
A Novel Meta-Analysis-Based Regularized Orthogonal Matching Pursuit Algorithm to Predict Lung Cancer with Selected Biomarkers


Abstract
:1. Introduction
2. The Regularized Orthogonal Matching Pursuit Algorithm for Biomarker Selection
| Algorithm 1 The MS-ROMP Algorithm for Biomarker Selection |
Input: Outcome values y, dataset X, stop condition ; Hyper-Parameters: ; Output: A subset of selected genes corresponding with its coefficient .
|
3. The MA-ROMP Algorithm for Biomarker Selection
| Algorithm 2 The MA-ROMP Algorithm for Biomarker Selection |
Input: Datasets , stop condition; Hyper-Parameters: ; Output: A subset of selected genes.
|
4. Experiments
4.1. Simulations
4.2. Real-Data Analysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolón-Canedo, V.; Sánchez-Maroño, N.; Alonso-Betanzos, A. Feature selection for high-dimensional data. Prog. Artif. Intell. 2016, 5, 65–75. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Dokeroglu, T.; Deniz, A.; Kiziloz, H.E. A comprehensive survey on recent metaheuristics for feature selection. Neurocomputing 2022, 494, 269–296. [Google Scholar] [CrossRef]
- Hu, L.; Yang, Y.; Tang, Z.; He, Y.; Luo, X. FCAN-MOPSO: An Improved Fuzzy-based Graph Clustering Algorithm for Complex Networks with Multi-objective Particle Swarm Optimization. IEEE Trans. Fuzzy Syst. 2023, 14, 1–16. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Fan, J.; Li, R. Variable selection via nonconcave penalized likelihood and its oracle properties. J. Am. Stat. Assoc. 2001, 96, 1348–1360. [Google Scholar] [CrossRef]
- Zhang, C.H. Nearly unbiased variable selection under minimax concave penalty. Ann. Stat. 2010, 38, 894–942. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Lin, Y. Model selection and estimation in regression with grouped variables. J. R. Stat. Soc. Ser. B Stat. Methodol. 2006, 68, 49–67. [Google Scholar] [CrossRef]
- Efron, B.; Hastie, T.; Johnstone, I.; Tibshirani, R. Least angle regression. Ann. Stat. 2004, 32, 407–499. [Google Scholar] [CrossRef]
- Mallat, S.; Zhang, Z. Matching pursuits with time-frequency dictionaries. IEEE Trans. Signal Process. 1993, 41, 3397–3415. [Google Scholar] [CrossRef]
- Tawfic, I.; Kayhan, S. Compressed sensing of ECG signal for wireless system with new fast iterative method. Comput. Methods Programs Biomed. 2015, 122, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhang, X. Reguladzation orthogonal matclling pursuit based on multiple support. Syst. Eng. Electron. 2020, 42, 8. [Google Scholar]
- Shi, X.; Xing, F.; Guo, Z.; Su, H.; Liu, F.; Yang, L. Structured orthogonal matching pursuit for feature selection. Neurocomputing 2019, 349, 164–172. [Google Scholar] [CrossRef]
- Tsagris, M.; Papadovasilakis, Z.; Lakiotaki, K.; Tsamardinos, I. The γ-OMP algorithm for feature selection with application to gene expression data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Toro-Domínguez, D.; Villatoro-García, J.A.; Martorell-Marugán, J.; Román-Montoya, Y.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. A survey of gene expression meta-analysis: Methods and applications. Brief. Bioinform. 2021, 22, 1694–1705. [Google Scholar] [CrossRef]
- Huang, H.H.; Rao, H.; Miao, R.; Liang, Y. A novel meta-analysis based on data augmentation and elastic data shared lasso regularization for gene expression. BMC Bioinform. 2022, 23, 353. [Google Scholar] [CrossRef]
- Li, Q.; Wang, S.; Huang, C.C.; Yu, M.; Shao, J. Meta-analysis based variable selection for gene expression data. Biometrics 2014, 70, 872–880. [Google Scholar] [CrossRef]
- Zhang, H.; Li, S.J.; Zhang, H.; Yang, Z.Y.; Ren, Y.Q.; Xia, L.Y.; Liang, Y. Meta-Analysis Based on Nonconvex Regularization. Sci. Rep. 2020, 10, 5755. [Google Scholar] [CrossRef]
- Hu, Z.; Zhou, Y.; Tong, T. Meta-Analyzing Multiple Omics Data With Robust Variable Selection. Front. Genet. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Khosravy, M.; Gupta, N.; Patel, N.; Duque, C.A. Recovery in compressive sensing: A review. Compressive Sens. Healthc. 2020, 2020, 25–42. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Dracheva, T.; Rotunno, M.; Figueroa, J.D.; Liu, H.; Dasgupta, A.; Mann, F.E.; Fukuoka, J.; Hames, M.; Bergen, A.W.; et al. Gene Expression Signature of Cigarette Smoking and Its Role in Lung Adenocarcinoma Development and Survival. PLoS ONE 2008, 3, e1651. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Aerts, J.; den Hamer, B.; van IJcken, W.; den Bakker, M.; Riegman, P.; van der Leest, C.; van der Spek, P.; Foekens, J.A.; Hoogsteden, H.C.; et al. Gene Expression-Based Classification of Non-Small Cell Lung Carcinomas and Survival Prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef]
- Lu, T.P.; Tsai, M.H.; Lee, J.M.; Hsu, C.P.; Chen, P.C.; Lin, C.W.; Shih, J.Y.; Yang, P.C.; Hsiao, C.K.; Lai, L.C.; et al. Identification of a novel biomarker, SEMA5A, for non-small cell lung carcinoma in nonsmoking women. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Donoho, D.L.; Maleki, A.; Montanari, A. Message passing algorithms for compressed sensing: I. motivation and construction. In Proceedings of the 2010 IEEE Information Theory Workshop on Information Theory (ITW 2010), Cairo, Egypt, 6–8 January 2010; pp. 1–5. [Google Scholar]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.J. Collagen Type XI Alpha 1 (COL11A1): A Novel Biomarker and a Key Player in Cancer. Cancers 2021, 13, 935. [Google Scholar] [CrossRef]
- Yi, X.; Luo, L.; Zhu, Y.; Deng, H.; Liao, H.; Shen, Y.; Zheng, Y. SPP1 facilitates cell migration and invasion by targeting COL11A1 in lung adenocarcinoma. Cancer Cell Int. 2022, 22, 324. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, G.; Dong, B.; Huang, L.; Zhang, C.; Sheng, Y.; Wu, B.; Han, L.; Wu, C.; Qi, Y. A pan-cancer analysis of the oncogenic role of secreted phosphoprotein 1 (SPP1) in human cancers. Ann. Transl. Med. 2022, 10, 279. [Google Scholar] [CrossRef]
- Tang, H.; Chen, J.; Han, X.; Feng, Y.; Wang, F. Upregulation of SPP1 Is a Marker for Poor Lung Cancer Prognosis and Contributes to Cancer Progression and Cisplatin Resistance. Front. Cell Dev. Biol. 2021, 9, 646390. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, W.; Chen, Z.; Xiang, C. Upregulation of PD-L1 by SPP1 mediates macrophage polarization and facilitates immune escape in lung adenocarcinoma. Exp. Cell Res. 2017, 359, 449–457. [Google Scholar] [CrossRef]
- Matsubara, E.; Yano, H.; Pan, C.; Komohara, Y.; Fujiwara, Y.; Zhao, S.; Shinchi, Y.; Kurotaki, D.; Suzuki, M. The Significance of SPP1 in Lung Cancers and Its Impact as a Marker for Protumor Tumor-Associated Macrophages. Cancers 2023, 15, 2250. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, K.; Qu, Z.; Xie, Z.; Tian, F. ADAMTS8 inhibited lung cancer progression through suppressing VEGFA. Biochem. Biophys. Res. Commun. 2022, 598, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Su, Q.; Li, C. Identidication of novel biomarkers in non-small cell lung cancer using machine learning. Sci. Rep. 2022, 12, 16693. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Chen, Z.S.; Zhou, W.D.; Shu, J.; Wang, X.H.; Jin, R.; Zhuang, L.L.; Hoda, M.A.; Zhang, H.; Zhou, G.P. Down-regulated GATA-1 up-regulates interferon regulatory factor 3 in lung adenocarcinoma. Sci. Rep. 2017, 7, 2551. [Google Scholar] [CrossRef] [PubMed]
- Falch, C.M.; Sundaram, A.Y.M.; Øystese, K.A.; Normann, K.R.; Lekva, T.; Silamikelis, I.; Eieland, A.K.; Andersen, M.; Bollerslev, J.; Olarescu, N.C. Gene expression profiling of fast- and slow-growing non-functioning gonadotroph pituitary adenomas. Eur. J. Endocrinol. 2018, 178, 295–307. [Google Scholar] [CrossRef]
- Zhang, Q.; Deng, S.; Li, Q.; Wang, G.; Guo, Z.; Zhu, D. Glycoprotein M6A Suppresses Lung Adenocarcinoma Progression via Inhibition of the PI3K/AKT Pathway. J. Oncol. 2022, 2022, 4601501. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | = 0.9 | = 0.5 | ||
|---|---|---|---|---|
| Sensitivity | 0.470 (0.021) | 0.400 (0.001) | 0.310 (0.011) | |
| SCAD | Specificity | 0.946 (0.011) | 0.942 (0.001) | 0.940 (0.002) |
| Accuracy | 0.941 (0.001) | 0.937 (0.001) | 0.934 (0.001) | |
| Sensitivity | 0.540 (0.049) | 0.520 (0.156) | 0.360 (0.197) | |
| lasso | Specificity | 0.947 (0.013) | 0.948 (0.011) | 0.945 (0.011) |
| Accuracy | 0.943 (0.013) | 0.944 (0.013) | 0.940 (0.013) | |
| Sensitivity | 0.560 (0.002) | 0.520 (0.001) | 0.320 (0.101) | |
| MCP | Specificity | 0.899 (0.001) | 0.894 (0.001) | 0.890 (0.001) |
| Accuracy | 0.896 (0.001) | 0.890 (0.001) | 0.885 (0.001) | |
| Sensitivity | 0.610 (0.002) | 0.570 (0.011) | 0.410 (0.002) | |
| MS-ROMP | Specificity | 0.944 (0.001) | 0.922 (0.001) | 0.943 (0.001) |
| Accuracy | 0.939 (0.001) | 0.917 (0.001) | 0.937 (0.001) | |
| Sensitivity | 0.890 (0.001) | 0.870 (0.001) | 0.690 (0.056) | |
| meta-SCAD | Specificity | 1 (0) | 0.999 (0.001) | 0.981 (0.004) |
| Accuracy | 0.999 (0.001) | 0.998 (0.001) | 0.978 (0.003) | |
| Sensitivity | 0.960 (0.001) | 0.860 (0.001) | 0.670 (0.048) | |
| meta-lasso | Specificity | 0.986 (0.001) | 0.984 (0.001) | 0.984 (0.001) |
| Accuracy | 0.986 (0.001) | 0.983 (0.001) | 0.981 (0.001) | |
| Sensitivity | 0.990 (0.032) | 0.930 (0.001) | 0.700 (0.067) | |
| meta-MCP | Specificity | 0.964 (0.002) | 0.999 (0.001) | 0.976 (0.004) |
| Accuracy | 0.965 (0.002) | 0.998 (0.001) | 0.975 (0.004) | |
| Sensitivity | 0.900 (0.001) | 1 (0) | 0.790 (0.001) | |
| MA-ROMP | Specificity | 0.994 (0.001) | 0.995 (0.001) | 0.993 (0.001) |
| Accuracy | 0.993 (0.001) | 0.995 (0.001) | 0.991 (0.001) |
| Datasets | GSE10072 | GSE19188 | GSE19804 |
|---|---|---|---|
| Platform | GPL96 | GPL570 | GPL570 |
| Total sample size | 107 | 156 | 120 |
| No. of genes | 22,283 | 54,675 | 54,675 |
| Methods | Training Data | ||
|---|---|---|---|
| Sensitivity | Specificity | Accuracy | |
| MA-ROMP | 0.9788 (0.001) | 0.9465 (0.001) | 0.9644 (0.001) |
| meta-lasso | 0.4722 (0.001) | 0.6667 (0.002) | 0.6683 (0.001) |
| meta-MCP | 0.4722 (0.001) | 0.6667 (0.001) | 0.6683 (0.002) |
| meta-SCAD | 0.5470 (0.004) | 0.9167 (0.001) | 0.7178 (0.001) |
| Methods | Testing data | ||
| Sensitivity | Specificity | Accuracy | |
| MA-ROMP | 1 (0) | 0.9188 (0.001) | 0.9563 (0.001) |
| meta-lasso | 0.4119 (0.003) | 0.6528 (0.002) | 0.5249 (0.011) |
| meta-MCP | 0.4050 (0.013) | 0.6528 (0.002) | 0.5215 (0.001) |
| meta-SCAD | 0.5278 (0.001) | 0.9231 (0.001) | 0.6571 (0.004) |
| Methods | GSE10072 | GSE19188 | GSE19804 |
|---|---|---|---|
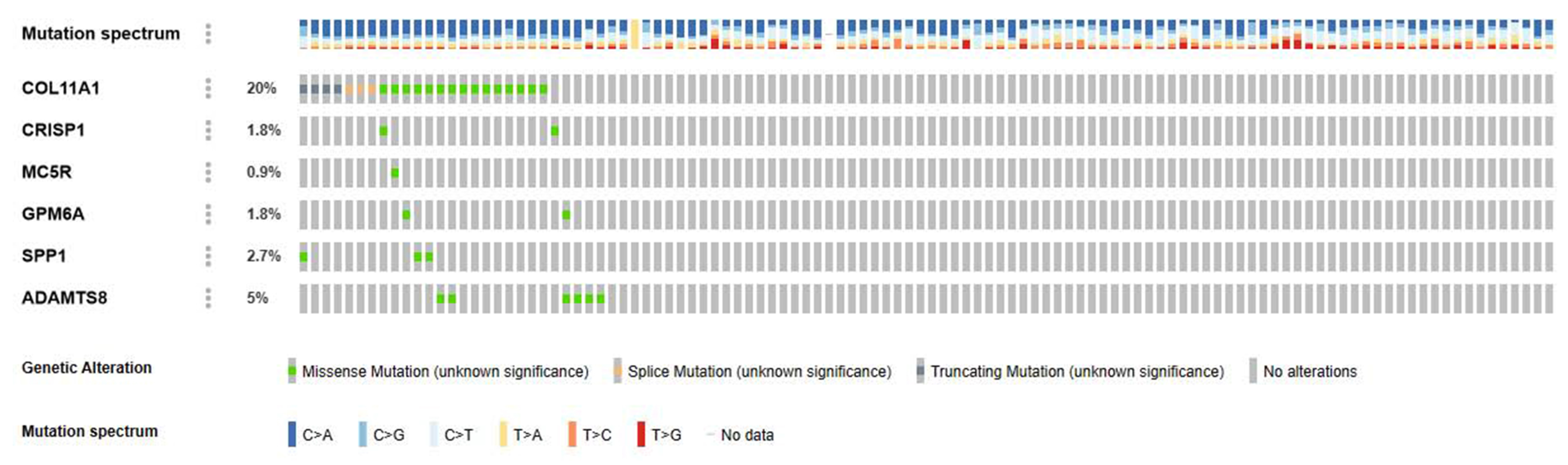
| MA-ROMP | COL11A1 | COL11A1 | COL11A1 |
| COL10A1 | COL10A1 | COL10A1 | |
| CRISP1 | CRISP1 | CRISP1 | |
| MC5R | MC5R | GPM6A | |
| GPM6A | GPM6A | SPP1 | |
| SPP1 | SPP1 | ADAMTS8 | |
| ADAMTS8 | ADAMTS8 | LINC00216 | |
| LINC00216 | LINC00216 | ||
| meta-MCP | EDNRB | EDNRB | EDNRB |
| CA4 | CA4 | CA4 | |
| GPM6A | GPM6A | GPM6A | |
| ADH1B | ADH1B | ADH1B | |
| TNNC1 | TNNC1 | TNNC1 | |
| AGER | AGER | AGER | |
| TMEM100 | TMEM100 | TMEM100 | |
| meta-SCAD | GREM1 | GREM1 | GREM1 |
| COL11A1 | COL11A1 | COL11A1 | |
| meta-lasso | GPM6A | GPM6A | GPM6A |
| AGER | AGER | AGER |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Wang, B.-Y.; Li, H.-F. A Novel Meta-Analysis-Based Regularized Orthogonal Matching Pursuit Algorithm to Predict Lung Cancer with Selected Biomarkers. Mathematics 2023, 11, 4171. https://doi.org/10.3390/math11194171
Wang S, Wang B-Y, Li H-F. A Novel Meta-Analysis-Based Regularized Orthogonal Matching Pursuit Algorithm to Predict Lung Cancer with Selected Biomarkers. Mathematics. 2023; 11(19):4171. https://doi.org/10.3390/math11194171
Chicago/Turabian StyleWang, Sai, Bin-Yuan Wang, and Hai-Fang Li. 2023. "A Novel Meta-Analysis-Based Regularized Orthogonal Matching Pursuit Algorithm to Predict Lung Cancer with Selected Biomarkers" Mathematics 11, no. 19: 4171. https://doi.org/10.3390/math11194171
APA StyleWang, S., Wang, B. -Y., & Li, H. -F. (2023). A Novel Meta-Analysis-Based Regularized Orthogonal Matching Pursuit Algorithm to Predict Lung Cancer with Selected Biomarkers. Mathematics, 11(19), 4171. https://doi.org/10.3390/math11194171