
Dopamine, Psychosis, and Symptom Fluctuation: A Narrative Review



{kind=link}
Abstract
:1. Introduction
2. Method
3. Molecular Pathways Underlying Schizophrenia Pathology
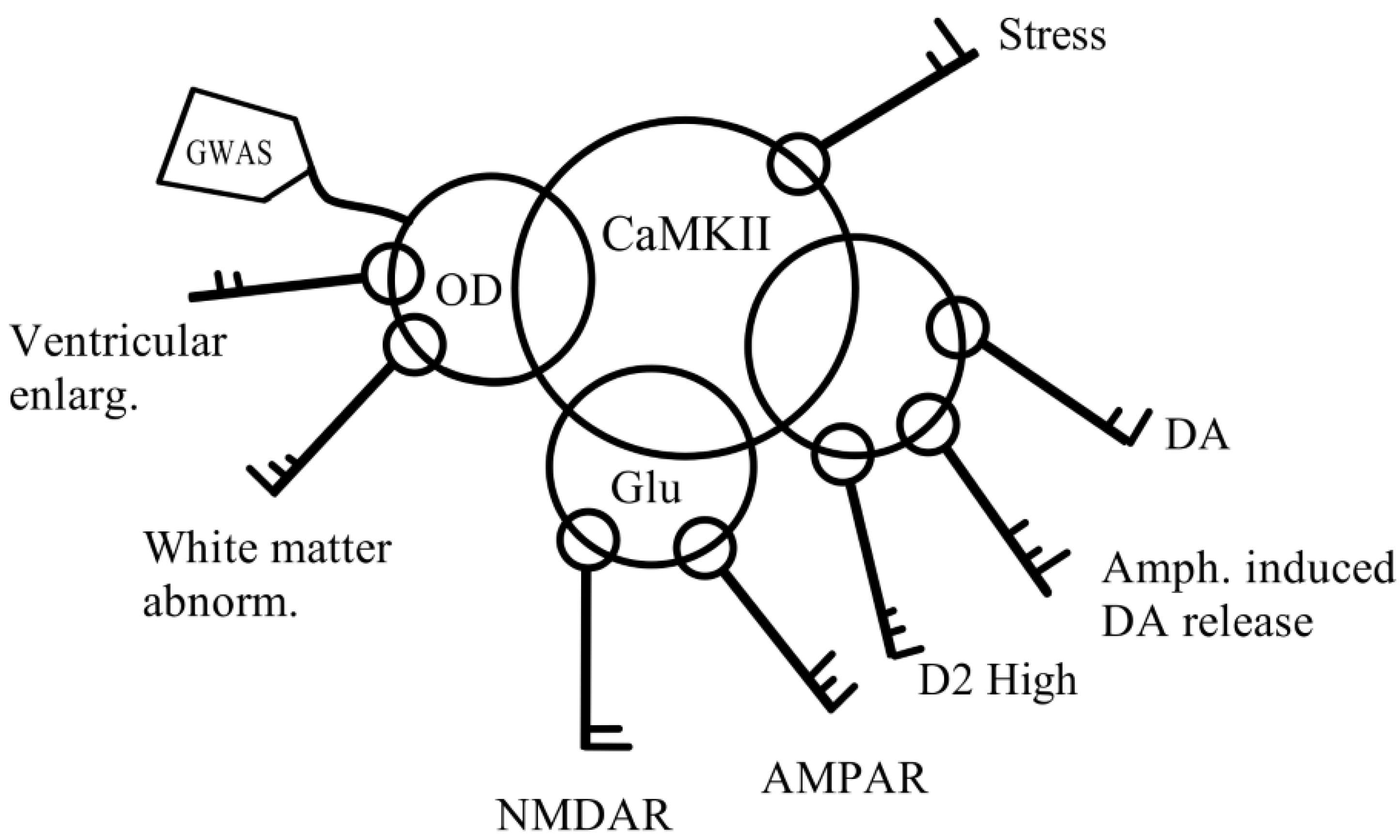
3.1. Dopamine and Schizophrenia
3.2. Schizophrenia Is a Result of the Synergistic Interaction between Genetic and Environmental Factors
3.3. The Involvement of the Striatum in Schizophrenia Pathology
3.4. Neurodevelopmental Process of the Striatum
3.5. Molecular Pathways Linking Schizophrenia Symptoms and Stress
4. Clinical Manifestations: Symptom Checks and Triggers as Evidenced by Patients
4.1. Schizophrenia Symptoms Fluctuate
4.2. Social Engagement
4.3. Role of Dopamine in Social Affiliation
4.4. Motor Activity
4.5. Dopamine and Exercise
4.6. Effects of Sleep
4.7. Effects of Distress
4.8. Dopamine in Sleep
4.9. Substance Use
4.10. Dopamine and Substance Use
5. Discussion
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMPAR | AMPA receptor |
| CaMKII | calcium/calmodulin-dependent protein kinase II |
| DA | dopamine |
| DAT | dopamine transporters (DAT) |
| DR | dopamine receptor |
| D2high | dopamine 2 receptor in high-affinity state |
| D2R | dopamine receptor type 2, or dopamine 2 receptor |
| EMA | ecological momentary assessments |
| GABA | gamma-aminobutyric acid |
| GABAR | GABA receptor |
| GWAS | genome-wide association studies |
| HVA | homovanillic acid |
| LOC | locus of control |
| MRI | magnetic resonance imaging |
| NMDA | N-methyl-D-aspartate |
| NMDAR | NMDA receptor |
| Nogo/RTN4 | Neurite outgrowth inhibitor/reticulon 4 |
| ODs | oligodendrocytes |
| PET | positron emission tomography |
| PFC | prefrontal cortex |
| SCN | suprachiasmatic nuclei |
| VTA | ventral tegmental area |
References
- Seeman, P. All roads to schizophrenia lead to dopamine supersensitivity and elevated dopamine d2high receptors. CNS Neurosci. Ther. 2011, 17, 118–132. [Google Scholar] [CrossRef]
- Hu, W.; Macdonald, M.L.; Elswick, D.E.; Sweet, R.A. The glutamate hypothesis of schizophrenia: Evidence from human brain tissue studies. Ann. N. Y. Acad. Sci. 2015, 1338, 38–57. [Google Scholar] [CrossRef]
- Emüller, N.; Eweidinger, E.; Eleitner, B.; Schwarz, M.J. The role of inflammation in schizophrenia. Front. Neurosci. 2015, 9, 372. [Google Scholar] [CrossRef]
- Murray, R.M.; Bhavsar, V.; Tripoli, G.; Howes, O. 30 years on: How the neurodevelopmental hypothesis of schizophrenia morphed into the developmental risk factor model of psychosis. Schizophr. Bull. 2017, 43, 1190–1196. [Google Scholar] [CrossRef]
- Schultze-Lutter, F. Subjective symptoms of schizophrenia in research and the clinic: The basic symptom concept. Schizophr. Bull. 2009, 35, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Bell, I.H.; Fielding-Smith, S.F.; Hayward, M.; Rossell, S.L.; Lim, M.H.; Farhall, J.; Thomas, N. Smartphone-based ecological momentary assessment and intervention in a blended coping-focused therapy for distressing voices: Development and case illustration. Internet Interv. 2018, 14, 18–25. [Google Scholar] [CrossRef]
- Myin-Germeys, I.; Kasanova, Z.; Vaessen, T.; Vachon, H.; Kirtley, O.; Viechtbauer, W.; Reininghaus, U. Experience sampling methodology in mental health research: New insights and technical developments. World Psychiatry 2018, 17, 123–132. [Google Scholar] [CrossRef]
- Oorschot, M.; Kwapil, T.; Delespaul, P.; Myin-Germeys, I. Momentary assessment research in psychosis. Psychol. Assess. 2009, 21, 498–505. [Google Scholar] [CrossRef]
- Myin-Germeys, I.; Marcelis, M.; Krabbendam, L.; Delespaul, P.; van Os, J. Subtle fluctuations in psychotic phenomena as functional states of abnormal dopamine reactivity in individuals at risk. Biol. Psychiatry 2005, 58, 105–110. [Google Scholar] [CrossRef]
- Novak, G. Upregulation of CaMKIIβ and Nogo-C mRNA in Schizophrenia and the Prevalence of CAA Insert in the 3′UTR of the Nogo Gene. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 2008. Available online: http://hdl.handle.net/1807/11240 (accessed on 6 September 2022).
- Fornaro, M.; Clementi, N.; Fornaro, P. Medicine and psychiatry in Western culture: Ancient Greek myths and modern prejudices. Ann. Gen. Psychiatry 2009, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clough, P.W. Reserpine in the treatment of neuropsychiatric disorders. Ann. Intern. Med. 1955, 43, 632–637. [Google Scholar] [CrossRef]
- Delay, J.; Deniker, P. Chlorpromazine and neuroleptic treatments in psychiatry. J. Clin. Exp. Psychopathol. 1956, 17, 19–24. [Google Scholar] [PubMed]
- Casey, J.F.; Lasky, J.J.; Klett, C.J.; Hollister, L.E. Treatment of schizophrenic reactions with phenothiazine derivatives. A comparative study of chlorpromazine, triflupromazine, mepazine, prochlorperazine, perphenazine, and phenobarbital. Am. J. Psychiatry 1960, 117, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Jahanshad, N.; Zalesky, A.; Kochunov, P.; Agartz, I.; Alloza, C.; Andreassen, O.; Arango, C.; Banaj, N.; Bouix, S.; et al. Widespread white matter microstructural differences in schizophrenia across 4322 individuals: Results from the ENIGMA Schizophrenia DTI Working Group. Mol. Psychiatry 2018, 23, 1261–1269. [Google Scholar] [CrossRef]
- Carlsson, A. A half-century of neurotransmitter research: Impact on neurology and psychiatry (Nobel lecture). ChemBioChem 2001, 2, 484–493. [Google Scholar] [CrossRef]
- van Rossum, J.M. The significance of dopamine-receptor blockade for the mechanism of action of neuroleptic drugs. Arch. Int. Pharmacodyn. Ther. 1966, 160, 492–494. [Google Scholar]
- Seeman, P.; Lee, T. Antipsychotic drugs: Direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 1975, 188, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Lee, T.; Chau-Wong, M.; Wong, K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature 1976, 261, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Rummel-Kluge, C.; Komossa, K.; Schwarz, S.; Hunger, H.; Schmid, F.; Kissling, W.; Davis, J.M.; Leucht, S. Second-generation antipsychotic drugs and extrapyramidal side effects: A systematic review and meta-analysis of head-to-head comparisons. Schizophr. Bull. 2012, 38, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Tamminga, C.A. Assessing striatal dopamine in schizophrenia. Biol. Psychiatry 2022, 91, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Cardno, A.G.; Marshall, E.J.; Coid, B.; Macdonald, A.M.; Ribchester, T.R.; Davies, N.J.; Venturi, P.; Jones, L.A.; Lewis, S.; Sham, P.C.; et al. Heritability estimates for psychotic disorders. Arch. Gen. Psychiatry 1999, 56, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Hilker, R.; Helenius, D.; Fagerlund, B.; Skytthe, A.; Christensen, K.; Werge, T.M.; Nordentoft, M.; Glenthøj, B. Heritability of schizophrenia and schizophrenia spectrum based on the nationwide danish twin register. Biol. Psychiatry 2018, 83, 492–498. [Google Scholar] [CrossRef]
- Marceau, K.; McMaster, M.T.B.; Smith, T.F.; Daams, J.G.; van Beijsterveldt, C.E.M.; Boomsma, D.I.; Knopik, V.S. The Prenatal Environment in Twin Studies: A Review on Chorionicity. Behav. Genet. 2016, 46, 286–303. [Google Scholar] [CrossRef] [PubMed]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kang, W.S.; Kim, J.W. Association between the promoter haplotype of RTN4 gene and schizophrenia in a Korean population. Psychiatry Res. 2021, 299, 113841. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.; Kim, D.; Seeman, P.; Tallerico, T. Schizophrenia and Nogo: Elevated mRNA in cortex, and high prevalence of a homozygous CAA insert. Mol. Brain Res. 2002, 107, 183–189. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Remington, L.; Maruschak, B.; Owens, T.; Brück, W. Nogo-A is a reliable oligodendroglial marker in adult human and mouse cns and in demyelinated lesions. J. Neuropathol. Exp. Neurol. 2007, 66, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Thornton, M.A.; Hughes, E.G. Neuron-oligodendroglia interactions: Activity-dependent regulation of cellular signaling. Neurosci. Lett. 2020, 727, 134916. [Google Scholar] [CrossRef] [PubMed]
- Cardno, A.G.; Gottesman, I.I. Twin studies of schizophrenia: From bow-and-arrow concordances to star wars Mx and functional genomics. Am. J. Med. Genet. 2000, 97, 12–17. [Google Scholar] [CrossRef]
- Welham, J.; Isohanni, M.; Jones, P.; McGrath, J. The antecedents of Schizophrenia: A review of birth cohort studies. Schizophr. Bull. 2009, 35, 603–623. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.N.; Frantom, L.V.; Strauss, G.P.; van Kammen, D.P. Differential patterns of premorbid academic and social deterioration in patients with schizophrenia. Schizophr. Res. 2005, 75, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Tarbox, S.I.; Pogue-Geile, M.F. Development of social functioning in preschizophrenia children and adolescents: A systematic review. Psychol. Bull. 2008, 134, 561–583. [Google Scholar] [CrossRef]
- Khandaker, G.; Barnett, J.H.; White, I.; Jones, P. A quantitative meta-analysis of population-based studies of premorbid intelligence and schizophrenia. Schizophr. Res. 2011, 132, 220–227. [Google Scholar] [CrossRef]
- Davis, K.L.; Stewart, D.G.; Friedman, J.I.; Buchsbaum, M.; Harvey, P.D.; Hof, P.R.; Buxbaum, J.; Haroutunian, V. White matter changes in schizophrenia. Arch. Gen. Psychiatry 2003, 60, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Bagary, M.S.; Symms, M.R.; Barker, G.J.; Mutsatsa, S.H.; Joyce, E.M.; Ron, M.A. Gray and white matter brain abnormalities in first-episode schizophrenia inferred from magnetization transfer imaging. Arch. Gen. Psychiatry 2003, 60, 779–788. [Google Scholar] [CrossRef]
- Roberts, G.W. Schizophrenia: The cellular biology of a functional psychosis. Trends Neurosci. 1990, 13, 207–211. [Google Scholar] [CrossRef]
- Breier, A.; Su, T.-P.; Saunders, R.; Carson, R.E.; Kolachana, B.S.; de Bartolomeis, A.; Weinberger, D.R.; Weisenfeld, N.; Malhotra, A.K.; Eckelman, W.C.; et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: Evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. USA 1997, 94, 2569–2574. [Google Scholar] [CrossRef]
- Laruelle, M. Imaging dopamine transmission in schizophrenia: A review and meta-analysis. Q. J. Nucl. Med. 1998, 42, 211. [Google Scholar] [PubMed]
- Abi-Dargham, A. Dopamine dysfunction in schizophrenia. Schizophr. Res. 2014, 160, e6–e7. [Google Scholar] [CrossRef]
- Weinberger, D.R.; Berman, K.F.; Zec, R.F. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia: I. regional cerebral blood flow evidence. Arch. Gen. Psychiatry 1986, 43, 114–124. [Google Scholar] [CrossRef]
- Farkas, T.; Wolf, A.P.; Jaeger, J.; Brodie, J.D.; Christman, D.R.; Fowler, J.S.; MacGregor, R.R.; Deleon, M.J.; Defina, P.; Goldman, A.; et al. Regional brain glucose metabolism in chronic schizophrenia. Arch. Gen. Psychiatry 1984, 41, 293–300. [Google Scholar] [CrossRef]
- Curran, C.; Byrappa, N.; McBride, A. Stimulant psychosis: Systematic review. Br. J. Psychiatry 2004, 185, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M.; Abi-Dargham, A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J. Psychopharmacol. 1999, 13, 358–371. [Google Scholar] [CrossRef]
- Kegeles, L.S.; Abi-Dargham, A.; Frankle, W.; Gil, R.; Cooper, T.B.; Slifstein, M.; Hwang, D.-R.; Huang, Y.; Haber, S.N.; Laruelle, M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch. Gen. Psychiatry 2010, 67, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.H.; Kellendonk, C. Insights about striatal circuit function and schizophrenia from a mouse model of dopamine d2 receptor upregulation. Biol. Psychiatry 2017, 81, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.H.; Gallo, E.F.; Balsam, P.D.; Javitch, J.A.; Kellendonk, C. How changes in dopamine D2 receptor levels alter striatal circuit function and motivation. Mol. Psychiatry 2022, 27, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Fervaha, G.; Foussias, G.; Agid, O.; Remington, G. Amotivation and functional outcomes in early schizophrenia. Psychiatry Res. 2013, 210, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Fusar-Poli, P.; Papanastasiou, E.; Stahl, D.; Rocchetti, M.; Carpenter, W.; Shergill, S.; McGuire, P. Treatments of negative symptoms in schizophrenia: Meta-analysis of 168 randomized placebo-controlled trials. Schizophr. Bull. 2015, 41, 892–899. [Google Scholar] [CrossRef]
- Goff, D.C.; Hill, M.; Barch, D. The treatment of cognitive impairment in schizophrenia. Pharmacol. Biochem. Behav. 2011, 99, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.H.; Kellendonk, C.; Kandel, E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron 2010, 65, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Haaf, M.; Leicht, G.; Curic, S.; Mulert, C. Glutamatergic deficits in schizophrenia—biomarkers and pharmacological interventions within the ketamine model. Curr. Pharm. Biotechnol. 2018, 19, 293–307. [Google Scholar] [CrossRef]
- Adams, D.H.; Zhang, L.; Millen, B.A.; Kinon, B.J.; Gomez, J.-C. Pomaglumetad methionil (ly2140023 monohydrate) and aripiprazole in patients with schizophrenia: A phase 3, multicenter, double-blind comparison. Schizophr. Res. Treat. 2014, 2014, 758212. [Google Scholar] [CrossRef] [PubMed]
- Bugarski-Kirola, D.; Iwata, N.; Sameljak, S.; Reid, C.; Blaettler, T.; Millar, L.; Marques, T.R.; Garibaldi, G.; Kapur, S. Efficacy and safety of adjunctive bitopertin versus placebo in patients with suboptimally controlled symptoms of schizophrenia treated with antipsychotics: Results from three phase 3, randomised, double-blind, parallel-group, placebo-controlled, multicentre studies in the SearchLyte clinical trial programme. Lancet Psychiatry 2016, 3, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, A.; Jacob, S.N. Doping the mind. Neuroscientist 2016, 22, 593–603. [Google Scholar] [CrossRef]
- Slifstein, M.; Van De Giessen, E.; Van Snellenberg, J.; Thompson, J.L.; Narendran, R.; Gil, R.; Hackett, E.; Girgis, R.; Ojeil, N.; Moore, H.; et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia. JAMA Psychiatry 2015, 72, 316–324. [Google Scholar] [CrossRef]
- Li, Y.-C.; Kellendonk, C.; Simpson, E.H.; Kandel, E.R.; Gao, W.-J. D2 receptor overexpression in the striatum leads to a deficit in inhibitory transmission and dopamine sensitivity in mouse prefrontal cortex. Proc. Natl. Acad. Sci. USA 2011, 108, 12107–12112. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; McCutcheon, R.; Owen, M.J.; Murray, R.M. The role of genes, stress, and dopamine in the development of schizophrenia. Biol. Psychiatry 2017, 81, 9–20. [Google Scholar] [CrossRef]
- Canetta, S.; Kellendonk, C. When time matters: An adolescent intervention to prevent adult brain dysfunction. Cell 2019, 178, 1282–1284. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.; Fan, T.; O’Dowd, B.F.; George, S.R. Striatal development involves a switch in gene expression networks, followed by a myelination event: Implications for neuropsychiatric disease. Synapse 2013, 67, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, B.; Darlington, R.; Finlay, B. Translating developmental time across mammalian species. Neuroscience 2001, 105, 7–17. [Google Scholar] [CrossRef]
- Cheli, V.T.; Correale, J.; Paez, P.M.; Pasquini, J.M. Iron metabolism in oligodendrocytes and astrocytes, implications for myelination and remyelination. ASN Neuro 2020, 12, 1759091420962681. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; Salmina, A.B.; Malinovskaya, N.A.; Noda, M.; Verkhratsky, A.; Moreira, J.C.F. The glial perspective of autism spectrum disorders. Neurosci. Biobehav. Rev. 2014, 38, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Koschnick, J.R.; Slegers, L.H.A.; Hof, P.R. Oligodendrocyte pathophysiology: A new view of schizophrenia. Int. J. Neuro Psychopharmacol. 2007, 10, 503–511. [Google Scholar] [CrossRef]
- Noori, R.; Park, D.; Griffiths, J.D.; Bells, S.; Frankland, P.W.; Mabbott, D.; Lefebvre, J. Activity-dependent myelination: A glial mechanism of oscillatory self-organization in large-scale brain networks. Proc. Natl. Acad. Sci. USA 2020, 117, 13227–13237. [Google Scholar] [CrossRef] [PubMed]
- Raabe, F.J.; Slapakova, L.; Rossner, M.J.; Cantuti-Castelvetri, L.; Simons, M.; Falkai, P.G.; Schmitt, A. Oligodendrocytes as a new therapeutic target in schizophrenia: From histopathological findings to neuron-oligodendrocyte interaction. Cells 2019, 8, 1496. [Google Scholar] [CrossRef]
- Fields, R.D. White matter in learning, cognition and psychiatric disorders. Trends Neurosci. 2008, 31, 361–370. [Google Scholar] [CrossRef]
- Johnstone, E.; Frith, C.; Crow, T.; Husband, J.; Kreel, L. Cerebral ventricular size and cognitive impairment in chronic schizophrenia. Lancet 1976, 308, 924–926. [Google Scholar] [CrossRef]
- Van Horn, J.D.; McManus, I.C. Ventricular enlargement in schizophrenia. Br. J. Psychiatry 1992, 160, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Kuperberg, G.R.; Broome, M.; McGuire, P.K.; David, A.S.; Eddy, M.; Ozawa, F.; Goff, D.; West, W.C.; Williams, S.C.R.; van der Kouwe, A.; et al. Regionally localized thinning of the cerebral cortex in schizophrenia. Arch. Gen. Psychiatry 2003, 60, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Konopaske, G.T.; Lange, N.; Coyle, J.T.; Benes, F.M. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 2014, 71, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Selemon, L.D.; Rajkowska, G.; Goldman-Rakic, P.S. Abnormally high neuronal density in the schizophrenic cortex. Arch. Gen. Psychiatry 1995, 52, 805–818. [Google Scholar] [CrossRef]
- Vostrikov, V.M.; Uranova, N.A.; Orlovskaya, D.D. Deficit of perineuronal oligodendrocytes in the prefrontal cortex in schizophrenia and mood disorders. Schizophr. Res. 2007, 94, 273–280. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [PubMed]
- Fink, C.C. Molecular mechanisms of CaMKII activation in neuronal plasticity. Curr. Opin. Neurobiol. 2002, 12, 293–299. [Google Scholar] [CrossRef]
- Yoshimuraa, Y.; Shinkawab, T.; Taoka, M.; Kobayashia, K.; Isobebc, T.; Yamauchia, T. Identification of protein substrates of Ca2+/calmodulin-dependent protein kinase II in the postsynaptic density by protein sequencing and mass spectrometry. Biochem. Biophys. Res. Commun. 2002, 290, 948–954. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Miyamoto, E.; Fukunaga, K. Activation of the rat dopamine D2 receptor promoter by mitogen-activated protein kinase and Ca2+/calmodulin-dependent protein kinase II pathways. J. Neurochem. 2002, 83, 784–796. [Google Scholar] [CrossRef]
- Aleisa, A.; Alzoubi, K.; Gerges, N.; Alkadhi, K. Chronic psychosocial stress-induced impairment of hippocampal LTP: Possible role of BDNF. Neurobiol. Dis. 2006, 22, 453–462. [Google Scholar] [CrossRef]
- Chameau, P.; Qin, Y.; Spijker, S.; Smit, G.; Joëls, M. Glucocorticoids Specifically Enhance L-Type Calcium Current Amplitude and Affect Calcium Channel Subunit Expression in the Mouse Hippocampus. J. Neurophysiol. 2007, 97, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, T.C.; Piedras-Renteria, E.S.; Tsien, R.W. α- and βCaMKII: Inverse Regulation by Neuronal Activity and Opposing Effects on Synaptic Strength. Neuron 2002, 36, 1103–1114. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, W.; Xiong, H.; Hoogenraad, C.; Krugers, H. Stress and excitatory synapses: From health to disease. Neuroscience 2013, 248, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.; Seeman, P.; Tallerico, T. Increased expression of calcium/calmodulin-dependent protein kinase IIβ in frontal cortex in schizophrenia and depression. Synapse 2006, 59, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.; Seeman, P.; Tallerico, T. Schizophrenia: Elevated mRNA for calcium-calmodulin-dependent protein kinase IIβ in frontal cortex. Mol. Brain Res. 2000, 82, 95–100. [Google Scholar] [CrossRef]
- Brocke, L.; Chiang, L.W.; Wagner, P.D.; Schulman, H. Functional implications of the subunit composition of neuronal CaM kinase II. J. Biol. Chem. 1999, 274, 22713–22722. [Google Scholar] [CrossRef]
- Mayford, M.; Wang, J.; Kandel, E.R.; O’Dell, T.J. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell 1995, 81, 891–904. [Google Scholar] [CrossRef]
- Mohanan, A.G.; Gunasekaran, S.; Jacob, R.S.; Omkumar, R.V. Role of Ca2+/Calmodulin-Dependent Protein Kinase Type II in Mediating Function and Dysfunction at Glutamatergic Synapses. Front. Mol. Neurosci. 2022, 15, 855752. [Google Scholar] [CrossRef]
- Ben-Ari, Y. GABAA, NMDA and AMPA receptors: A developmentally regulated ‘ménage à trois’. Trends Neurosci. 1997, 20, 523–529. [Google Scholar] [CrossRef]
- Novak, G.; Fan, T.; O’Dowd, B.F.; George, S.R. Postnatal maternal deprivation and pubertal stress have additive effects on dopamine D2 receptor and CaMKII beta expression in the striatum. Int. J. Dev. Neurosci. 2013, 31, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-E. Impairing the amphetamine conditioning in rats through the inhibition of hippocampal calcium/calmodulin-dependent protein kinase II activity. Neuropharmacology 2002, 42, 540–547. [Google Scholar] [CrossRef]
- Kantor, L.; Hewlett, G.H.K.; Gnegy, M.E. Enhanced amphetamine- and K+-mediated dopamine release in rat striatum after repeated amphetamine: Differential requirements for Ca2+- and calmodulin-dependent phosphorylation and synaptic vesicles. J. Neurosci. 1999, 19, 3801–3808. [Google Scholar] [CrossRef] [Green Version]
- Kosten, T.A.; Zhang, X.Y.; Kehoe, P. Chronic neonatal isolation stress enhances cocaine-induced increases in ventral striatal dopamine levels in rat pups. Dev. Brain Res. 2003, 141, 109–116. [Google Scholar] [CrossRef]
- Novak, G.; Seeman, P. Hyperactive mice show elevated D2Highreceptors, a model for schizophrenia: Calcium/calmodulin-dependent kinase II alpha knockouts. Synapse 2010, 64, 794–800. [Google Scholar] [CrossRef]
- Greenstein, R.; Novak, G.; Seeman, P. Amphetamine sensitization elevates CaMKIIß mRNA. Synapse 2007, 61, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Lebek, S.; Plößl, A.; Baier, M.; Mustroph, J.; Tarnowski, D.; Lücht, C.; Schopka, S.; Flörchinger, B.; Schmid, C.; Zausig, Y.; et al. The novel CaMKII inhibitor GS-680 reduces diastolic SR Ca leak and prevents CaMKII-dependent pro-arrhythmic activity. J. Mol. Cell. Cardiol. 2018, 118, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Báthori, N.; Polner, B.; Simor, P. Schizotypy unfolding into the night? Schizotypal traits and daytime psychotic-like experiences predict negative and salient dreams. Schizophr. Res. 2022, 246, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, H.S. Schizophrenia as a Human Process; W.W. Norton: New York, NY, USA, 1974. [Google Scholar]
- Strauss, J.S. Subjective Experiences of Schizophrenia: Toward a New Dynamic Psychiatry--II. Schizophr. Bull. 1989, 15, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Marley, J.A. People Matter: Client-reported interpersonal interaction and its impact on symptoms of schizophrenia. Soc. Work 1998, 43, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Colori, S. My Experience with hallucinations. Schizophr. Bull. 2018, 46, 227–228. [Google Scholar] [CrossRef]
- Delespaul, P.; Devries, M.; Van Os, J. Determinants of occurrence and recovery from hallucinations in daily life. Soc. Psychiatry Psychiatr. Epidemiol. 2002, 37, 97–104. [Google Scholar] [CrossRef]
- Crosier, B.S.; Brian, R.M.; Ben-Zeev, D.; Yellowlees, P.; Chan, S.; Gipson, S.; Teo, A. Using facebook to reach people who experience auditory hallucinations. J. Med. Internet Res. 2016, 18, e160. [Google Scholar] [CrossRef] [Green Version]
- Collip, D.; Oorschot, M.; Thewissen, V.; Van Os, J.; Bentall, R.; Myin-Germeys, I. Social world interactions: How company connects to paranoia. Psychol. Med. 2011, 41, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Husky, M.; Tournier, M.; Swendsen, J.D. Social environments and daily life occurrence of psychotic symptoms. Soc. Psychiatry 2003, 38, 654–661. [Google Scholar] [CrossRef]
- Myin-Germeys, I.; Nicolson, N.A.; Delespaul, P.A.E.G. The context of delusional experiences in the daily life of patients with schizophrenia. Psychol. Med. 2001, 31, 489–498. [Google Scholar] [CrossRef]
- Krijnen, L.J.G.; Lemmers-Jansen, I.L.J.; Fett, A.-K.J.; Krabbendam, L. Benefits of social contact in individuals with psychotic symptoms: Do closeness of the contact and empathic skills make the difference? Front. Psychol. 2021, 12, 769091. [Google Scholar] [CrossRef]
- Leonhardt, B.L.; Kukla, M.; Belanger, E.; Chaudoin-Patzoldt, K.A.; Buck, K.D.; Minor, K.S.; Vohs, J.L.; Hamm, J.A.; Lysaker, P.H. Emergence of psychotic content in psychotherapy: An exploratory qualitative analysis of content, process, and therapist variables in a single case study. Psychother. Res. 2018, 28, 264–280. [Google Scholar] [CrossRef] [PubMed]
- Badal, V.D.; Parrish, E.M.; Holden, J.L.; Depp, C.A.; Granholm, E. Dynamic contextual influences on social motivation and behavior in schizophrenia: A case-control network analysis. NPJ Schizophr. 2021, 7, 62. [Google Scholar] [CrossRef]
- Ludwig, L.; Mehl, S.; Krkovic, K.; Lincoln, T.M. Effectiveness of emotion regulation in daily life in individuals with psychosis and nonclinical controls—An experience-sampling study. J. Abnorm. Psychol. 2020, 129, 408–421. [Google Scholar] [CrossRef]
- Galliot, G.; Gozé, T. Schizophrénie et rétablissement: Analyse phénoménologique d’un cas de retrait positif. Ann. Médico-Psychol. Rev. Psychiatr. 2021, 179, 401–408. [Google Scholar] [CrossRef]
- Seeman, M.V. Solitude and schizophrenia. Psychosis 2017, 9, 176–183. [Google Scholar] [CrossRef]
- Fett, A.-K.J.; Hanssen, E.; Eemers, M.; Peters, E.; Shergill, S.S. Social isolation and psychosis: An investigation of social interactions and paranoia in daily life. Eur. Arch. Psychiatry Clin. Neurosci. 2022, 272, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Schlier, B.; Winkler, K.; Jaya, E.S.; Lincoln, T.M. Fluctuations in hallucination spectrum experiences co-vary with social defeat but not with social deafferentation. A 3-week daily assessment study. Cogn. Ther. Res. 2018, 42, 92–102. [Google Scholar] [CrossRef]
- Deserno, L.; Schlagenhauf, F.; Heinz, A. Striatal dopamine, reward, and decision making in schizophrenia. Dialog. Clin. Neurosci. 2016, 18, 77–89. [Google Scholar] [CrossRef]
- Goodwin, N.; Lopez, S.A.; Lee, N.S.; Beery, A.K. Comparative role of reward in long-term peer and mate relationships in voles. Horm. Behav. 2019, 111, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Bariselli, S.; Hörnberg, H.; Prévost-Solié, C.; Musardo, S.; Hatstatt-Burklé, L.; Scheiffele, P.; Bellone, C. Role of VTA dopamine neurons and neuroligin 3 in sociability traits related to nonfamiliar conspecific interaction. Nat. Commun. 2018, 9, 3173. [Google Scholar] [CrossRef] [PubMed]
- Solié, C.; Girard, B.; Righetti, B.; Tapparel, M.; Bellone, C. VTA dopamine neuron activity encodes social interaction and promotes reinforcement learning through social prediction error. Nat. Neurosci. 2022, 25, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.F.; Soares, M.C.; Taborsky, M. Dopamine modulates social behaviour in cooperatively breeding fish. Mol. Cell. Endocrinol. 2022, 550, 111649. [Google Scholar] [CrossRef]
- Atzil, S.; Touroutoglou, A.; Rudy, T.; Salcedo, S.; Feldman, R.; Hooker, J.M.; Dickerson, B.C.; Catana, C.; Barrett, L.F. Dopamine in the medial amygdala network mediates human bonding. Proc. Natl. Acad. Sci. USA 2017, 114, 2361–2366. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, S.; Hedman-Lagerlöf, E.; Ikoma, Y.; Djurfeldt, D.; Rück, C.; Halldin, C.; Lindefors, N. Changes in dopamine D2-receptor binding are associated to symptom reduction after psychotherapy in social anxiety disorder. Transl. Psychiatry 2012, 2, e120. [Google Scholar] [CrossRef] [PubMed]
- Plavén-Sigray, P.; Hedman, E.; Victorsson, P.; Matheson, G.J.; Forsberg, A.; Djurfeldt, D.R.; Rück, C.; Halldin, C.; Lindefors, N.; Cervenka, S. Extrastriatal dopamine D2-receptor availability in social anxiety disorder. Eur. Neuro Psychopharmacol. 2017, 27, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Gellner, A.-K.; Voelter, J.; Schmidt, U.; Beins, E.C.; Stein, V.; Philipsen, A.; Hurlemann, R. Molecular and neurocircuitry mechanisms of social avoidance. Cell Mol. Life Sci. 2020, 78, 1163–1189. [Google Scholar] [CrossRef] [PubMed]
- Kimhy, D.; Tay, C.; Vakhrusheva, J.; Beck-Felts, K.; Ospina, L.H.; Ifrah, C.; Parvaz, M.; Gross, J.J.; Bartels, M.N. Enhancement of aerobic fitness improves social functioning in individuals with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 367–376. [Google Scholar] [CrossRef]
- Kimhy, D.; Vakhrusheva, J.; Bartels, M.N.; Armstrong, H.F.; Ballon, J.S.; Khan, S.; Chang, R.W.; Hansen, M.C.; Ayanruoh, L.; Lister, A.; et al. The impact of aerobic exercise on brain-derived neurotrophic factor and neurocognition in individuals with schizophrenia: A single-blind, randomized clinical trial. Schizophr. Bull. 2015, 41, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Chamove, A.S. Positive short-term effects of activity on behaviour in chronic schizophrenic patients. Br. J. Clin. Psychol. 1986, 25, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Dauwan, M.; Begemann, M.J.H.; Heringa, S.M.; Sommer, I. Exercise improves clinical symptoms, quality of life, global functioning, and depression in schizophrenia: A systematic review and meta-analysis. Schizophr. Bull. 2016, 42, 588–599. [Google Scholar] [CrossRef]
- Mittal, V.A.; Vargas, T.; Osborne, K.J.; Dean, D.; Gupta, T.; Ristanovic, I.; Hooker, C.I.; Shankman, S.A. Exercise treatments for psychosis: A review. Curr. Treat. Options Psychiatry 2017, 4, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, Y.; Fang, Y.; He, Y.; Wang, X.; So, B.C.L.; Shum, D.H.K.; Yan, C. The effect of physical activity on anhedonia in individuals with depressive symptoms. PsyCh J. 2022, 11, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.J.; Merwin, R.M. The role of exercise in management of mental health disorders: An integrative review. Annu. Rev. Med. 2021, 72, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Girdler, S.J.; Confino, J.E.; Woesner, M.E. Exercise as a Treatment for Schizophrenia: A Review. Psychopharmacol. Bull. 2019, 49, 56–69. [Google Scholar] [PubMed]
- Maurus, I.; Hasan, A.; Röh, A.; Takahashi, S.; Rauchmann, B.; Keeser, D.; Malchow, B.; Schmitt, A.; Falkai, P. Neurobiological effects of aerobic exercise, with a focus on patients with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.; Marconcin, P.; Werneck, A.; Ferrari, G.; Gouveia, É.R.; Kliegel, M.; Peralta, M.; Ihle, A. Bidirectional association between physical activity and dopamine across adulthood—A systematic review. Brain Sci. 2021, 11, 829. [Google Scholar] [CrossRef]
- Andorko, N.D.; Mittal, V.; Thompson, E.; Denenny, D.; Epstein, G.; Demro, C.; Wilson, C.; Sun, S.; Klingaman, E.A.; DeVylder, J.; et al. The association between sleep dysfunction and psychosis-like experiences among college students. Psychiatry Res. 2017, 248, 6–12. [Google Scholar] [CrossRef]
- Thompson, E.C.; Jay, S.Y.; Andorko, N.D.; Millman, Z.B.; Rouhakhtar, P.R.; Sagun, K.; Han, S.C.; Herman, B.; Schiffman, J. Sleep quality moderates the association between psychotic-like experiences and suicidal ideation among help-seeking university students. Psychiatry Res. 2021, 296, 113668. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.; Hanna, D.; Davidson, S.; Shannon, C.; Mulholland, C. The association between sleep quality and attenuated psychotic symptoms. Early Interv. Psychiatry 2021, 15, 837–848. [Google Scholar] [CrossRef]
- Andorko, N.D. Stress or sleep: Using Ecological Momentary Assessment Methods to Examine the Pathway to Daily Psychotic Experiences within an Undergraduate Sample. Ph.D. Thesis, University of Maryland, College Park, MD, USA, 2020. [Google Scholar]
- Kimhy, D.; Wall, M.M.; Hansen, M.C.; Vakhrusheva, J.; Choi, C.J.; Delespaul, P.; Tarrier, N.; Sloan, R.P.; Malaspina, L. Autonomic regulation and auditory hallucinations in individuals with schizophrenia: An experience sampling study. Schizophr. Bull. 2017, 43, 754–763. [Google Scholar] [CrossRef]
- Lincoln, T.M.; Peter, N.; Schäfer, M.; Moritz, S. Impact of stress on paranoia: An experimental investigation of moderators and mediators. Psychol. Med. 2009, 39, 1129–1139. [Google Scholar] [CrossRef]
- Vassena, E.; Van Opstal, F.; Goethals, I.; Verguts, T. Striatal dopamine D2 binding correlates with locus of control: Preliminary evidence from [11C] raclopride Positron Emission Tomography. Int. J. Psychophysiol. 2019, 146, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ashton, A.; Jagannath, A. Disrupted sleep and circadian rhythms in schizophrenia and their interaction with dopamine signaling. Front. Neurosci. 2020, 14, 636. [Google Scholar] [CrossRef] [PubMed]
- Eban-Rothschild, A.; Appelbaum, L.; de Lecea, L. Neuronal Mechanisms for Sleep/Wake Regulation and Modulatory Drive. Neuro Psychopharmacol. 2018, 43, 937–952. [Google Scholar] [CrossRef]
- Yates, N.J. Schizophrenia: The role of sleep and circadian rhythms in regulating dopamine and psychosis. Rev. Neurosci. 2016, 27, 669–687. [Google Scholar] [CrossRef]
- Waite, F.; Myers, E.; Harvey, A.G.; Espie, C.A.; Startup, H.; Sheaves, B.; Freeman, D. Treating Sleep Problems in Patients with Schizophrenia. Behav. Cogn. Psychother. 2016, 44, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Leeuwen, J.; Vink, M.; Joëls, M.; Kahn, R.; Hermans, E.; Vinkers, C. Increased responses of the reward circuitry to positive task feedback following acute stress in healthy controls but not in siblings of schizophrenia patients. NeuroImage 2019, 184, 547–554. [Google Scholar] [CrossRef]
- van Leeuwen, J.M.C.; Vinkers, C.H.; Vink, M.; Kahn, R.S.; Joëls, M.; Hermans, E.J. Disrupted upregulation of salience network connectivity during acute stress in siblings of schizophrenia patients. Psychol. Med. 2021, 51, 1038–1048. [Google Scholar] [CrossRef]
- Laruelle, M. The role of endogenous sensitization in the pathophysiology of schizophrenia: Implications from recent brain imaging studies. Brain Res. Rev. 2000, 31, 371–384. [Google Scholar] [CrossRef]
- Gray, R.; Bressington, D.; Hughes, E.; Ivanecka, A. A systematic review of the effects of novel psychoactive substances ‘legal highs’ on people with severe mental illness. J. Psychiatr. Ment. Health Nurs. 2016, 23, 267–281. [Google Scholar] [CrossRef]
- D’Souza, D.C.; Gil, R.B.; Madonick, S.; Perry, E.B.; Forselius-Bielen, K.; Braley, G.; Donahue, L.; Tellioglu, T.; Zimolo, Z.; Gueorguieva, R.; et al. Enhanced sensitivity to the euphoric effects of alcohol in schizophrenia. Neuro Psychopharmacol. 2006, 31, 2767–2775. [Google Scholar] [CrossRef]
- Henquet, C.; van Os, J.; Kuepper, R.; Delespaul, P.; Smits, M.; Campo, J.; Myin-Germeys, I. Psychosis reactivity to cannabis use in daily life: An experience sampling study. Br. J. Psychiatry 2010, 196, 447–453. [Google Scholar] [CrossRef]
- van Os, J.; Pries, L.-K.; Have, M.T.; de Graaf, R.; van Dorsselaer, S.; Bak, M.; Wittchen, H.-U.; Rutten, B.P.F.; Guloksuz, S. Schizophrenia and the environment:Within-person analyses may be required to yield evidence of unconfounded and causal association—The example of cannabis and psychosis. Schizophr. Bull. 2021, 47, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Menne, V.; Chesworth, R. Schizophrenia and drug addiction comorbidity: Recent advances in our understanding of behavioural susceptibility and neural mechanisms. Neuroanat. Behav. 2020, 2, e10. [Google Scholar] [CrossRef]
- Spasova, V.; Mehmood, S.; Minhas, A.; Azhar, R.; Anand, S.; Abdelaal, S.; Sham, S.; Chauhan, T.M.; Dragas, D. Impact of Nicotine on Cognition in Patients with Schizophrenia: A Narrative Review. Cureus 2022, 14, e24306. [Google Scholar] [CrossRef] [PubMed]
- Heinz, A.; Schlagenhauf, F. Dopaminergic dysfunction in schizophrenia: Salience attribution revisited. Schizophr. Bull. 2010, 36, 472–485. [Google Scholar] [CrossRef] [Green Version]
- Heinz, A. Dopaminergic dysfunction in alcoholism and schizophrenia—Psychopathological and behavioral correlates. Eur. Psychiatry 2002, 17, 9–16. [Google Scholar] [CrossRef]
- Proebstl, L.; Kamp, F.; Manz, K.; Krause, D.; Adorjan, K.; Pogarell, O.; Koller, G.; Soyka, M.; Falkai, P.; Kambeitz, J. Effects of stimulant drug use on the dopaminergic system: A systematic review and meta-analysis of in vivo neuroimaging studies. Eur. Psychiatry 2019, 59, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Waggener, C.T.; DuPree, J.L.; Elgersma, Y.; Fuss, B. CaMKII Regulates oligodendrocyte maturation and CNS myelination. J. Neurosci. 2013, 33, 10453–10458. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novak, G.; Seeman, M.V. Dopamine, Psychosis, and Symptom Fluctuation: A Narrative Review. Healthcare 2022, 10, 1713. https://doi.org/10.3390/healthcare10091713
Novak G, Seeman MV. Dopamine, Psychosis, and Symptom Fluctuation: A Narrative Review. Healthcare. 2022; 10(9):1713. https://doi.org/10.3390/healthcare10091713
Chicago/Turabian StyleNovak, Gabriela, and Mary V. Seeman. 2022. "Dopamine, Psychosis, and Symptom Fluctuation: A Narrative Review" Healthcare 10, no. 9: 1713. https://doi.org/10.3390/healthcare10091713
APA StyleNovak, G., & Seeman, M. V. (2022). Dopamine, Psychosis, and Symptom Fluctuation: A Narrative Review. Healthcare, 10(9), 1713. https://doi.org/10.3390/healthcare10091713