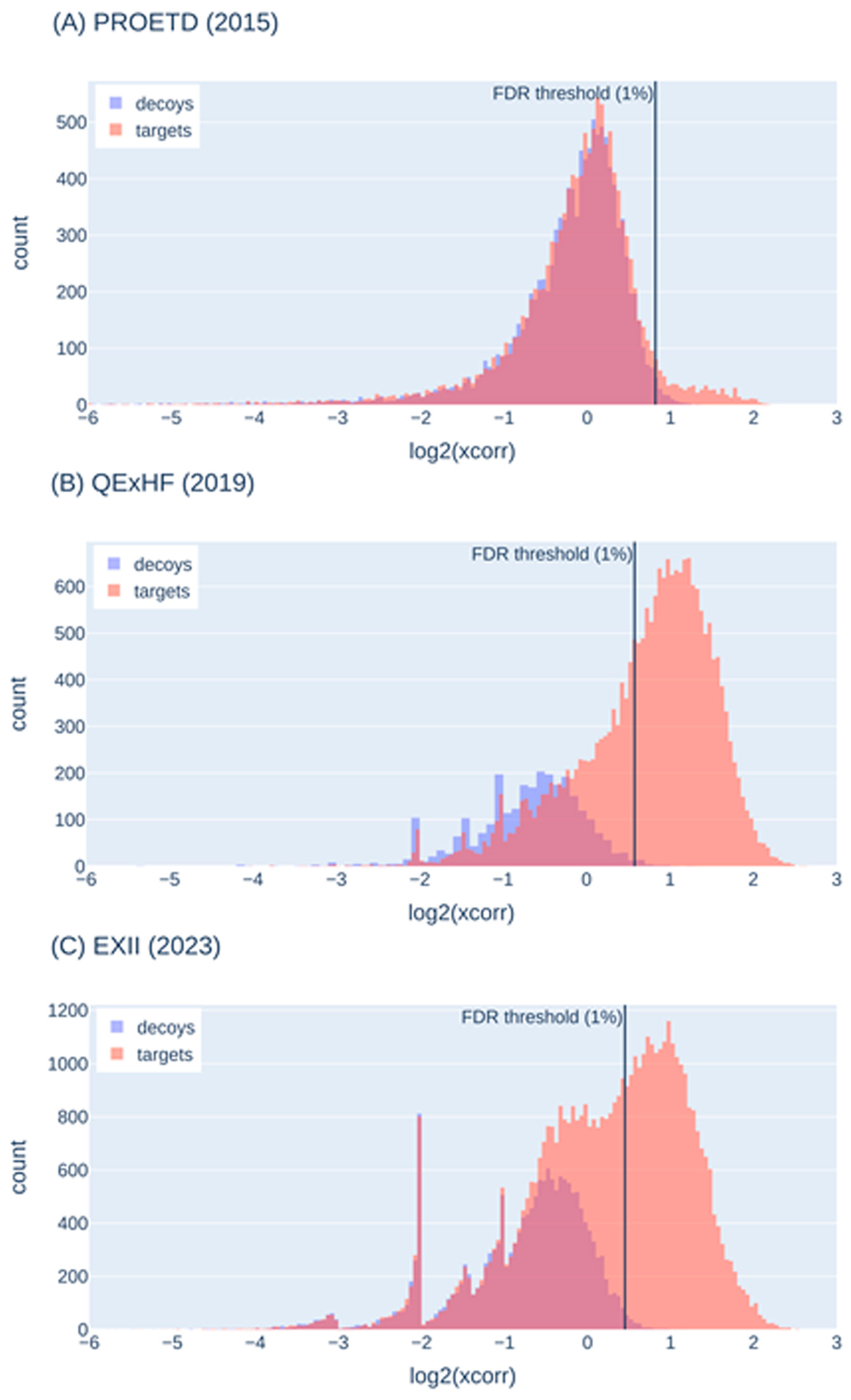
2.5.2. False-Discovery Estimation with Decoy Sequences
3. Materials and Methods
3.1. Description of ISA Files
3.2. QC Tool
3.3. Description of Decoy Workflow
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duncan, M. Good mass spectrometry and its place in good science. J. Mass Spectrom. 2012, 47, 795–809. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Yergey, A.L. Proteomics is analytical chemistry: Fitness-for-purpose in the application of top-down and bottom-up analyses. Proteomes 2015, 3, 440–453. [Google Scholar] [CrossRef] [PubMed]
- König, S. Spectral quality overrides software score—A brief tutorial on the analysis of peptide fragmentation data for mass spectrometry laymen. J. Mass Spectrom. 2021, 56, e4616. [Google Scholar] [CrossRef] [PubMed]
- Gegner, H.M.; Naake, T.; Dugourd, A.; Müller, T.; Czernilofsky, F.; Kliewer, G.; Jäger, E.; Helm, B.; Kunze-Rohrbach, N.; Klingmüller, U.; et al. Pre-analytical processing of plasma and serum samples for combined proteome and metabolome analysis. Front. Mol. Biosci. 2022, 9, 961448. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Atherton, J.; Costelloe, S.J.; Pourmahram, G.; Stretton, A.; Cornes, M. Preanalytical errors in medical laboratories: A review of the available methodologies of data collection and analysis. Ann. Clin. Biochem. 2017, 54, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Hassis, M.E.; Niles, R.K.; Braten, M.N.; Albertolle, M.E.; Witkowska, E.H.; Hubel, C.A.; Fisher, S.J.; Williams, K.E. Evaluating the effects of preanalytical variables on the stability of the human plasma proteome. Anal. Biochem. 2015, 478, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Bayer, M.; König, S. A vote for robustness: Monitoring serum enzyme activity by thin-layer chromatography of dabsylated bradykinin products. J. Pharmaceut. Biomed. Anal. 2017, 143, 199–203. [Google Scholar] [CrossRef]
- CDC Hemolysis Palette. Available online: https://www.cdc.gov/ncezid/dvbd/specimensub/hemolysis-palette.html (accessed on 9 September 2023).
- Aziz, S.; Rasheed, F.; Zahra, R.; König, S. Gastric cancer pre-stage detection and early diagnosis of gastritis using serum protein signatures. Molecules 2022, 27, 2857. [Google Scholar] [CrossRef]
- Ni, X.; Tan, Z.; Ding, C.; Zhang, C.; Song, L.; Yang, S.; Liu, M.; Jia, R.; Zhao, C.; Song, L.; et al. A region-resolved mucosa proteome of the human stomach. Nat. Commun. 2019, 10, 39. [Google Scholar] [CrossRef]
- Tepasse, P.-R.; Vollenberg, R.; Steinebrey, N.; König, S. The dysregulation of the renin-angiotensin-system in COVID-19 studied by serum proteomics: Angiotensinogen increases with disease severity. Molecules 2022, 27, 2495. [Google Scholar] [CrossRef]
- Brett, D.; Pospisil, H.; Valcárcel, J.; Reich, J.; Bork, P. Alternative splicing and genome complexity. Nat. Genet. 2002, 30, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fang, L.; Wu, C. Alternative splicing and isoforms: From mechanisms to diseases. Genes 2022, 13, 401. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L. Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cook, K.D. Oxidation artifacts in the electrospray mass spectrometry of Abeta peptide. Anal. Chem. 2007, 79, 2031–2036. [Google Scholar] [CrossRef] [PubMed]
- Morand, K.; Talbo, G.; Mann, M. Oxidation of peptides during electrospray ionization. Rapid Commun. Mass Spectrom. 1993, 7, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Unimod Database. Available online: www.unimod.org (accessed on 22 January 2024).
- Delta Mass Database. Available online: www.abrf.org/delta-mass (accessed on 22 January 2024).
- Tang, M.; Huang, P.; Wu, L.; Zhou, P.; Gong, P.; Liu, X.; Wei, Q.; Hou, X.; Hu, H.; Zhang, A.; et al. Comprehensive evaluation and optimization of the data-dependent LC-MS/MS workflow for deep proteome profiling. Anal. Chem. 2023, 95, 7897–7905. [Google Scholar] [CrossRef] [PubMed]
- Huffmann, R.G.; Chen, A.; Specht, H.; Slavov, N. DO-MS: Data-driven mass spectrometry methods. J. Proteome Res. 2019, 18, 2493–2500. [Google Scholar] [CrossRef]
- Distler, U.; Kuharev, J.; Navarro, P.; Tenzer, S. Label-free quantification in ion mobility-enhanced data-independent acquisition proteomics. Nat. Protoc. 2016, 11, 795–812. [Google Scholar] [CrossRef]
- Taylor, C.F.; Paton, N.W.; Lilley, K.S.; Binz, P.A.; Julian, R.K., Jr.; Jones, A.R.; Zhu, W.; Apweiler, R.; Aebersold, R.; Deutsch, E.W.; et al. The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 2007, 25, 887–893. [Google Scholar] [CrossRef]
- Walzer, M.; Pernas, L.E.; Nasso, S.; Bittremieux, W.; Nahnsen, S.; Kelchtermans, P.; Pichler, P.; van den Toorn, H.W.; Staes, A.; Vandenbussche, J.; et al. qcML: An exchange format for quality control metrics from mass spectrometry experiments. Mol. Cell. Proteom. 2014, 13, 1905–1913. [Google Scholar] [CrossRef]
- Pichler, P.; Mazanek, M.; Dusberger, F.; Weilnböck, L.; Huber, C.G.; Stingl, C.; Luider, T.M.; Straube, W.L.; Köcher, T.; Mechtler, K. SIMPATIQCO: A server-based software suite which facilitates monitoring the time course of LC-MS performance metrics on Orbitrap instruments. J. Proteome Res. 2012, 11, 5540–5547. [Google Scholar] [CrossRef]
- Rudnick, P.A.; Clauser, K.R.; Kilpatrick, L.E.; Tchekhovskoi, D.V.; Neta, P.; Blonder, N.; Billheimer, D.D.; Blackman, R.K.; Bunk, D.M.; Cardasis, H.L.; et al. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol. Cell. Proteom. 2010, 9, 225–241. [Google Scholar] [CrossRef]
- Rozanova, S.; Uszkoreit, J.; Schork, K.; Serschnitzki, B.; Eisenacher, M.; Tönges, L.; Barkovits-Boeddinghaus, K.; Marcus, K. Quality control—A stepchild in quantitative proteomics: A case study for the human CSF proteome. Biomolecules 2023, 13, 491. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef]
- Reidegeld, K.A.; Eisenacher, M.; Kohl, M.; Chamrad, D.; Körting, G.; Blüggel, M.; Meyer, H.E.; Stephan, C. An easy-to-use decoy database builder software tool, implementing different decoy strategies for false discovery rate calculation in automated MS/MS protein identifications. Proteomics 2008, 8, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Keich, U.; Noble, W.S. On the importance of well-calibrated scores for identifying shotgun proteomics spectra. J. Proteome Res. 2015, 14, 1147–1160. [Google Scholar] [CrossRef]
- Debrie, E.; Malfait, M.; Gabriels, R.; Declerq, A.; Sticker, A.; Martens, L.; Clement, L. Quality control for the target decoy approach for peptide identification. J. Proteome Res. 2023, 22, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Mao, J.; Yao, Y.; Wang, K.; Qiao, Q.; Fang, Z.; Ye, M. Sensitive profiling of cell surface proteome by using an optimized biotinylation method. J. Proteom. 2019, 196, 33–41. [Google Scholar] [CrossRef] [PubMed]
- May, C.; Serschnitzki, B.; Marcus, K. Good old-fashioned protein concentration determination by amino acid analysis. Methods Mol. Biol. 2021, 2228, 21–28. [Google Scholar]
- Kley, R.A.; Maerkens, A.; Leber, Y.; Theis, V.; Schreiner, A.; van der Ven, P.F.; Uszkoreit, J.; Stephan, C.; Eulitz, S.; Euler, N.; et al. A combined laser microdissection and mass spectrometry approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy patients. Mol. Cell. Proteom. 2013, 12, 215–227. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, W.; Meysman, P.; Martens, L.; Valkenborg, D.; Laukens, K. Unsupervised quality assessment of mass spectrometry proteomics experiments by multivariate quality control metrics. J. Proteome Res. 2016, 15, 1300–1307. [Google Scholar] [CrossRef]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef]
- Weisser, H.; Nahnsen, S.; Grossmann, J.; Nilse, L.; Quandt, A.; Brauer, H.; Sturm, M.; Kenar, E.; Kohlbacher, O.; Aebersold, R.; et al. An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. 2013, 12, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Uszkoreit, J.; Maerkens, A.; Perez-Riverol, Y.; Meyer, H.E.; Marcus, K.; Stephan, C.; Kohlbacher, O.; Eisenacher, M. PIA: An intuitive protein inference engine with a web-based user interface. J. Proteome Res. 2015, 14, 2988–2997. [Google Scholar] [CrossRef]
- Uszkoreit, J.; Perez-Riverol, Y.; Eggers, B.; Marcus, K.; Eisenacher, M. Protein inference using PIA workflows and PSI standard file formats. J. Proteome Res. 2019, 18, 741–747. [Google Scholar] [CrossRef]
- Plotly Technologies Inc. Collaborative Data Science. Montréal, QC. 2015. Available online: https://plot.ly (accessed on 19 December 2023).
- Dost, B.; Bandeira, N.; Li, X.; Shen, Z.; Briggs, S.P.; Bafna, V. Accurate mass spectrometry based protein quantification via shared peptides. J. Comput. Biol. 2012, 19, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Schork, K.; Turewicz, M.; Uszkoreit, J.; Rahnenführer, J.; Eisenacher, M. Characterization of peptide-protein relationships in protein ambiguity groups via bipartite graphs. PLoS ONE 2022, 17, e0276401. [Google Scholar] [CrossRef]
- The, M.; Edfors, F.; Perez-Riverol, Y.; Payne, S.H.; Hoopmann, M.R.; Palmblad, M.; Forsström, B.; Käll, L. A protein standard that emulates homology for the characterization of protein inference algorithms. J. Proteome Res. 2018, 17, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Chiva, C.; Elhamraoui, Z.; Solé, A.; Serret, M.; Wilhelm, M.; Sabidó, E. Assessment and prediction of human proteotypic peptide stability for proteomics quantification. Anal. Chem. 2023, 95, 13746–13749. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | 2015 | 2019 | 2023 |
|---|---|---|---|
| Instrument code | PROETD | QExHF | EXII |
| Nanoelectrospray mass spectrometer (Thermo Fisher) | Velos Plus | Q Excactive HF | Orbitrap Exploris 480 |
| Analyzer | radial ejection linear ion trap | quadrupole | quadrupole |
| Detector | electron multiplier | inductive detector | inductive detector |
| Number of raw files | 3172 | 1930 | 3287 |
| Project Code | Tolerance | ||
|---|---|---|---|
| Peptide Mass | Fragment Bin | Fragment Bin Offset | |
| PROETD (2015) | 10 ppm | 1.0005 | 0.4 |
| QExHF (2019) | 10 ppm | 0.02 | 0 |
| EXII (2023) | 5 ppm | 0.02 | 0 |
| Observed | Solution |
|---|---|
| Pre-analysis | |
| Poor project design, underpowered analyses, poor group assembly | Communication between PI, collaborators (e.g., surgeons) and AC; joint agreement on study design and sample requirements (SOPs) |
| Exaggerated expectations on the possible outcome of the analysis when searching for a particular target | Prior to analysis, literature and database searches are made for the known parameters of the target, like its concentration in the cell or its physico-chemical properties. Plausibility consideration: Can the target be expected to be detected with this method at all based on the instrumentation dynamic range and measurement principle? Consideration of enrichment or depletion steps |
| Lack of standardized sampling, information loss in large projects with many collaborators, free interpretation of SOPs, varying use of consumables and chemicals from different vendors within a project, use of devices such as centrifuges arbitrarily, accidental contamination of labware by colleagues | AC: Training of the PI in sample requirementsPI: Communication of this information to the project group and enforcement of SOPs, proper training of all involved personnel |
| Analysis | |
| Unexpected difficulties due to sample quality, unknown protein concentration, matrix issues | Running of test samples and adjustment of procedure |
| Outliers identified in QC visualization (e.g., TIC, PCA) or with QC software | Routine control of the instrument setup with benchmarks to ensure that it is working properly and that outliers result from the sample quality. Instrument maintenance and re-measurement |
| Post-analysis | |
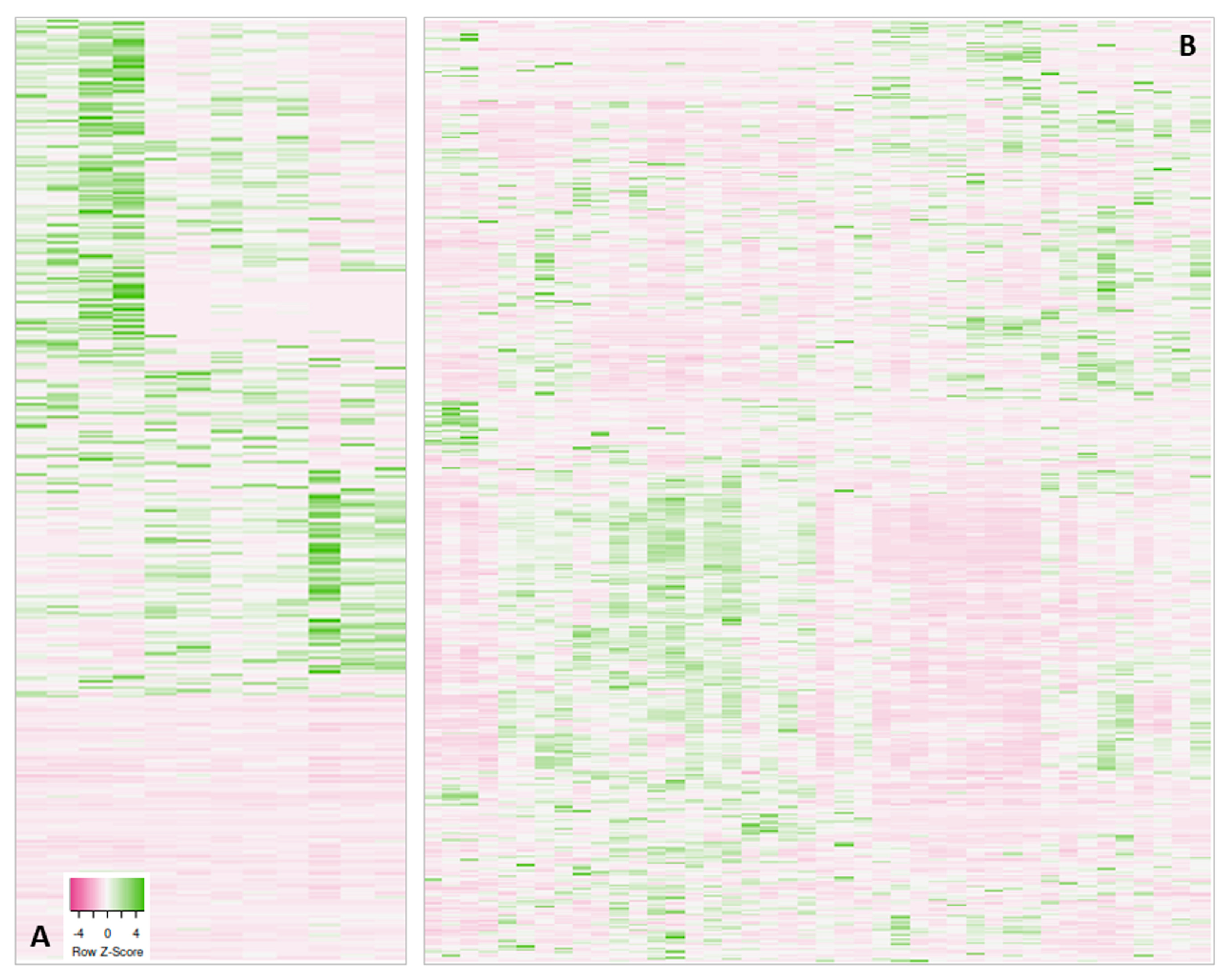
| Visualization of the protein abundancies (heatmaps) within a project shows distinct sample subgroups | Search for reasons, possible re-measurement or removal of outlier samples, data re-analysis considering subgroups |
| Hypothesis generation based on weak spectra | Conservative use of the results output from automatic experiments, no undue trust in software-generated Excel tables. Every person working in proteomics should be able to analyze peptide fragment ion spectra manually to have the knowledge to evaluate results from software algorithms. Manually re-check spectral data for potentially important proteins. Familiarization with the meaning of the peptide score in automatic experiments by running target MS/MS on the same peptides in the same sample. |
| Not a single protein form is assigned, but rather several proteins or isoforms sharing peptide sequences. | Unless a certain protein of interest is separately validated, conclusions should be carefully phrased. The truly functional protein (e.g., modified protein or isoform) may be present sub-stoichiometrically and individually designed analyses may be required. |
| Overlooked artifacts, e.g., from sample preparation or the analytical process | Careful manual interrogation of spectral evidence, plausibility consideration, validation by orthogonal methods |
| Excessive use of allowed modifications in database searches, leading to increased false-positive hits | Tailoring of sample preparation and analyses to modification of interest such as phosphorylation (e.g., enrichment), validation |
| Use of inappropriate databases for analysis of minor species | Plausibility consideration: Is it at all possible that proteins of species can be detected in the bulk sample? |
| Use of databases of related species in case of unsequenced species | Awareness that the analyte protein will have a (slightly) differing sequence; validation of the peptides by target MS/MS, Edman or other methods |
| Not finding abundantly present proteins or finding proteins that are not present | Possible software issues; tests of database, algorithm, and search parameters |
| Decoy histogram does not show sufficient decoys for representation of false-positive target identifications | Optimization of MS parameters (e.g., collision energy) and search engine parameters (e.g., mass tolerances), change of search engine, variation of the protein sequence database (not too large or too small) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
König, S.; Schork, K.; Eisenacher, M. Observations from the Proteomics Bench. Proteomes 2024, 12, 6. https://doi.org/10.3390/proteomes12010006
König S, Schork K, Eisenacher M. Observations from the Proteomics Bench. Proteomes. 2024; 12(1):6. https://doi.org/10.3390/proteomes12010006
Chicago/Turabian StyleKönig, Simone, Karin Schork, and Martin Eisenacher. 2024. "Observations from the Proteomics Bench" Proteomes 12, no. 1: 6. https://doi.org/10.3390/proteomes12010006
APA StyleKönig, S., Schork, K., & Eisenacher, M. (2024). Observations from the Proteomics Bench. Proteomes, 12(1), 6. https://doi.org/10.3390/proteomes12010006