Modeling Melanoma In Vitro and In Vivo

Abstract
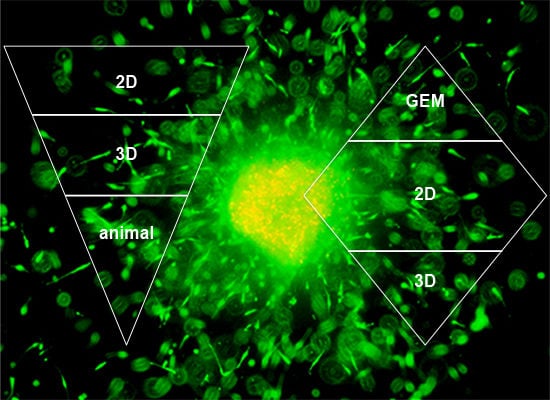
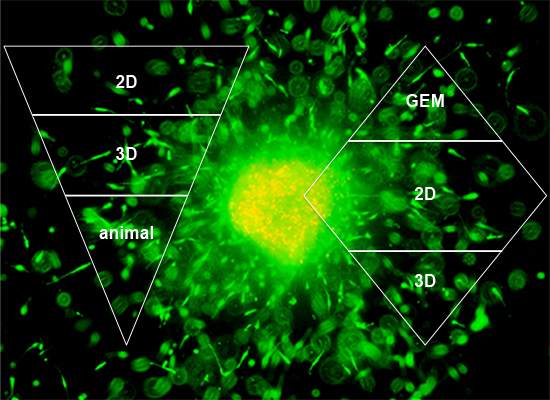
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Two-Dimensional Cell Culture

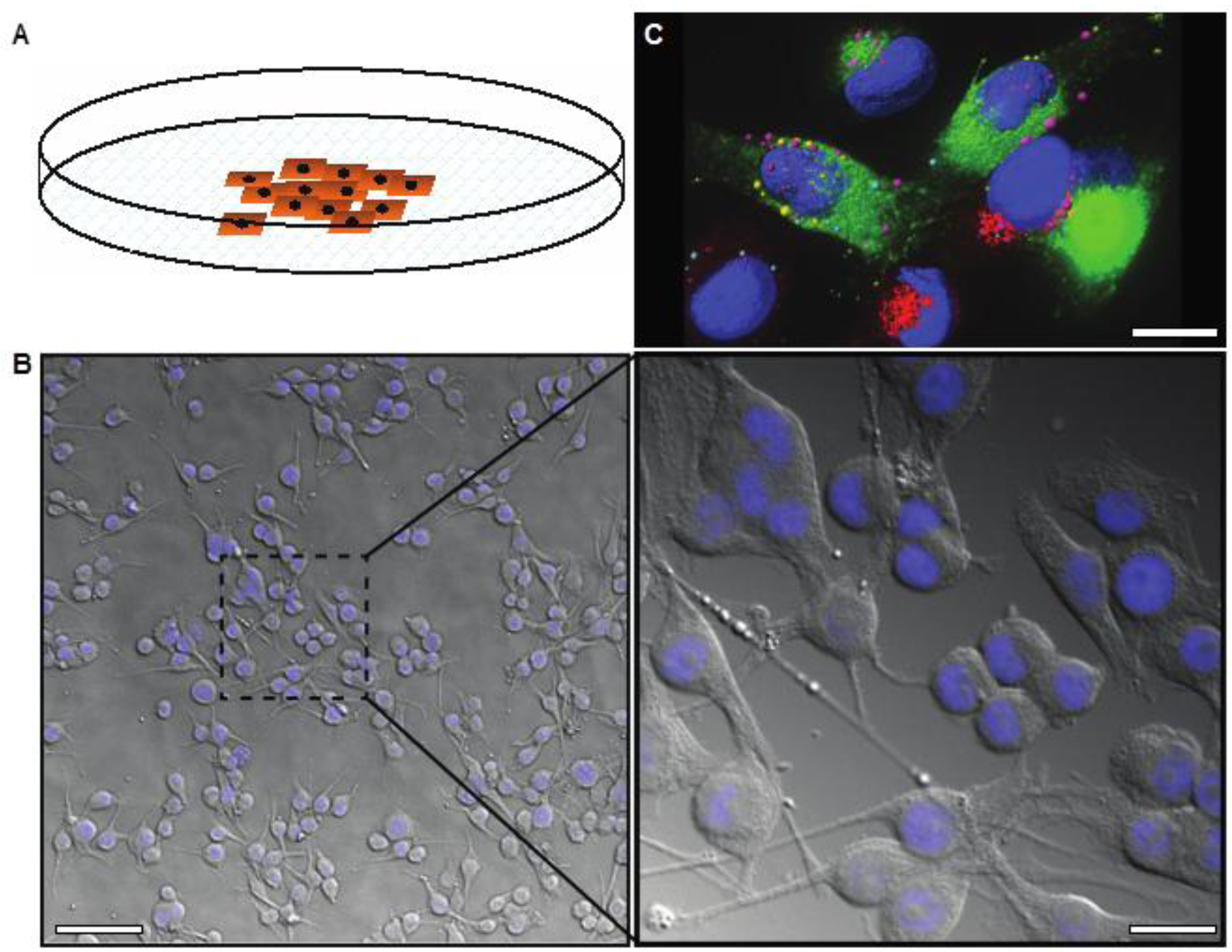
3. Three-Dimensional Cell Culture
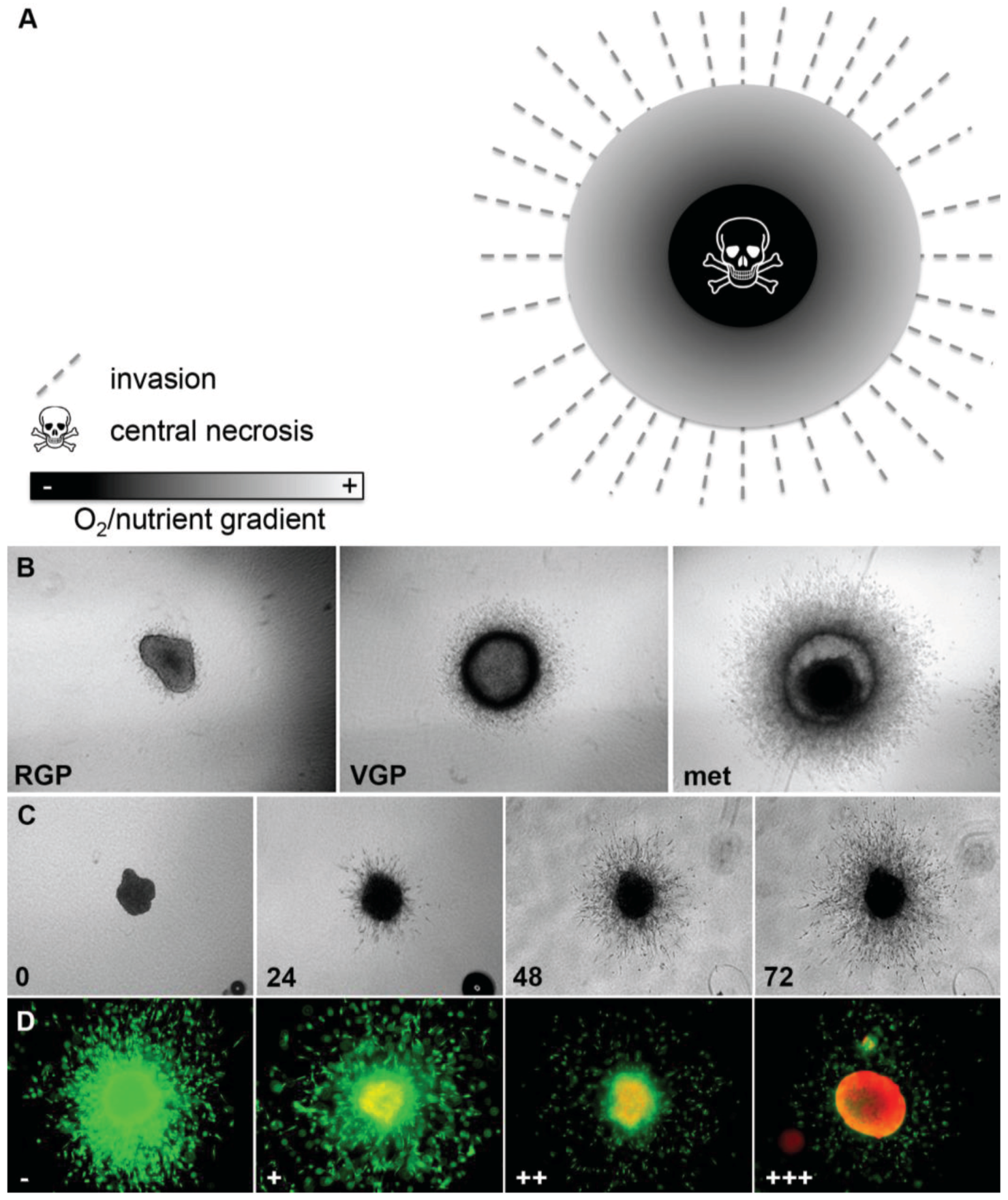
3.1. 3D Spheroid Model
3.2. Tumor Sphere Model

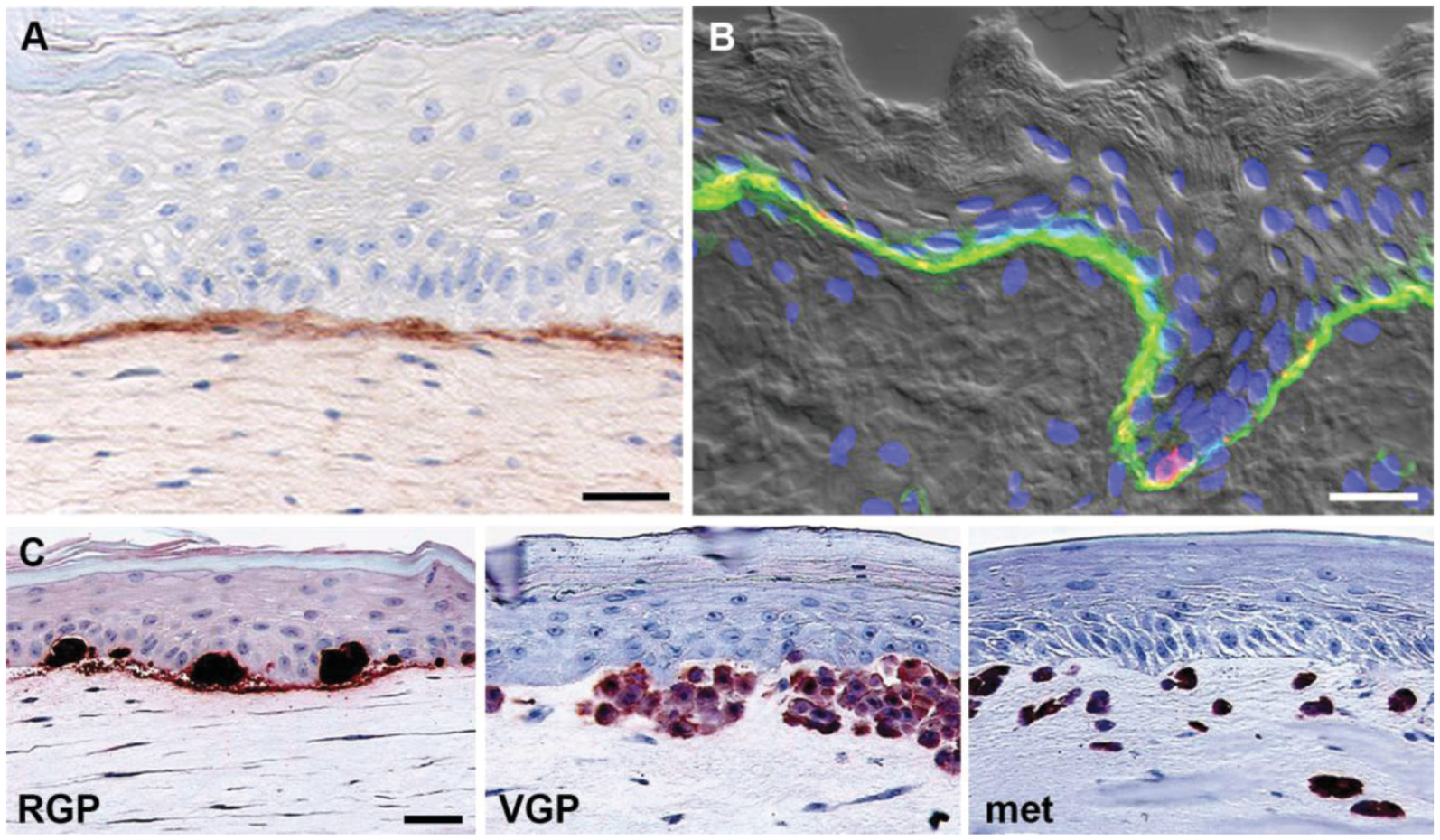
3.3. 3D Skin Reconstruct Model

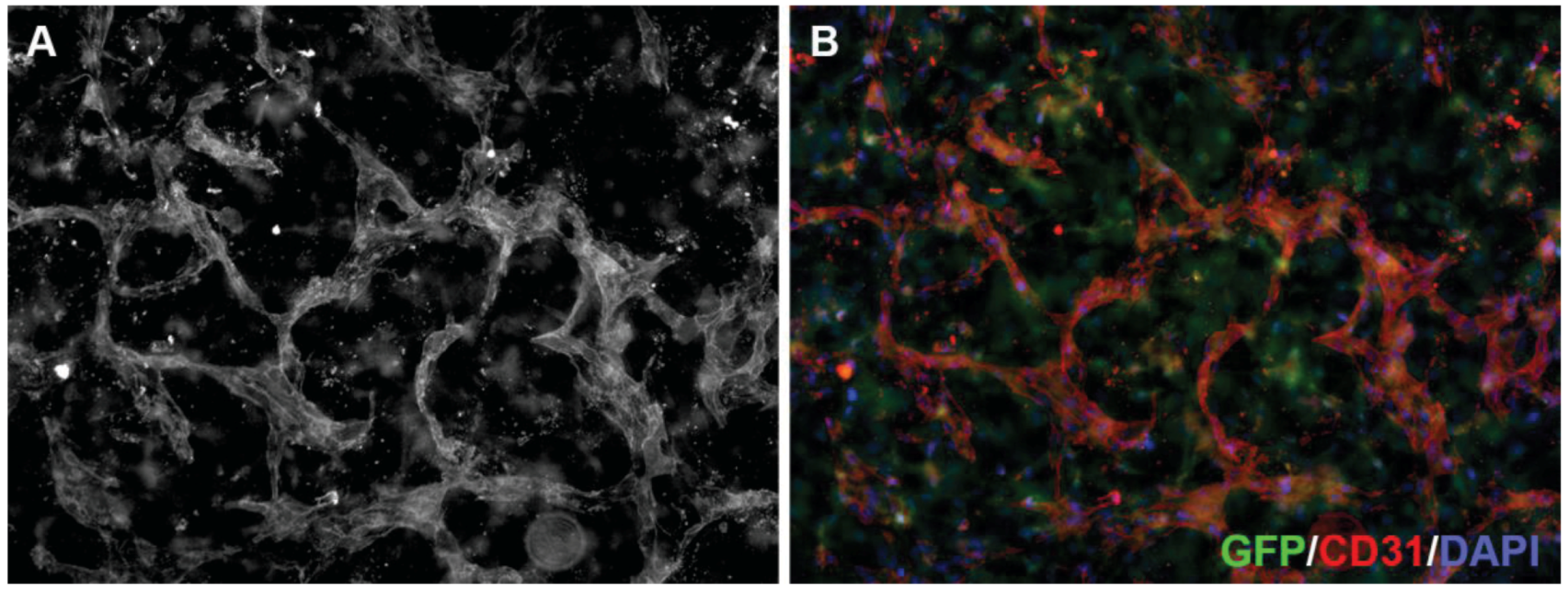
3.4. 3D Neoangiogenesis Model

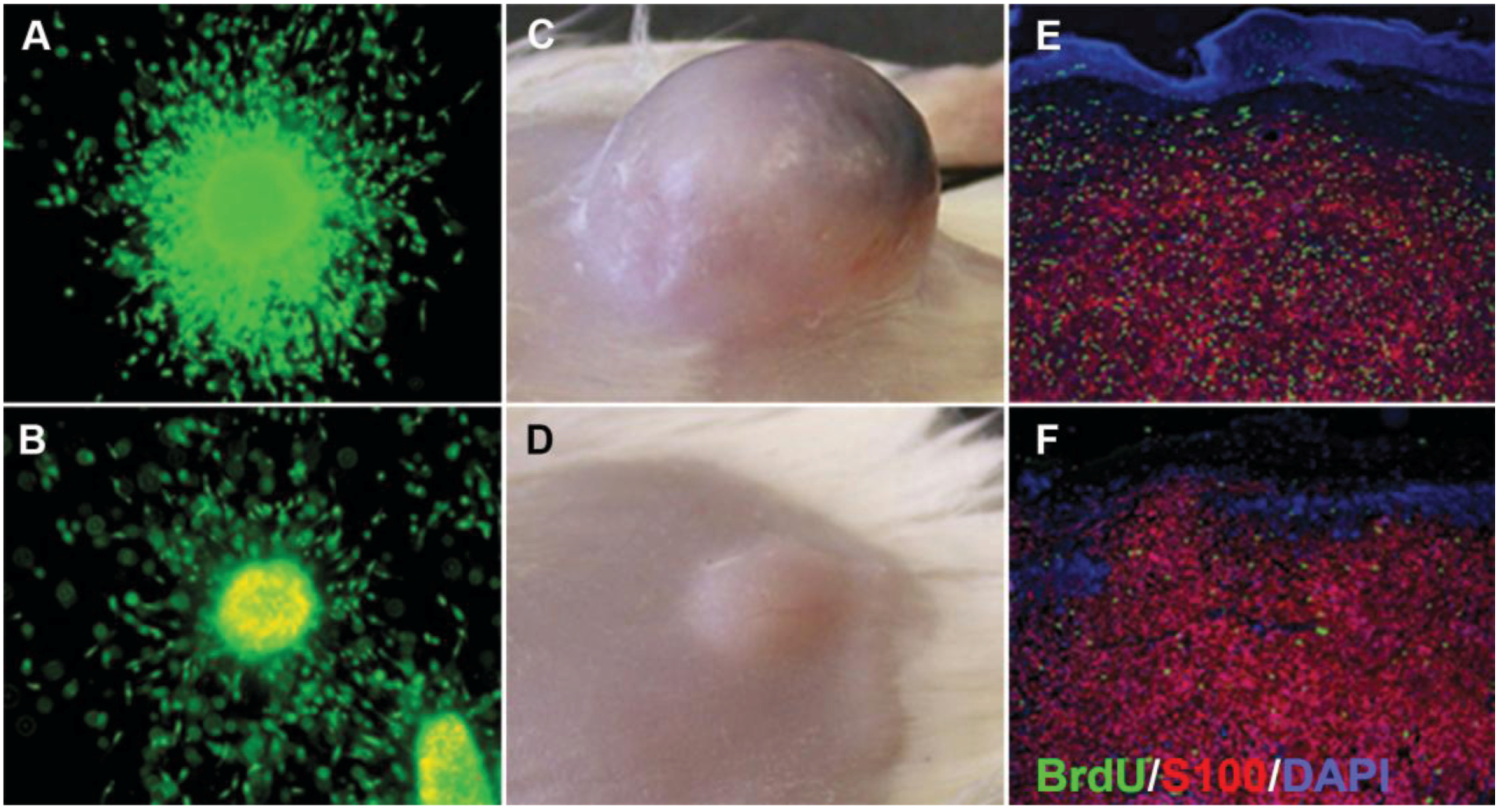
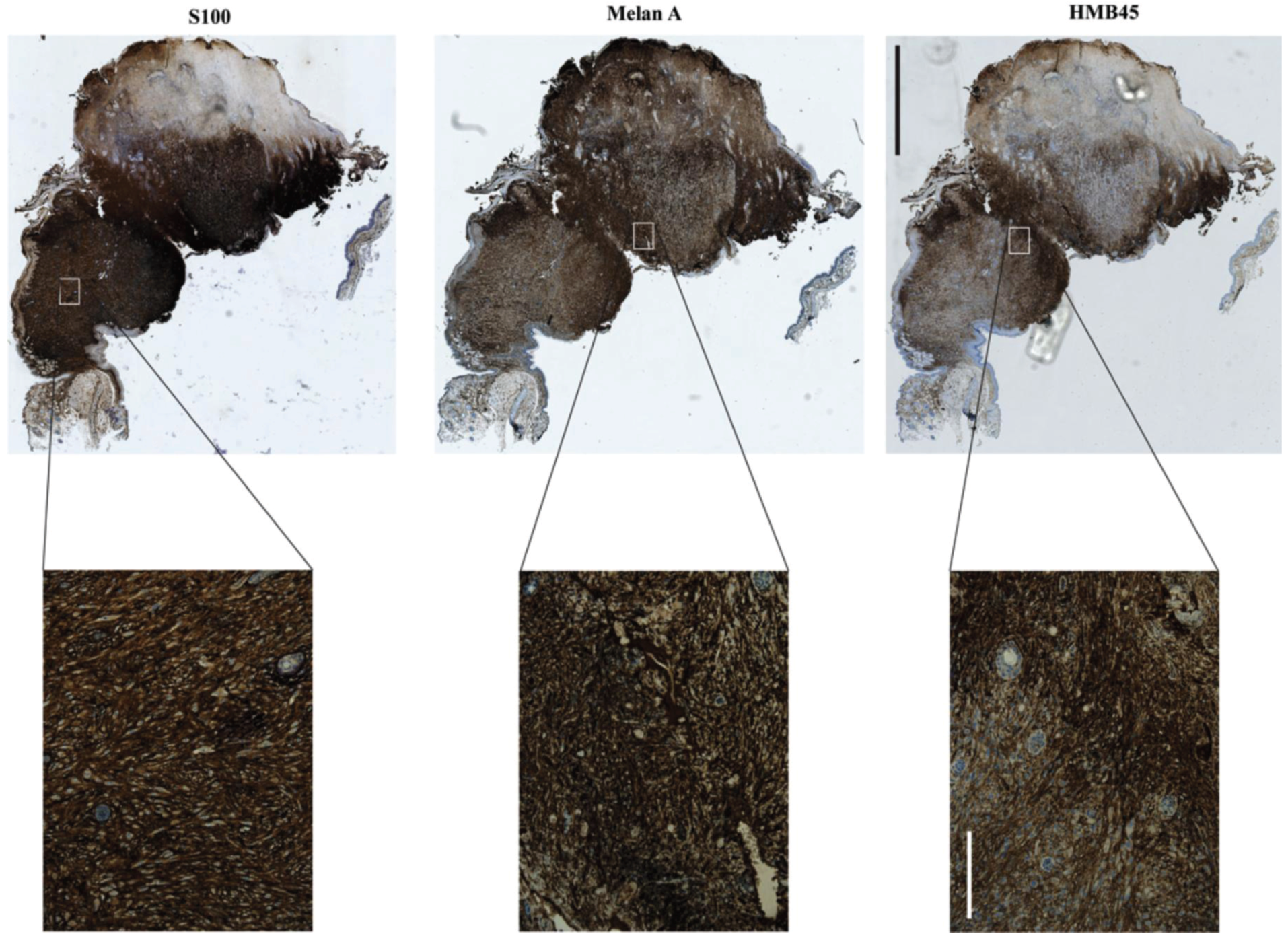
4. Xenograft Models

5. Genetically Engineered Mouse Models (GEM)

6. Other Animal Models
6.1. Fish Models
6.2. Avian and More Mammalian Models
7. Conclusions

Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Smigal, C.; Thun, M.J. Cancer statistics, 2006. CA Cancer J. Clin. 2006, 56, 106–130. [Google Scholar] [CrossRef]
- Balch, C.M.; Soong, S.J.; Gershenwald, J.E.; Thompson, J.F.; Reintgen, D.S.; Cascinelli, N.; Urist, M.; McMasters, K.M.; Ross, M.I.; Kirkwood, J.M.; et al. Prognostic factors analysis of 17,600 melanoma patients: Validation of the American Joint Committee on Cancer melanoma staging system. J. Clin. Oncol. 2001, 19, 3622–3634. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.; Sosman, J.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef]
- Lee, J.T.; Li, L.; Brafford, P.A.; van den Eijnden, M.; Halloran, M.B.; Sproesser, K.; Haass, N.K.; Smalley, K.S.; Tsai, J.; Bollag, G.; et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010, 23, 820–827. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Brandner, J.M.; Haass, N.K. Melanoma’s connections to the tumour microenvironment. Pathology 2013, 45, 443–452. [Google Scholar] [CrossRef]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef]
- Santiago-Walker, A.; Li, L.; Haass, N.K.; Herlyn, M. Melanocytes: From morphology to application. Skin Pharmacol. Physiol. 2009, 22, 114–121. [Google Scholar] [CrossRef]
- Haass, N.K.; Smalley, K.S.; Li, L.; Herlyn, M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef]
- Kuphal, S.; Haass, N.K. Cell-cell and cell-matrix contacts in melanoma and the tumor microenvironment. In Melanoma Development—Molecular Biology, Genetics and Clinical Application; Bosserhoff, A.K., Ed.; Springer-Verlag: Wien, Austria, 2011; pp. 181–215. [Google Scholar]
- Parachoniak, C.A.; Park, M. Dynamics of receptor trafficking in tumorigenicity. Trends Cell Biol. 2012, 22, 231–240. [Google Scholar] [CrossRef]
- Hung, M.C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef]
- Hsu, M.Y.; Meier, F.E.; Nesbit, M.; Hsu, J.Y.; van Belle, P.; Elder, D.E.; Herlyn, M. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am. J. Pathol. 2000, 156, 1515–1525. [Google Scholar] [CrossRef]
- Fukunaga-Kalabis, M.; Martinez, G.; Liu, Z.J.; Kalabis, J.; Mrass, P.; Weninger, W.; Firth, S.M.; Planque, N.; Perbal, B.; Herlyn, M. CCN3 controls 3D spatial localization of melanocytes in the human skin through DDR1. J. Cell Biol. 2006, 175, 563–569. [Google Scholar] [CrossRef]
- Smalley, K.S.; Lioni, M.; Noma, K.; Haass, N.K.; Herlyn, M. In vitro three-dimensional tumor microenvironment models for anticancer drug discovery. Expert Opin. Drug Discov. 2008, 3, 1–10. [Google Scholar] [CrossRef]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef]
- Lucas, K.M.; Mohana-Kumaran, N.; Lau, D.; Zhang, X.D.; Hersey, P.; Huang, D.C.; Weninger, W.; Haass, N.K.; Allen, J.D. Modulation of NOXA and MCL-1 as a strategy for sensitizing melanoma cells to the BH3-mimetic ABT-737. Clin. Cancer Res. 2012, 18, 783–795. [Google Scholar] [CrossRef]
- Wroblewski, D.; Mijatov, B.; Mohana-Kumaran, N.; Lai, F.; Gallagher, S.J.; Haass, N.K.; Zhang, X.D.; Hersey, P. The BH3-mimetic ABT-737 sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors but does not reverse acquired resistance. Carcinogenesis 2013, 34, 237–247. [Google Scholar] [CrossRef]
- Vorsmann, H.; Groeber, F.; Walles, H.; Busch, S.; Beissert, S.; Walczak, H.; Kulms, D. Development of a human three-dimensional organotypic skin-melanoma spheroid model for in vitro drug testing. Cell Death Dis. 2013, 4, e719. [Google Scholar] [CrossRef]
- Smalley, K.S.; Haass, N.K.; Brafford, P.A.; Lioni, M.; Flaherty, K.T.; Herlyn, M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol. Cancer Ther. 2006, 5, 1136–1144. [Google Scholar]
- Haass, N.K.; Sproesser, K.; Nguyen, T.K.; Contractor, R.; Medina, C.A.; Nathanson, K.L.; Herlyn, M.; Smalley, K.S. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin. Cancer Res. 2008, 14, 230–239. [Google Scholar] [CrossRef]
- Zhuang, L.; Lee, C.S.; Scolyer, R.A.; McCarthy, S.W.; Palmer, A.A.; Zhang, X.D.; Thompson, J.F.; Bron, L.P.; Hersey, P. Activation of the extracellular signal regulated kinase (ERK) pathway in human melanoma. J. Clin. Pathol. 2005, 58, 1163–1169. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Smalley, K.S.; Brafford, P.; Haass, N.K.; Brandner, J.M.; Brown, E.; Herlyn, M. Up-regulated expression of zonula occludens protein-1 in human melanoma associates with N-cadherin and contributes to invasion and adhesion. Am. J. Pathol. 2005, 166, 1541–1554. [Google Scholar] [CrossRef]
- Stehn, J.R.; Haass, N.K.; Bonello, T.; Desouza, M.; Kottyan, G.; Treutlein, H.; Zeng, J.; Nascimento, P.R.; Sequeira, V.B.; Butler, T.L.; et al. A novel class of anticancer compounds targets the actin cytoskeleton in tumor cells. Cancer Res. 2013, 73, 5169–5182. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschlager, M.; Dolznig, H. In vitro cell migration and invasion assays. Mutat. Res. 2013, 752, 10–24. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Suresh, V.; Peyton, S.R.; Raub, C.B.; Meyskens, F.L., Jr.; George, S.C.; Putnam, A.J. A novel three-dimensional model to quantify metastatic melanoma invasion. Mol. Cancer Ther. 2007, 6, 552–561. [Google Scholar] [CrossRef]
- Flach, E.H.; Rebecca, V.W.; Herlyn, M.; Smalley, K.S.; Anderson, A.R. Fibroblasts contribute to melanoma tumor growth and drug resistance. Mol. Pharm. 2011, 8, 2039–2049. [Google Scholar] [CrossRef]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef]
- Mo, J.; Sun, B.; Zhao, X.; Gu, Q.; Dong, X.; Liu, Z.; Ma, Y.; Zhao, N.; Liu, Y.; Chi, J.; Sun, R. The in-vitro spheroid culture induces a more highly differentiated but tumorigenic population from melanoma cell lines. Melanoma Res. 2013, 23, 254–263. [Google Scholar]
- Thurber, A.E.; Douglas, G.; Sturm, E.C.; Zabierowski, S.E.; Smit, D.J.; Ramakrishnan, S.N.; Hacker, E.; Leonard, J.H.; Herlyn, M.; Sturm, R.A. Inverse expression states of the BRN2 and MITF transcription factors in melanoma spheres and tumour xenografts regulate the NOTCH pathway. Oncogene 2011, 30, 3036–3048. [Google Scholar] [CrossRef] [Green Version]
- Berking, C.; Herlyn, M. Human skin reconstruct models: A new application for studies of melanocyte and melanoma biology. Histol. Histopathol. 2001, 16, 669–674. [Google Scholar]
- Meier, F.; Nesbit, M.; Hsu, M.Y.; Martin, B.; van Belle, P.; Elder, D.E.; Schaumburg-Lever, G.; Garbe, C.; Walz, T.M.; Donatien, P.; et al. Human melanoma progression in skin reconstructs: Biological significance of bFGF. Am. J. Pathol. 2000, 156, 193–200. [Google Scholar] [CrossRef]
- Meier, F.; Busch, S.; Lasithiotakis, K.; Kulms, D.; Garbe, C.; Maczey, E.; Herlyn, M.; Schittek, B. Combined targeting of MAPK and AKT signalling pathways is a promising strategy for melanoma treatment. Br. J. Dermatol. 2007, 156, 1204–1213. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Velazquez, O.C.; Snyder, R.; Liu, Z.J.; Fairman, R.M.; Herlyn, M. Fibroblast-dependent differentiation of human microvascular endothelial cells into capillary-like 3-dimensional networks. FASEB J. 2002, 16, 1316–1318. [Google Scholar]
- Merlino, G.; Flaherty, K.; Acquavella, N.; Day, C.P.; Aplin, A.; Holmen, S.; Topalian, S.; van Dyke, T.; Herlyn, M. Meeting report: The future of preclinical mouse models in melanoma treatment is now. Pigment Cell Melanoma Res. 2013, 26, E8–E14. [Google Scholar] [CrossRef]
- Herlyn, D.; Iliopoulos, D.; Jensen, P.J.; Parmiter, A.; Baird, J.; Hotta, H.; Adachi, K.; Ross, A.H.; Jambrosic, J.; Koprowski, H.; et al. In vitro properties of human melanoma cells metastatic in nude mice. Cancer Res. 1990, 50, 2296–2302. [Google Scholar]
- Juhasz, I.; Albelda, S.M.; Elder, D.E.; Murphy, G.F.; Adachi, K.; Herlyn, D.; Valyi-Nagy, I.T.; Herlyn, M. Growth and invasion of human melanomas in human skin grafted to immunodeficient mice. Am. J. Pathol. 1993, 143, 528–537. [Google Scholar]
- Khanna, C.; Hunter, K. Modeling metastasis in vivo. Carcinogenesis 2005, 26, 513–523. [Google Scholar] [CrossRef]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 2010, 18, 510–523. [Google Scholar] [CrossRef]
- Fidler, I.J.; Nicolson, G.L. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J. Natl. Cancer Inst. 1976, 57, 1199–1202. [Google Scholar]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Lower, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Herraiz, C.; Journe, F.; Abdel-Malek, Z.; Ghanem, G.; Jimenez-Cervantes, C.; Garcia-Borron, J.C. Signaling from the human melanocortin 1 receptor to ERK1 and ERK2 mitogen-activated protein kinases involves transactivation of cKIT. Mol. Endocrinol. 2011, 25, 138–156. [Google Scholar] [CrossRef]
- Larue, L.; Beermann, F. Cutaneous melanoma in genetically modified animals. Pigment Cell Res. 2007, 20, 485–497. [Google Scholar] [CrossRef]
- Walker, G.J.; Soyer, H.P.; Terzian, T.; Box, N.F. Modelling melanoma in mice. Pigment Cell Melanoma Res. 2011, 24, 1158–1176. [Google Scholar] [CrossRef]
- Mintz, B.; Silvers, W.K. Transgenic mouse model of malignant skin melanoma. Proc. Natl. Acad. Sci. USA 1993, 90, 8817–8821. [Google Scholar] [CrossRef]
- Chin, L.; Pomerantz, J.; Polsky, D.; Jacobson, M.; Cohen, C.; Cordon-Cardo, C.; Horner, J.W., 2nd; DePinho, R.A. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997, 11, 2822–2834. [Google Scholar] [CrossRef]
- Serrano, M.; Lee, H.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef]
- Dhomen, N.; Reis-Filho, J.S.; da Rocha Dias, S.; Hayward, R.; Savage, K.; Delmas, V.; Larue, L.; Pritchard, C.; Marais, R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 2009, 15, 294–303. [Google Scholar] [CrossRef]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Gould Rothberg, B.E.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. beta-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef]
- Recio, J.A.; Noonan, F.P.; Takayama, H.; Anver, M.R.; Duray, P.; Rush, W.L.; Lindner, G.; de Fabo, E.C.; DePinho, R.A.; Merlino, G. Ink4a/arf deficiency promotes ultraviolet radiation-induced melanomagenesis. Cancer Res. 2002, 62, 6724–6730. [Google Scholar]
- Ackermann, J.; Frutschi, M.; Kaloulis, K.; McKee, T.; Trumpp, A.; Beermann, F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005, 65, 4005–4011. [Google Scholar] [CrossRef]
- McKinney, A.J.; Holmen, S.L. Animal models of melanoma: A somatic cell gene delivery mouse model allows rapid evaluation of genes implicated in human melanoma. Chin. J. Cancer 2011, 30, 153–162. [Google Scholar] [CrossRef]
- Patton, E.E.; Mitchell, D.L.; Nairn, R.S. Genetic and environmental melanoma models in fish. Pigment Cell Melanoma Res. 2010, 23, 314–337. [Google Scholar] [CrossRef]
- Nairn, R.S.; Kazianis, S.; Della Coletta, L.; Trono, D.; Butler, A.P.; Walter, R.B.; Morizot, D.C. Genetic analysis of susceptibility to spontaneous and UV-induced carcinogenesis in Xiphophorus hybrid fish. Mar. Biotechnol. 2001, 3, S24–S36. [Google Scholar] [CrossRef]
- Kazianis, S.; Gimenez-Conti, I.; Trono, D.; Pedroza, A.; Chovanec, L.B.; Morizot, D.C.; Nairn, R.S.; Walter, R.B. Genetic analysis of neoplasia induced by N-nitroso-N-methylurea in Xiphophorus hybrid fish. Mar. Biotechnol. 2001, 3, S37–S43. [Google Scholar] [CrossRef]
- Walter, R.B.; Kazianis, S. Xiphophorus interspecies hybrids as genetic models of induced neoplasia. ILAR J. 2001, 42, 299–321. [Google Scholar] [CrossRef]
- Haass, N.K. From Xiphophorus to melanoma—A tribute to Steven Kazianis (1966–2008). Zebrafish 2008, 5, 91–92. [Google Scholar] [CrossRef]
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopani, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.D.; et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr. Biol. 2005, 15, 249–254. [Google Scholar] [CrossRef]
- Schartl, M.; Wilde, B.; Laisney, J.A.; Taniguchi, Y.; Takeda, S.; Meierjohann, S. A mutated EGFR is sufficient to induce malignant melanoma with genetic background-dependent histopathologies. J. Invest. Dermatol. 2010, 130, 249–258. [Google Scholar] [CrossRef]
- Patton, E.E.; Zon, L.I. The art and design of genetic screens: Zebrafish. Nat. Rev. Genet. 2001, 2, 956–966. [Google Scholar] [CrossRef]
- Spitsbergen, J. Imaging neoplasia in zebrafish. Nat. Methods 2007, 4, 548–549. [Google Scholar] [CrossRef]
- Stern, H.M.; Zon, L.I. Cancer genetics and drug discovery in the zebrafish. Nat. Rev. Cancer 2003, 3, 533–539. [Google Scholar] [CrossRef]
- Bardeesy, N.; Wong, K.K.; DePinho, R.A.; Chin, L. Animal models of melanoma: Recent advances and future prospects. Adv. Cancer Res. 2000, 79, 123–156. [Google Scholar] [CrossRef]
- Greene, J.F., Jr.; Morgan, C.D.; Rao, A.; Amoss, M.S., Jr.; Arguello, F. Regression by differentiation in the Sinclair swine model of cutaneous melanoma. Melanoma Res. 1997, 7, 471–477. [Google Scholar] [CrossRef]
- Millikan, L.E.; Boylon, J.L.; Hook, R.R.; Manning, P.J. Melanoma in Sinclair swine: A new animal model. J. Invest. Dermatol. 1974, 62, 20–30. [Google Scholar]
- Fleury, C.; Berard, F.; Leblond, A.; Faure, C.; Ganem, N.; Thomas, L. The study of cutaneous melanomas in Camargue-type gray-skinned horses (2): Epidemiological survey. Pigment Cell Res. 2000, 13, 47–51. [Google Scholar]
- Fleury, C.; Berard, F.; Balme, B.; Thomas, L. The study of cutaneous melanomas in Camargue-typegray-skinned horses (1): Clinical-pathological characterization. Pigment Cell Res. 2000, 13, 39–46. [Google Scholar]
- Rosengren Pielberg, G.; Golovko, A.; Sundstrom, E.; Curik, I.; Lennartsson, J.; Seltenhammer, M.H.; Druml, T.; Binns, M.; Fitzsimmons, C.; Lindgren, G.; et al. A cis-acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nat. Genet. 2008, 40, 1004–1009. [Google Scholar] [CrossRef]
- Ley, R.D. Animal models of ultraviolet radiation (UVR)-induced cutaneous melanoma. Front. Biosci. 2002, 7, d1531–d1534. [Google Scholar] [CrossRef]
- Schriek, G.; Oppitz, M.; Busch, C.; Just, L.; Drews, U. Human SK-Mel 28 melanoma cells resume neural crest cell migration after transplantation into the chick embryo. Melanoma Res. 2005, 15, 225–234. [Google Scholar] [CrossRef]
- Oppitz, M.; Busch, C.; Schriek, G.; Metzger, M.; Just, L.; Drews, U. Non-malignant migration of B16 mouse melanoma cells in the neural crest and invasive growth in the eye cup of the chick embryo. Melanoma Res. 2007, 17, 17–30. [Google Scholar] [CrossRef]
- Busch, C.; Drews, U.; Garbe, C.; Eisele, S.R.; Oppitz, M. Neural crest cell migration of mouse B16-F1 melanoma cells transplanted into the chick embryo is inhibited by the BMP-antagonist noggin. Int. J. Oncol. 2007, 31, 1367–1378. [Google Scholar]
- Busch, C.; Drews, U.; Eisele, S.R.; Garbe, C.; Oppitz, M. Noggin blocks invasive growth of murine B16-F1 melanoma cells in the optic cup of the chick embryo. Int. J. Cancer 2008, 122, 526–533. [Google Scholar] [CrossRef]
- Busch, C.; Krochmann, J.; Drews, U. The chick embryo as an experimental system for melanoma cell invasion. PLoS One 2013, 8, e53970. [Google Scholar] [CrossRef]
- Krochmann, J.; Sinnberg, T.; Meier, F.; Garbe, C.; Busch, C. Melanoma cells in distinct growth phases retain specific invasive qualities during brain metastasis in vivo. Pigment Cell Melanoma Res. 2012, 25, 113–114. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Beaumont, K.A.; Mohana-Kumaran, N.; Haass, N.K. Modeling Melanoma In Vitro and In Vivo. Healthcare 2014, 2, 27-46. https://doi.org/10.3390/healthcare2010027
Beaumont KA, Mohana-Kumaran N, Haass NK. Modeling Melanoma In Vitro and In Vivo. Healthcare. 2014; 2(1):27-46. https://doi.org/10.3390/healthcare2010027
Chicago/Turabian StyleBeaumont, Kimberley A., Nethia Mohana-Kumaran, and Nikolas K. Haass. 2014. "Modeling Melanoma In Vitro and In Vivo" Healthcare 2, no. 1: 27-46. https://doi.org/10.3390/healthcare2010027
APA StyleBeaumont, K. A., Mohana-Kumaran, N., & Haass, N. K. (2014). Modeling Melanoma In Vitro and In Vivo. Healthcare, 2(1), 27-46. https://doi.org/10.3390/healthcare2010027