1. Introduction
News outlets report daily numbers about COVID-19 spread, evolution, and consequences. In the very first months, the media was hectically running after data published by authorities, that often were confused, or related to measures that were taken with different criteria, or reported with different delays and missing proper timestamping. The crisis found the international community dis-aligned and without unique modus operandi for data collection, metadata organization, validation, or information extraction: moreover, the behavior and the reactions of governments has been quite different and without coordination. In some cases, in which local authorities are directly in charge of decisions about COVID-19 management, the situation became fragmented and odd phenomena emerged, causing confusion, misleading information, chaotic management of the situation, and delays in decisions and in enacting solution strategies. Politicians turned to scientists for advice, but there was no unanimous response, and a certain spectacularization of scientific communication caused doubts and bewilderment in public opinion. Lockdowns mitigated the consequences of the initial circulation of the virus but, in turn, had severe consequences on the economy of many countries, endangering many enterprises and putting small businesses and shops that depend on normal circulation of people in serious trouble because they cannot survive the costs of prevention and the change of habits. For example, the development of the events in the case of Italy has been briefly, but richly, resumed in Reference [
1], in which a detailed chronological description of the events is reported, together with the main implications and a model of COVID-19 epidemic and consequent interventions in Italy founded on an extended Susceptible-Infected-Recovered (SIR) approach.
These months produced an enormous volume of scientific literature about whatever is related to COVID-19, from clinical reports to policy-oriented prescriptions in social sciences: what seems to emerge as a fact is that COVID-19 is still here to stay and that quantitative modeling of COVID-19 is crucial to cope with the management of the crisis and to support resource allocation at every level and planning of reactions. Providing predictive models to governments and international organization is of paramount importance to control damages and delay the second wave, that is documented in many countries, until enough clinical knowledge will be assessed and consolidated to produce optimal protocols for treatment, proper clinical setups for patient management, and effective medicines, hopefully including preventative means.
Modeling the spread of the virus to provide predictive tools should consider many aspects. The first aspect to be considered is the evolution in time of the phenomenon: anyway, it does not capture its dynamics, which are not completely described by levels that change in time and thresholds, but need to take into account state changes, that document relations between the conditions of a same individual of the population in different time frames. The analyses reported in References [
2,
3] provide good insights and data for COVID-19. Read et al. [
2] used simulation to evaluate the virus impact on Wuhan and other cities of China. Anastassopoulou et al. [
3] estimate case fatality and recovery ratios using data on Wuhan outbreak and a variant of an SIR model.
Scientific literature largely considered the problem of mathematical models for the description of infectious diseases and epidemiological dynamics. The topic is object of comprehensive analyses and discussions that we recommend to interested readers as general introduction references. For example, Diekmann and Heesterbeek [
4] propose translating biological assumptions into mathematics, focusing on a perspective of mathematical analysis aided by interpretation and about obtaining insight into epidemic phenomena when translating mathematical results back into population biology. Hethcote [
5] presents endemic models with either continuous age or age groups parameters and also considers various diseases. Brauer and Castillo-Chavez [
6] consider simple and analyzable models and unsolvable models to address the population explosion under different conditions. Finally, Cordero et al. [
7] describe a signal transduction pathway involved in a biological process by a detailed representation which results in mathematical models with a lower computational and structural complexity.
As also evidenced by the previous citations, a popular family of models is that of SIR models [
8], which are based on the classification of individuals of the population into categories (the name derives from a classification into three groups: Susceptible, Infected, and Recovered) that can be considered as states between which individuals move according to given probabilities that are inferred by existing statistical data, or more complex mechanisms, including contact networks, which are, anyway, a problem worth of investigation that literature dealt with, as the availability of contact data is generally rare and not fully reliable. In the following, we list some works focused on contact data: Machens et al. [
9] show that a contact matrix that only contains average contact durations between role classes fails to reproduce the size of the epidemic obtained using the high-resolution contact data and also fails to identify the most at-risk classes; Sapiezynski et al. [
10] and Huang et al. [
11] provide multiple types of communication and contact networks, respectively; McCaw et al. [
12] study different mathematical models of infection that are dependent on the assumed mixing patterns of the population; Salathe et al. [
13] analyze infectious diseases in an American college spread by close proximity interactions. Moreover, the Italian government promoted an app for smartphones, namely Immuni [
14], to collect anonymous information about contacts based on Bluetooth proximity, with some minor results, and there is a request for properly organizing the existing data about the pandemics and its treatment in the spirit of the OpenData initiative, to disclose information for community-based research efforts, including network-based modeling. SIR is a family of models that is widely explored and used in literature, and is the base for many variants, such as SIS, after Susceptible, Infected, and Susceptible, SEIR, that adds a Exposed stage, or, for example, the recent SIDARTHE [
1] (Susceptible, Infected, Diagnosed, Ailing, Recognized, Threatened, Healed, and Extinct) and SECIHURD [
15] (Susceptible, Exposed, Carrier, Infected, Hospitalized, in intensive care Unit, Dead, and Recovered) specifically developed for COVID-19. The model described in Reference [
1] deals with data about Italy, uses a larger number of stages for the evolution of COVID-19 to provide a closer representation of the phenomenon, and is a case that represents how researchers evolved the basic idea into different custom variants, while the model described by Großmann et al. [
15] shows how an SIR-like model can be translated into a meanfield model for solution. Another recent example of application of SIR models, using data from China, is provided by Chen et al. [
16], in which interested readers can find a good step-by-step description of the approach, that we found useful to get insights into this modeling technique, while Reference [
8] provides a very good introduction to modeling of epidemic processes in complex networks in general.
An important feature that models can leverage to increase their modeling power and significance with respect to the transmission dynamics of the virus is their accounting for spatial features. In fact, considering all individuals as equally subject to contagion with no respect for their presence in places with a higher or lower concentration of infection vectors, that is, in a densely populated environment rather than in a convenient and prudential isolation condition, going beyond contact networks, increases the realism. The movements of infected subjects on the considered territory and their presence on board of public transportation, or the speed of their movement across the territory, is a relevant aspect to be considered: the spread of infection in Italy during the first phase was largely asymmetric across different regions because contagion was concentrated in northern areas and traveling was controlled, but a sudden and early announcement of regional lockdown caused southern people working in northern regions to assault the last southbound trains, paving the way for a rise of infection cases in the south, as well. Please note that regions in Italy have local autonomy and administrative powers, that include the possibility of limitations of free movements between regions and even smaller administrative subdivisions: in the most critical phase of the lockdown, people was forbidden to exit the borders of their municipalities without a certificated need.
Spatial models may account for these factors. Spatial models consider locality when describing the probability of virus transmission, and describe subjects as agents that may move from one place to another, may get in contact in different places with different agents density, or may be subject to a different exposition to the virus in places that represent situations in which higher or lower risk mitigation actions are taken. Considering agents evolution in time and space and their actual contacts (that is, coexistence in the same place in the same time) may provide a more realistic evaluation of specific behaviors proper of different agents classes (e.g., different categories of workers, different perception of the need for respecting the rules, different requirements in visiting some specific places of the model, different frequency of presence or persistence in specific places). In this paper, we adopt a modeling technique that has spatial features, namely Markovian Agents [
17], that well matches SIR-like models: first of all, it is based on agents that stochastically evolve between states coherently with the SIR logic, in which compartments can be associated to the agent model states. Furthermore, individual behaviors, which are affected by global interactions with other individuals and time-related location-dependent events, are well suited to be defined with a parametric grain of the interactions in the space (an example of such an application of the modeling technique is provided by Gribaudo et al. [
18]). To highlight the contribution of this work to the literature, please refer to the works presented in References [
1,
15]. The former take into account the Italian contagion as we did in our work, while the latter develop an approach starting from the mean field analysis that we have adopted as a solution technique to calculate the results we presented. A relevant difference is that our study is able to consider the spatial location of the population and, in particular, the impact of the infection among the Italian regions, beyond the overall national evolution of the pandemic presented in Reference [
1] that our model can also represent. Furthermore, the goal of Großmann et al. [
15] is to point out that stochastic models offer a more reliable epidemic prediction tool than deterministic ones. We exploit a formalism, namely the Markovian agent-based models, and the provided results show a good fit to the real data provided by the government.
The aim of this work was to show how these formalisms, as agent-based Markov models, are suitable for describing and evaluating systems characterised by enormous quantities of entities, such as countries where the population is affected by a pandemic. In this case, the evolution of the COVID-19 epidemic was studied with the support of real data to understand the applicability of such approaches.
2. Materials and Methods
To prove the increase potential of spatial-based modeling in cases, like the study of the evolution of COVID-19, we start from the extended SIR model presented in Reference [
15], and we tailor it to match the measurements available in Reference [
19]. In particular, we consider the following states for each individual:
S—Susceptible: the individual has never been in contact with the virus and can still be infected.
I—Infected: the individual has been infected by COVID-19. However, she is still asymptomatic and remains undetected showing no evidence of the infection. An individual in this state can infect other people and can either be recognized as infected, recover autonomously, or die without knowing that his death was caused by the virus.
Q—Home treatment: the individual has been detected as infected, but he/she is still in a condition that can be managed from his/her own home.
H—Hospitalized: the health conditions become more severe, requiring hospitalization.
C—Intensive Care: the health conditions are critical, requiring intensive care treatment.
R and R′—Recovered: the individual has been recognized as recovered, and he/she is no longer in danger or contagious. Measurements only account for recovered individuals that have been detected. To match available data, we split the recovered state into two: the first (R) accounts for the recovery of individual who were discovered, while the second (R′) considers the recovered individuals which remained undetected.
D and D′—Dead: the individual who died due to the virus. In addition, in this case, the state is split into two, to consider both the ones that have been detected (state D) and the ones that died before their infection was discovered (state D′).
The main purpose of this model is to match data that we can find in the public available data set, which report the evolution of the number of people in states Q, H, C, R, and D for each day of the evolution of the spread of the disease.
The proposed model can be directly mapped on the Markovian Agent shown in
Figure 1. Each state of the SIR model is directly mapped into an agent state. Agents are further divided into classes, all with the same structure shown in
Figure 1 but with different parameters: in particular, we used one agent class for each Italian region. Note that transitions among states are determined by specific actions that are indicated in the diagram by arcs with different colors; the text in the figure shows the association action/arc color.
The transition rate from state
S to state
I is proportional to the total number of infected people the agent might come by, which includes both his local neighbors, plus agents in other regions that might come in contact with him. In particular, let us focus on a given region
c. We call
the transition rate from state
S to state
I at time
t, and we defined it as:
where parameter
is a location-independent, but time-dependent, parameter that accounts for the infection rate. We assume it to be independent from the location, since it is connected to the specific infectiveness of the considered virus, which we assume being constant over all the considered territory. Instead, we make it time-dependent, to include the reduction of exposure due to countermeasures applied by social distancing and hygiene policies.
represents the total number of individual which might become in contact with the considered agent in location
c at time
t: we will return on its definition later. Let us define the following rates:
the rate at which an infected person gets recognized,
the death rate, and
the rate at which an individual recovers. Moreover, a recognized agent can be cared for at home with probability
, hospitalized with probability
, and requiring intensive care with probability
. The transition rates out of the infected state
I, leading to states
R′ (recovered without having been discovered),
D′ (death without having been discovered),
Q (home treatment),
H (Hospitalized), and
C (requiring intensive care), can be, respectively, defined as follows:
Rates
and
and probabilities
,
, and
have been considered time-dependent to model the improvements in the treatment that reduces the recovery time and chance of death as time passes. Rates
and
are also used to describe the recovery and death from states different from
I, i.e.,
Worsening of the disease is modeled with time dependent parameters
and
, by defining jump rates from state
Q (home treatment) to either
H or
C (hospitalized or intensive care), and from
H to
C, as follows:
The key variable of this model is
: the total number of individual which might become in contact with the considered agent. If we call
the number of agents in state
S of region
c at time
t, we used:
Equation (
12) accounts for the fact that an agent can be infected if it comes in contact with an agent in states
I,
Q,
H, or
C. Coefficient
is used to give a different weight to agents that are known to be infected since they are either in states
Q,
H, or
C. Factor
accounts also for agents in other regions, according to a weight
, which depends on the considered region
c and each neighbor
d. In particular,
accounts for the agents in the same area, while
as neighbor regions become more distant. This weight
is also time dependent to model the effect of confinement and lock-down policies.
2.1. Parameter Definition
We have focused on eight Italian regions and determined the model parameters with either a fitting procedure on the data available in the local civil protection service or from public national databases. Note that, by fitting the parameters with data sets of other countries, the considered model may be used for different geographical areas.
In the following, we will present the obtained values, and the fitting procedure will be explained at the end of this section. The considered regions, namely Piemonte (denoted with PI), Valle d’Aosta (VA), Lombardia (LO), Trentino Alto Adige (TA), Veneto (VE), Friuli (FR), Liguria (LI), and Emilia Romagna (ER), are the ones who played a very important role in the beginning of the virus spreading process. We started the analysis on 24 February 2020 on day 0, and then we modulated the parameters of the model according to the different time phases foreseen by the Italian government, reported in
Table 1.
As introduced in Equation (
1), infection rate parameter
is time dependent and, in particular, its values vary according to the considered time phases.
Table 1 illustrates the temporal values obtained by the fitting procedure described below.
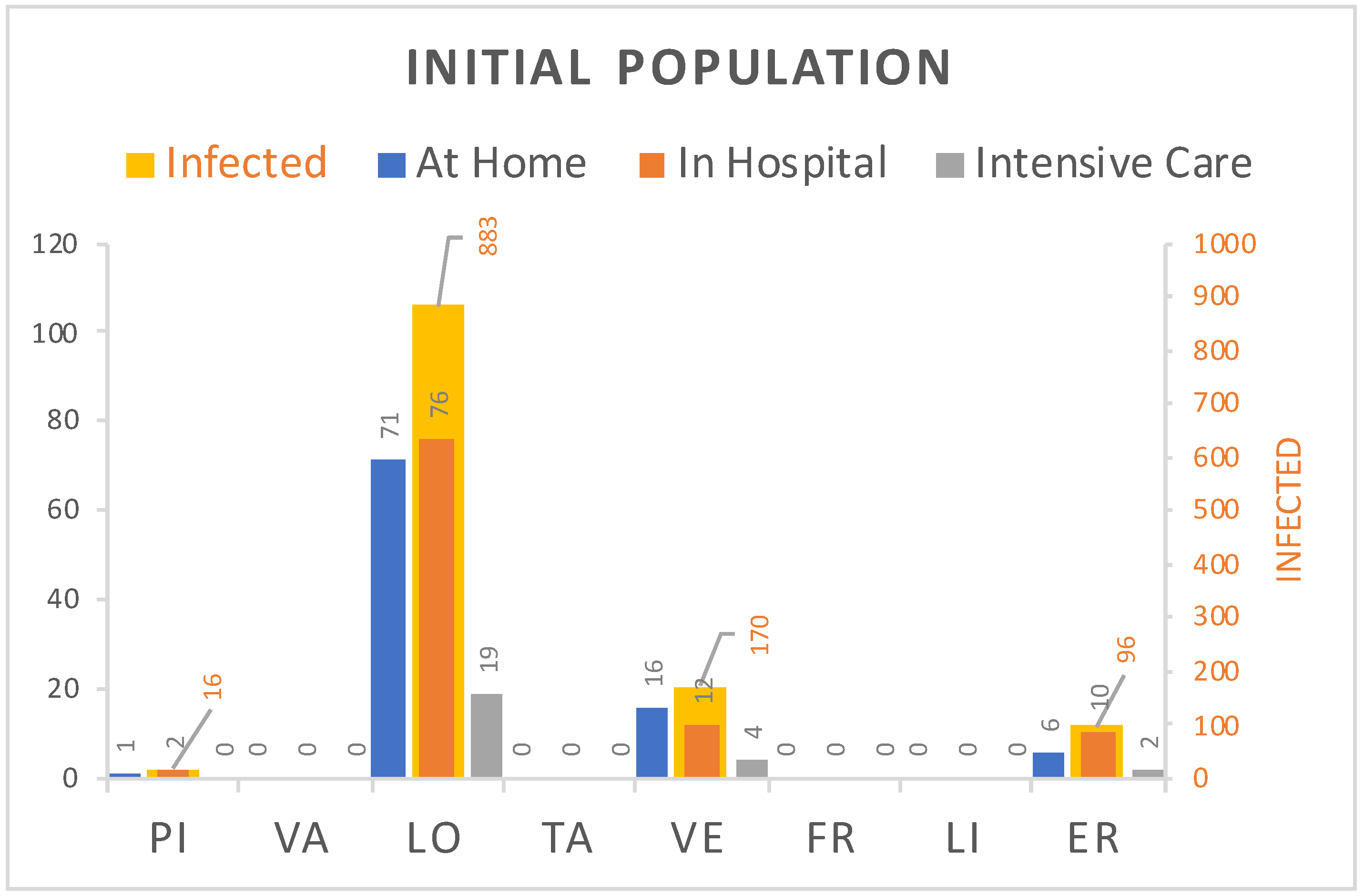
Figure 2 shows the estimated initial number of contagions for each region, including the no-diagnosed (yellow bar labeled with the value), and also the diagnosed divided in home treated (blue), hospitalized (orange), and in intensive care (gray). The diagnosed are taken from Reference [
19], while the no-diagnosed are computed by multiplying the number of diagnosed with a coefficient
determined with fitting. In particular, we have set it to
. Note that this is not too different to the value 6, which was supposed in most of the literature, e.g., Reference [
20]. The initial number of susceptible has been derived subtracting the number of infected individuals accounted in the other states from the number of inhabitants of the region, reported in official documents and shown in
Table 2.
As mentioned above, contagion in a given region
c also counts for agents in neighboring regions
d according to a weight of
. We have obtained these weights as the product of two factors:
where
considers the geographical distance, and, in some cases, we have set them by fitting to take into account particular correlations, as for LO, which in fact strongly determined the spread of the virus in the neighboring regions. The resulting coefficients
are reported in
Table 3. The effect of lockdown is modeled by
and is set to 1 in time phases without lockdown, and to
otherwise.
By using the fitting technique which will be introduced in the next section, we also define the following parameters. The rate at which an infected person gets recognized has been set to . This speed is modulated by two sets of probabilities depending on the date: values , , and are used to determine if diagnosed contagions become home treated, hospitalized, or in intensive care before the lockdown, and , , and are used for the same cases later.
Rates that account for the disease worsening, i.e., and , have been, respectively, set to and before the lockdown, and to and later. The first parameter, , defines the speed at which home treated become hospitalized, and the second one, , accounts for both home treated and hospitalized going in intensive care.
The recovery rate and death rate depend on time to consider the changes in the logistical situation of the hospitals and the improvement in medical care that occurred during the pandemic. has been set to from day zero to the beginning of the Italian Lockdown, then to until Phase Two started, and, finally, to . Death rate has been set to until the beginning of the Italian Lockdown, and to until the end of analysis.
2.2. Parameter Fitting
To show the potentiality of the spatial agent-based modeling formalism, we fit the parameters with the measurements available from the Italian Civil Protection. We used a custom genetic algorithm implemented in MathWork’s MATLAB R2020a Update 3 (May 27 2020), which focuses on a single time-varying parameter at a time. We used the time instants defined in
Table 1 and considered each interval
(for a total of
intervals). For each parameter
, we identified which state
y and region
c were more influenced by it, as well as minimized the distance between real measures
and model prediction
at the end of considered interval
. Algorithm 1 shows a simplified version of the procedure used.
| Algorithm 1 Fit parameter against real measures using reference times |
| Set current error |
| for k=1...MaxIterations do |
| for do |
| Set a random perturbation of : |
| |
| Solve the model with |
| |
| if then |
| , |
| end if |
| end for |
| end for |
We run several executions of the algorithm to perform a random restart, to avoid that the procedure may converge to a local minimum. In particular, we restarted the algorithm 10 times: in each execution, setting was sufficient to find a good set of values. The random perturbation was performed by multiplying the previous value of the parameter with a uniformly distributed value in a range with and , and a constraint to keep the resulting value in an admissible range to avoid finding results that are too far from the previous versions.
4. Discussion
The model we developed simulates the agents evolution in time and space, and it allowed us to represent a realistic scenario where the probability of a virus transmission in different places can be considered. The proposed technique is based on mean field analysis that provides a more precise evaluation for the dynamics of a large population of agents as in the case of a pandemic. We computed the spread of the COVID-19 for eight Italian regions taking into account the contagion among all their populations (see
Figure 3); however, the model can be easily extended to consider all twenty-one regions. Moreover, the contagion in the whole country was also simulated (see
Figure 5). All results present a good accuracy when compared with the available information, as it can be seen in
Figure 4, where the average relative error of the model results against the real life data is plotted.
This approach could lead to new experiments in which the effect of vaccine inoculation can be analyzed both locally and nationwide, and it can also be used to observe the impact of outbreaks.
Simulations were conducted solving a Markovian Agent model, which represents a system as a collection of Markovian Agents (MAs) spread over a geographical space which, in this case, corresponds to the set of Italian regions with . Each MA is described by a Continuous Time Markov Chain, in which an infinitesimal generator describes the transition rates among N different states.
Agents are analyzed by studying the evolution of their
counting process and exploiting mean field approximation [
21,
22]. In particular, the average number agents in state
s for a region
at time
t is denoted as
. To simplify the notation, let us collect the terms into row vectors
that represent the state distribution of a MA belonging to region
c at time
t. The dynamic evolution of the average number of agents can be computed solving the system of ordinary differential equations in (
14):
Here, the elements of matrix may depend on the count of the population of region c, and of other neighbor regions, as well as from time t. In particular, represents the transition rate from state i to state j in a region c at time t.
The considered scenario leads to a small system of well conditioned differential equations, where N is the number of states and K the number of regions. We performed its solution in MATLAB using ode45(...) solver, and it required only few milliseconds.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}