


Self-Immobilizing Quinone Methides for the Fluorescent Sensing of Enzyme Activity


Abstract
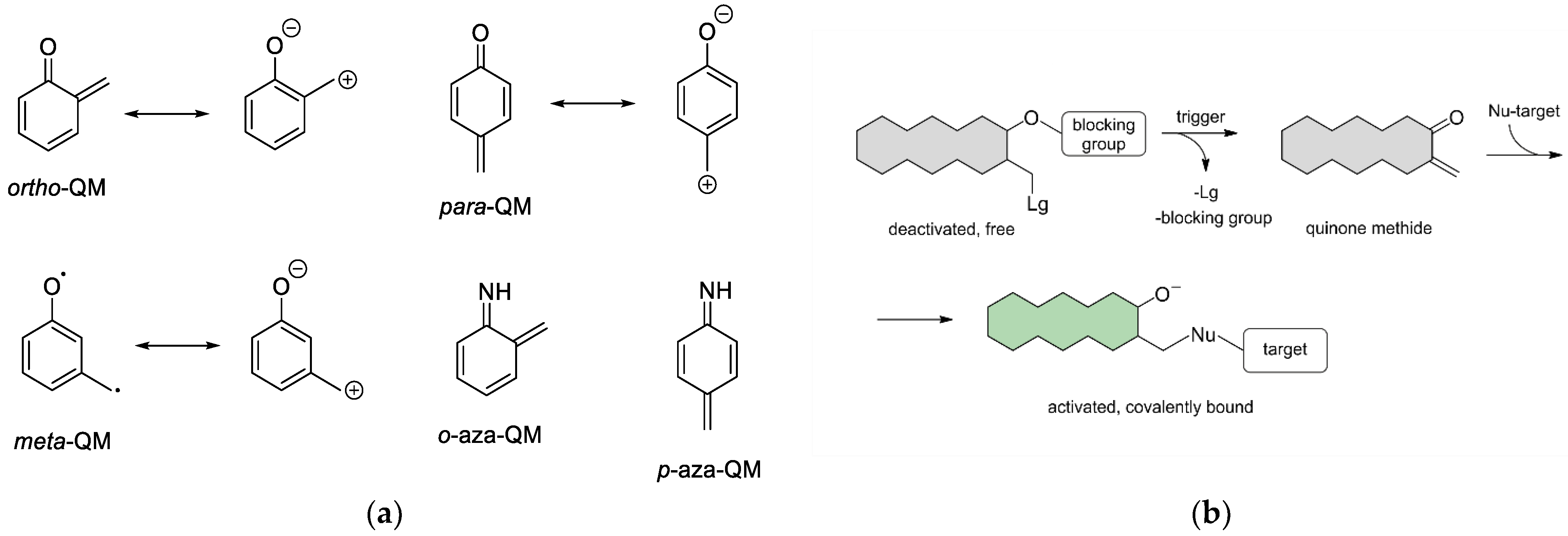
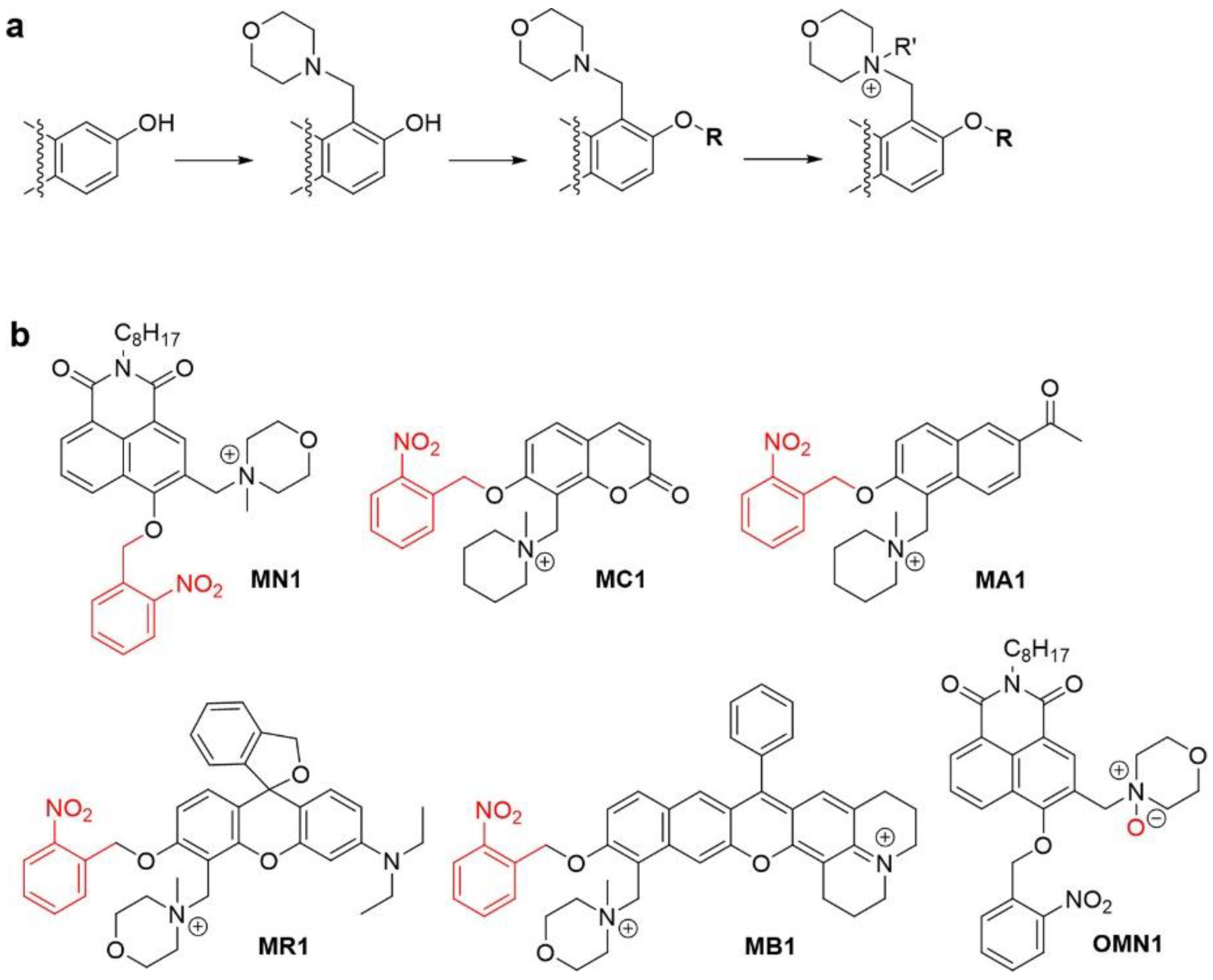
:1. Introduction
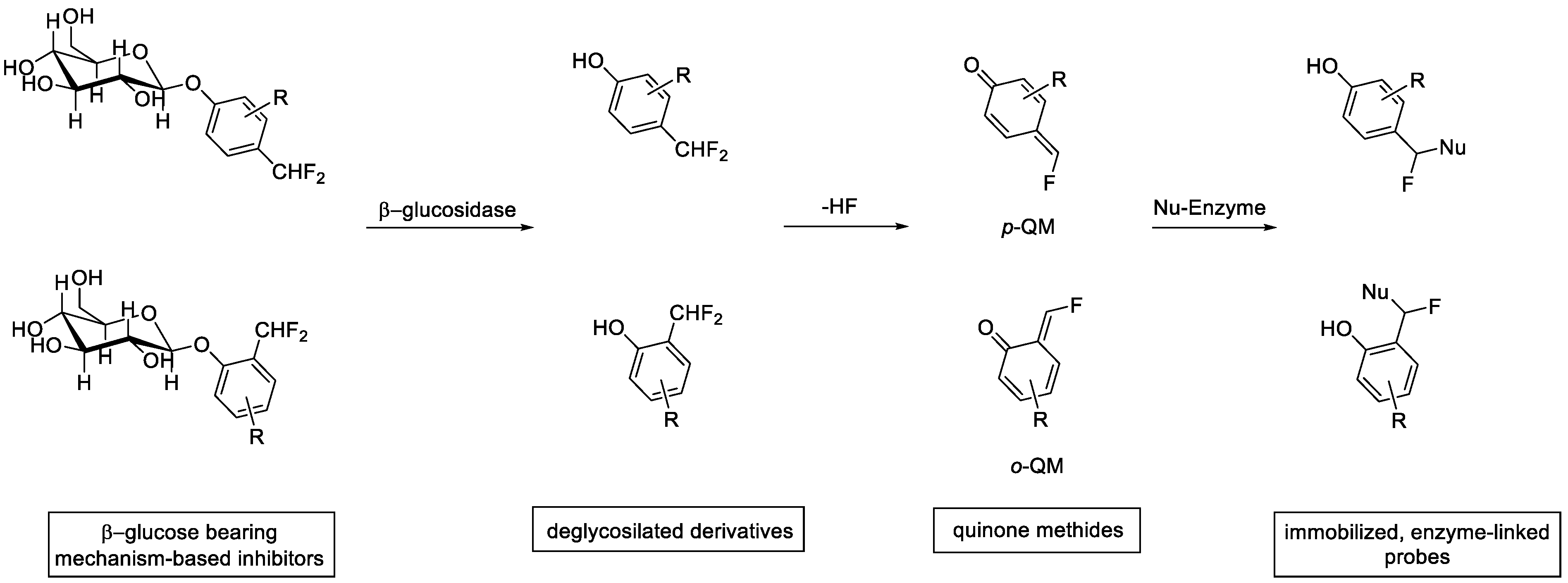
2. Enzyme Inhibition
3. Activity-Based Probes
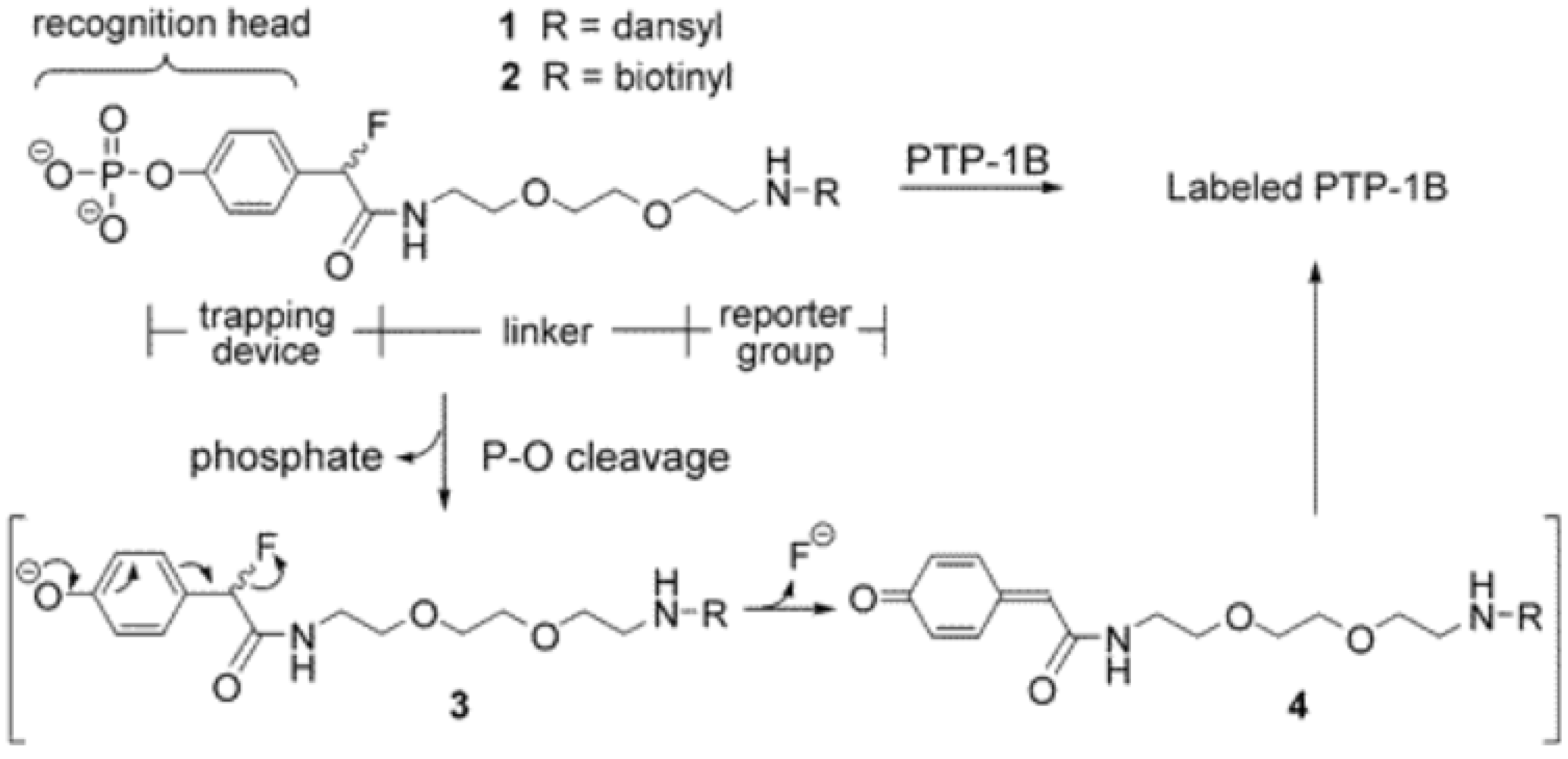
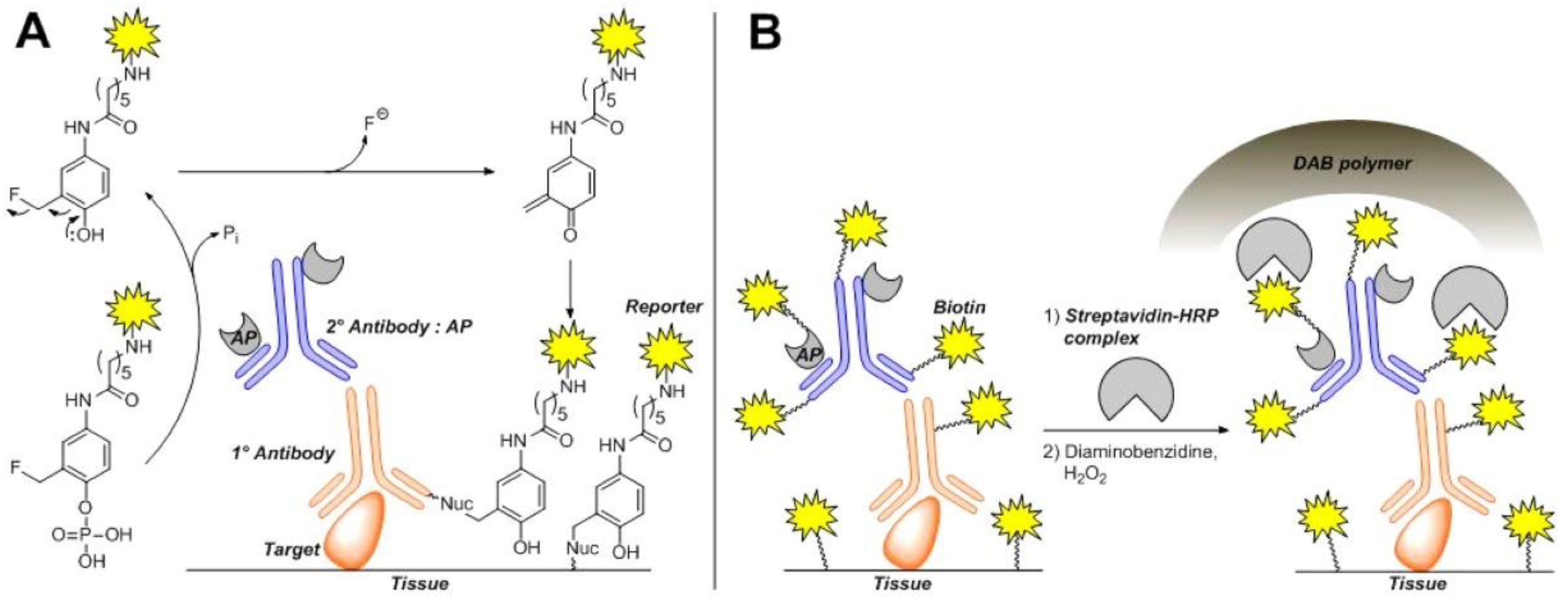
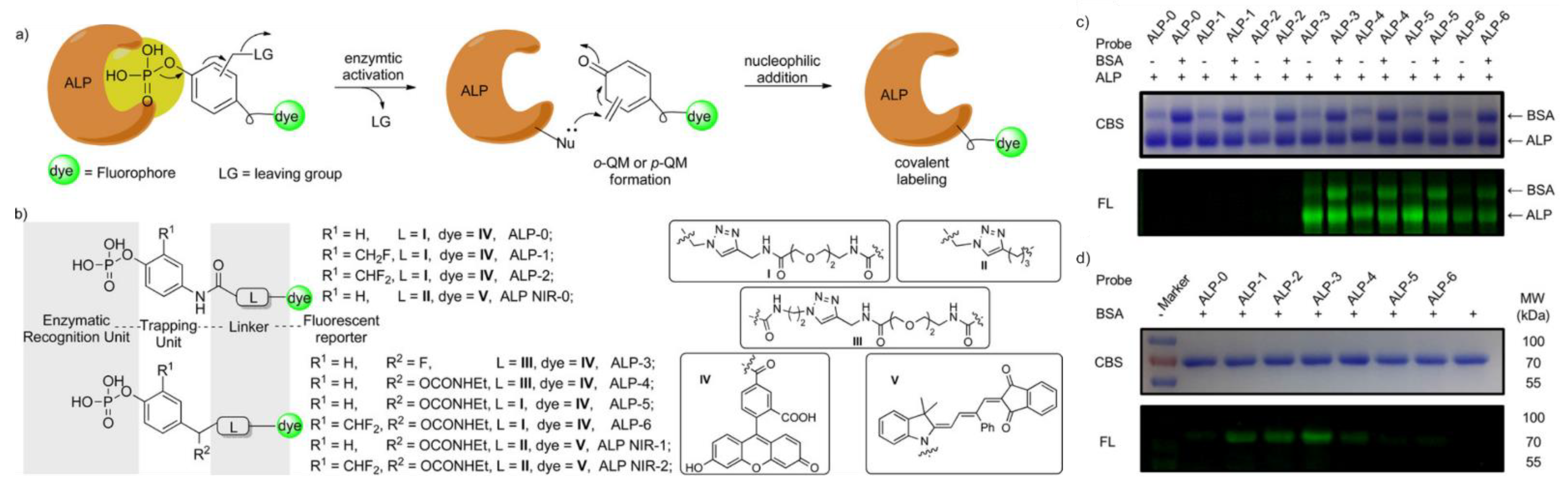
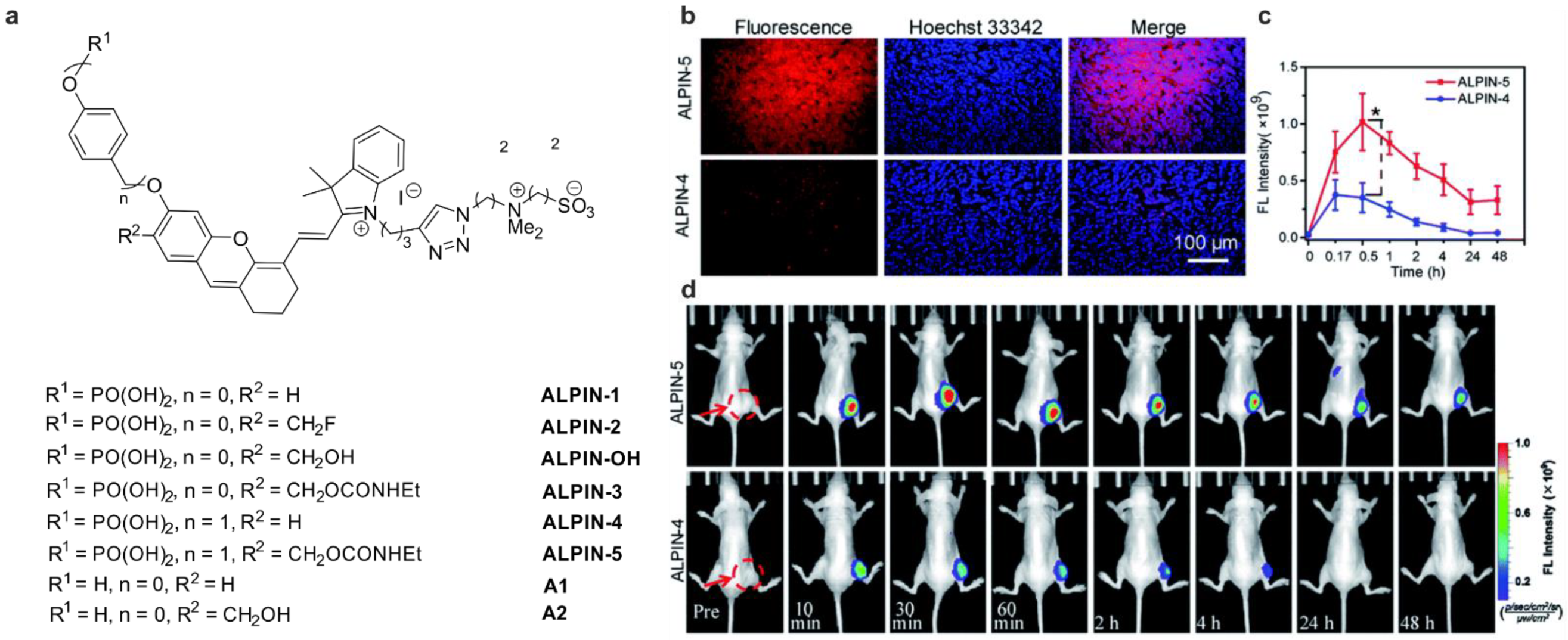
3.1. Phosphatases
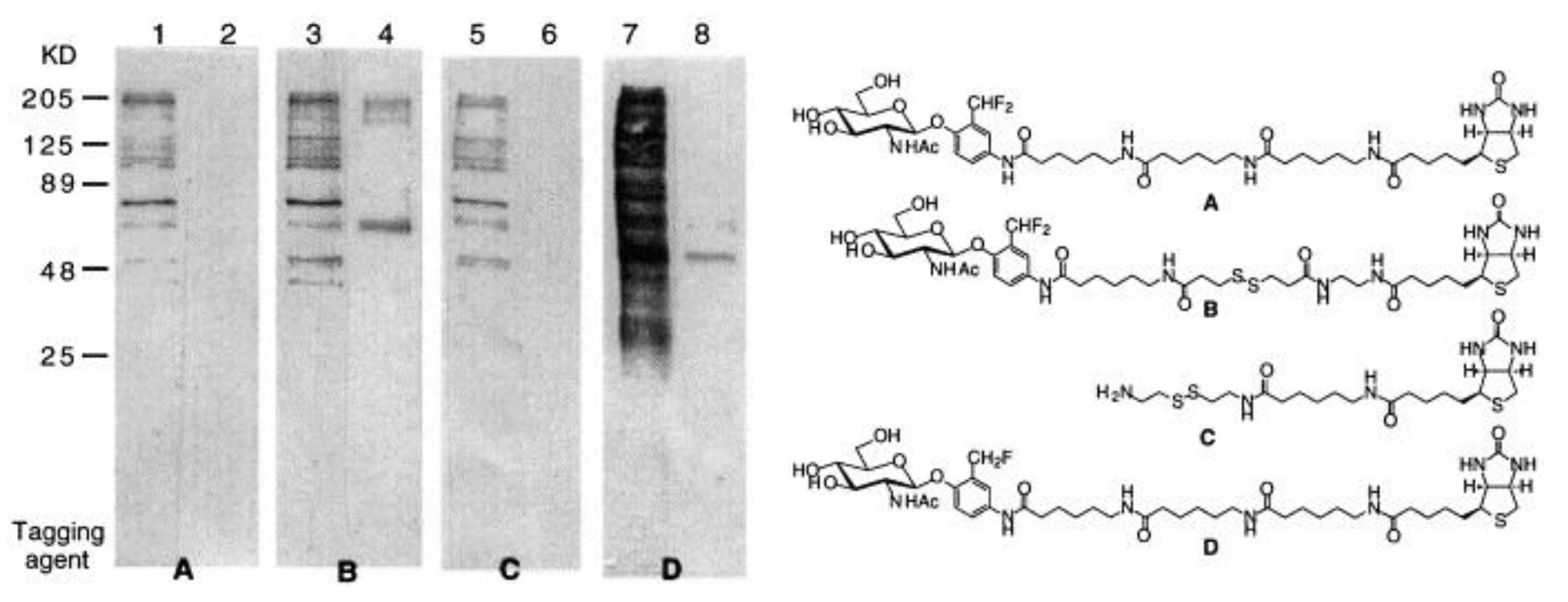
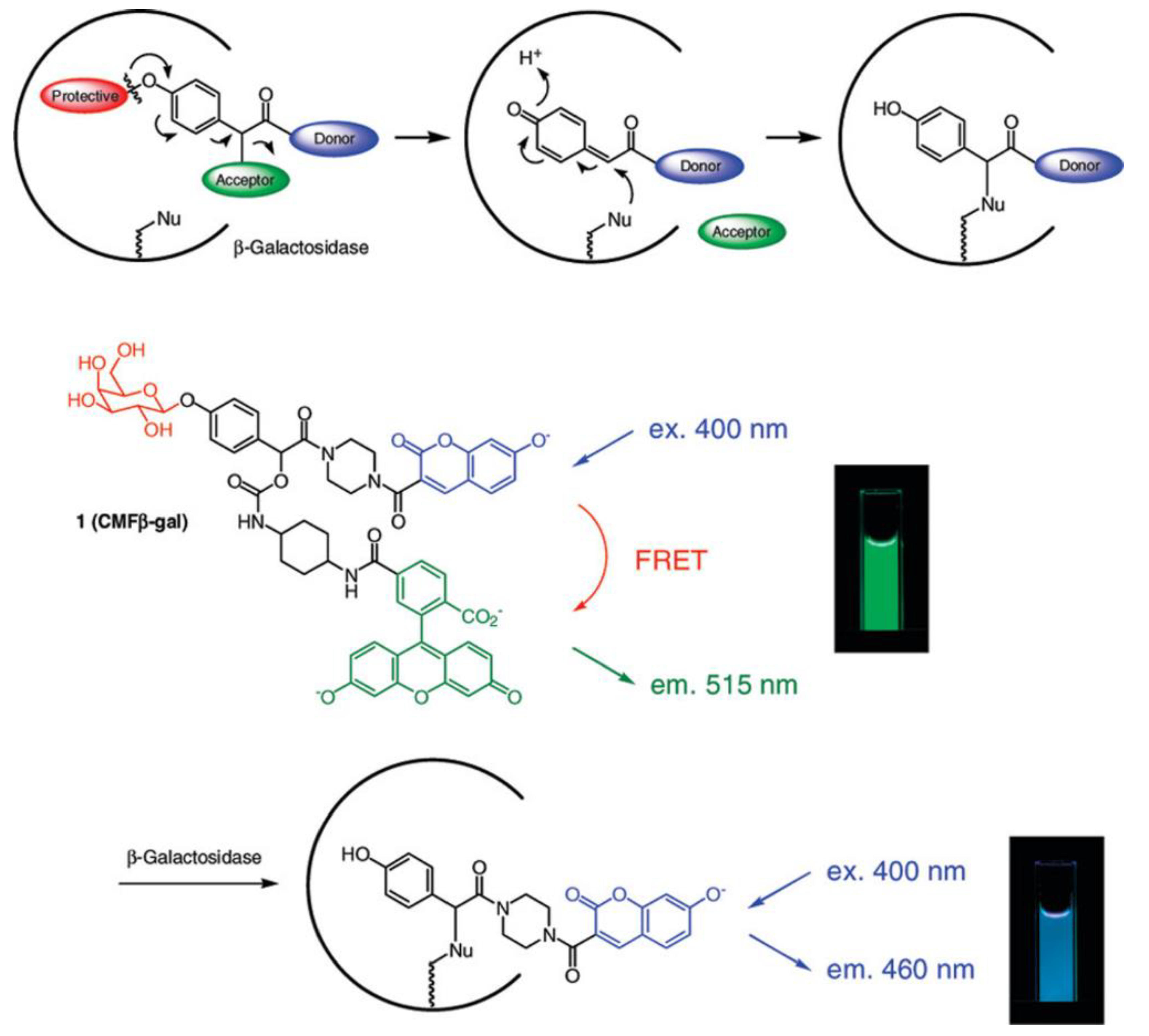
3.2. Glycoside Hydrolases
3.3. γ-Glutamyl-Transpeptidase
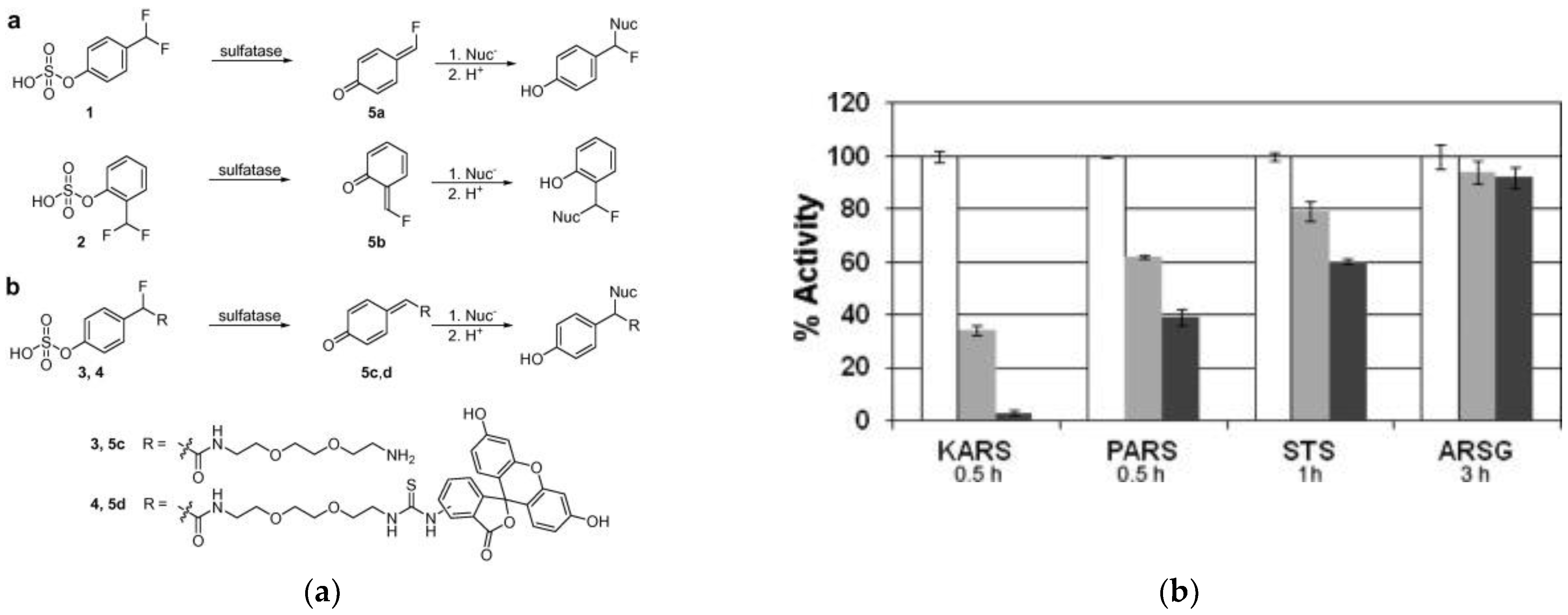
3.4. Sulfatases
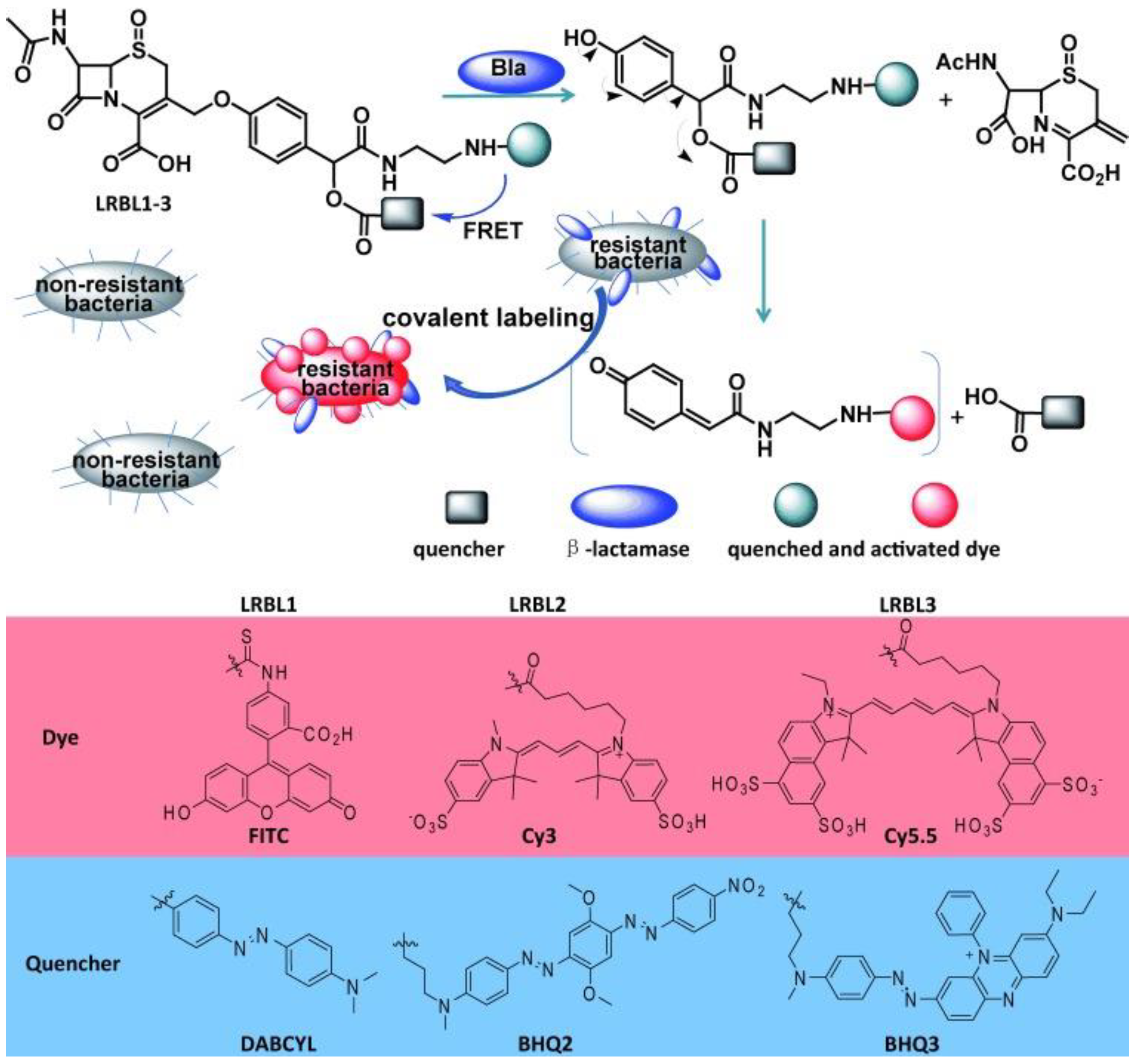
3.5. β-Lactamases
3.6. Other Enzymes
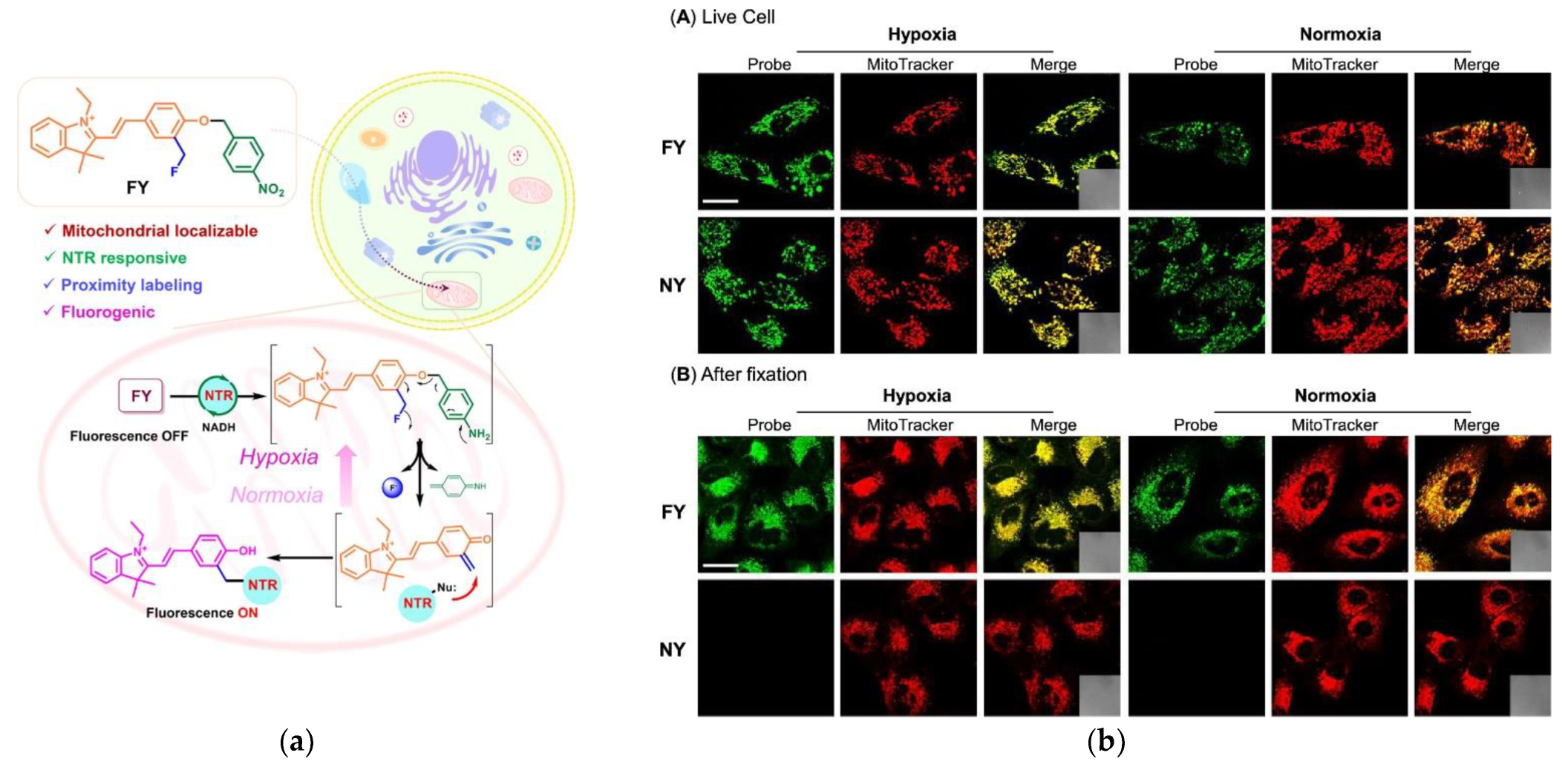
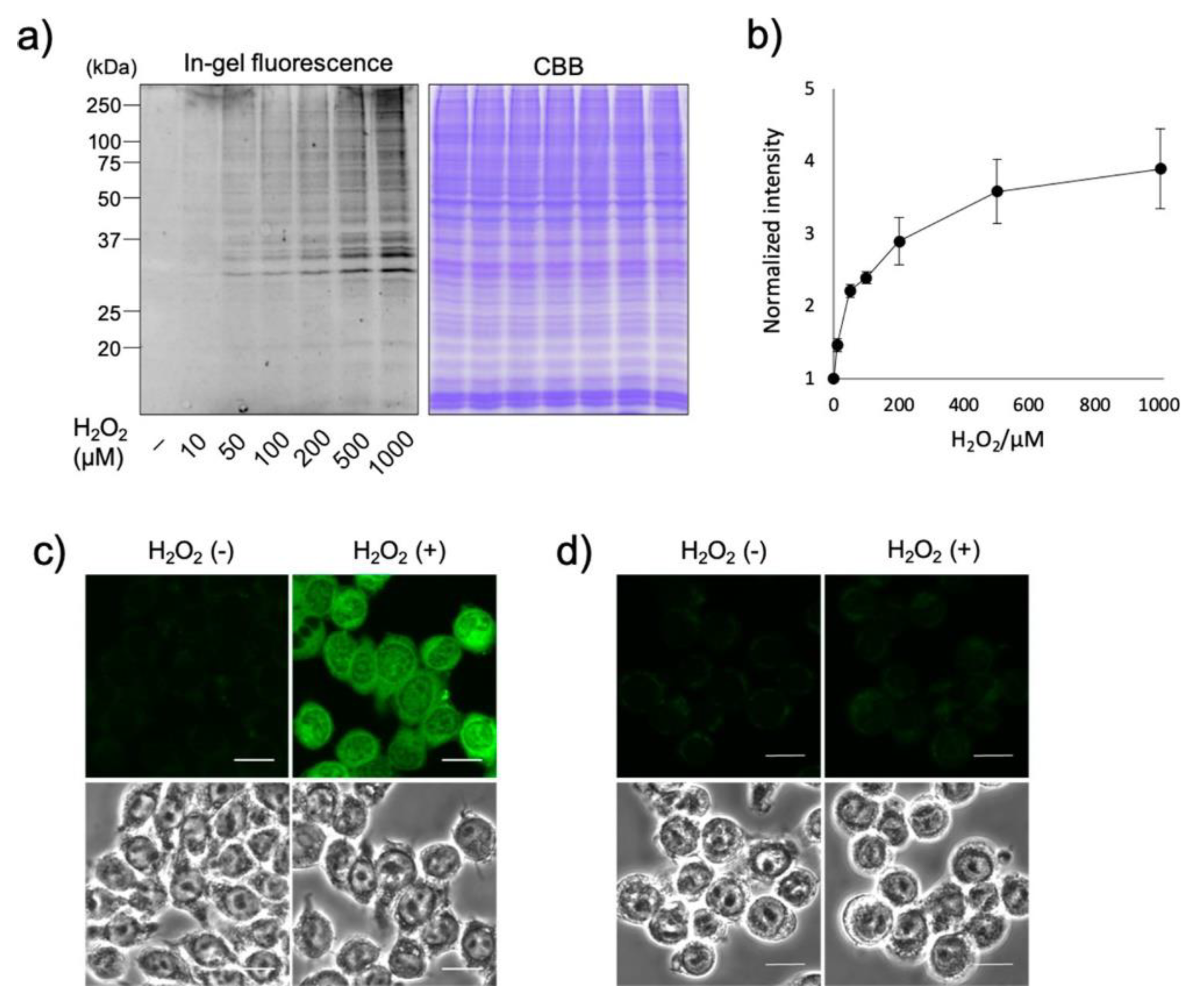
3.7. Reactive Oxygen Species (ROS)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ravikumar, Y.; Nadarajan, S.P.; Yoo, T.H.; Lee, C.-S.; Yun, H. Unnatural amino acid mutagenesis-based enzyme engineering. Trends Biotechnol. 2015, 33, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Rawale, D.G.; Thakur, K.; Adusumalli, S.R.; Rai, V. Chemical Methods for Selective Labeling of Proteins. Eur. J. Org. Chem. 2019, 2019, 6749–6763. [Google Scholar] [CrossRef]
- Scinto, S.L.; Bilodeau, D.A.; Hincapie, R.; Lee, W.; Nguyen, S.S.; Xu, M.; am Ende, C.W.; Finn, M.G.; Lang, K.; Lin, Q.; et al. Bioorthogonal chemistry. Nat. Rev. Methods Prim. 2021, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Singh, J. The Ascension of Targeted Covalent Inhibitors. J. Med. Chem. 2022, 65, 5886–5901. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zou, P. Photocatalytic proximity labeling for profiling the subcellular organization of biomolecules. ChemBioChem 2023. [Google Scholar] [CrossRef]
- Fischer, N.H.; Oliveira, M.T.; Diness, F. Chemical modification of proteins–challenges and trends at the start of the 2020s. Biomater. Sci. 2023. [Google Scholar] [CrossRef]
- Turner, A.B. Quinone methides. Q. Rev. Chem. Soc. 1964, 18, 347–360. [Google Scholar] [CrossRef]
- Toteva, M.M.; Richard, J.P. The Generation and Reactions of Quinone Methides. Adv. Phys. Org. Chem. 2011, 45, 39–91. [Google Scholar]
- Goodman, J.L.; Peters, K.S.; Lahti, P.M.; Berson, J.A. Picosecond Absorption Studies on m-Naphthoquinomethane. Singlet-Triplet Intersystem Crossing. J. Am. Chem. Soc. 1985, 107, 276–277. [Google Scholar] [CrossRef]
- Béchet, J.-J.; Dupaix, A.; Yon, J.; Wakselman, M.; Robert, J.-C.; Vilkas, M. Inactivation of α-Chymotrypsin by a Bifunctional Reagent, 3,4-Dihydro-3,4-dibromo-6-bromomethylcoumarin. Eur. J. Biochem. 1973, 35, 527–539. [Google Scholar] [CrossRef]
- Wakselman, M. 1,4- and 1,6-Eliminations from Hydroxy- and Amino-Substituted Benzyl Systems: Chemical and Biochemical Applications. Nouv. J. Chim. 1983, 7, 439–447. [Google Scholar] [CrossRef]
- Harper, J.W.; Powers, J.C. 3-Alkoxy-7-amino-4-chloroisocoumarins: A New Class of Suicide Substrates for Serine Proteases. J. Am. Chem. Soc. 1984, 106, 7619–7621. [Google Scholar] [CrossRef]
- Halazy, S.; Danzin, C.; Ehrhard, A.; Gerhart, F. 1,1-Difluoroalkyl glucosides: A new class of enzyme-activated irreversible inhibitors of α-glucosidases. J. Am. Chem. Soc. 1989, 111, 3484–3485. [Google Scholar] [CrossRef]
- Halazy, S.; Berges, V.; Ehrhard, A.; Danzin, C. Ortho- and para-(difluoromethyl)aryl-β-d-glucosidases. Bioorg. Chem. 1990, 18, 330–344. [Google Scholar] [CrossRef]
- Wakselman, M.; Cerutti, I.; Chany, C. Nitrobenzyl esters as potential conjugated alkylating and differentiation promoting agents: Antitumor effect in vivo. Eur. J. Med. Chem. 1990, 25, 519–526. [Google Scholar] [CrossRef]
- Wakselman, M.; Xie, J.; Mazaleyrat, J.-P. New Mechanism-Based Inactivators of Trypsin-like Proteinases. Selective Inactivation of Urokinase by Functionalized Cyclopeptides Incorporating a Sulfoniomethyl-Substituted m-Aminobenzoic Acid Residue. J. Med. Chem. 1993, 36, 1539–1547. [Google Scholar] [CrossRef]
- Myers, J.K.; Widlanski, T.S. Mechanism-based inactivation of prostatic acid phosphatase. Science 1993, 262, 1451–1453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dechert, U.; Jirik, F.; Withers, S.G. Suicide Inactivation of Human Prostatic Acid Phosphatase and a Phosphotyrosine Phosphatase. Biochem. Biophys. Res. Commun. 1994, 200, 577–583. [Google Scholar] [CrossRef]
- Born, T.L.; Myers, J.K.; Widlanski, T.S.; Rusnak, F. 4-(Fluoromethyl)phenyl phosphate acts as a mechanism-based inhibitor of calcineurin. J. Biol. Chem. 1995, 270, 25651–25655. [Google Scholar] [CrossRef] [Green Version]
- Myers, J.K.; Cohen, J.D.; Widlanski, T.S. Substituent Effects on the Mechanism-Based Inactivation of Prostatic Acid Phosphatase. J. Am. Chem. Soc. 1995, 117, 11049–11054. [Google Scholar] [CrossRef]
- Massière, F.; Badet-Denisot, M.-A.; René, L.; Badet, B. Design, Synthesis and Evaluation of the First Mechanism-Based Inhibitor of Glucosamine 6-Phosphate Synthase. J. Am. Chem. Soc. 1997, 119, 5748–5749. [Google Scholar] [CrossRef]
- Aráoz, R.; Anhalt, E.; René, L.; Badet-Denisot, M.-A.; Courvalin, P.; Badet, B. Mechanism-Based Inactivation of VanX, a d-Alanyl-d-alanine Dipeptidase Necessary for Vancomycin Resistance. Biochemistry 2000, 39, 15971–15979. [Google Scholar] [CrossRef] [PubMed]
- Yaouancq, L.; Anissimova, M.; Badet-Denisot, M.-A.; Badet, B. Design and Evaluation of Mechanism-Based Inhibitors of d-Alanyl-d-alanine Dipeptidase VanX. Eur. J. Org. Chem. 2002, 2002, 3573–3579. [Google Scholar] [CrossRef]
- Janda, K.D.; Lo, L.-C.; Lo, C.-H.L.; Sim, M.-M.; Wang, R.; Wong, C.-H.; Lerner, R.A. Chemical Selection for Catalysis in Combinatorial Antibody Libraries. Science 1997, 275, 945–948. [Google Scholar] [CrossRef]
- Betley, J.R.; Cesaro-Tadic, S.; Mekhalfia, A.; Rickard, J.H.; Denham, H.; Partridge, L.J.; Plückthun, A.; Blackburn, G.M. Direct Screening for Phosphatase Activity by Turnover-Based Capture of Protein Catalysts. Angew. Chem. Int. Ed. 2002, 41, 775–777. [Google Scholar] [CrossRef]
- Cesaro-Tardic, S.; Lagos, D.; Honegger, A.; Rickard, J.H.; Partridge, L.J.; Blackburn, G.M.; Plückthun, A. Turnover-based in vitro selection and evolution of biocatalysts from a fully synthetic antibody library. Nat. Biotechnol. 2003, 21, 679–685. [Google Scholar] [CrossRef]
- Lo, L.-C.; Lo, C.-H.L.; Janda, K.D. A Versatile Mechanism-Based Reaction Probe for the Direct Selection of Biocatalysts. Bioorg. Med. Chem. Lett. 1996, 6, 2117–2120. [Google Scholar] [CrossRef]
- Lo, L.-C.; Chiang, Y.-L.; Kuo, C.-H.; Liao, H.-K.; Chen, Y.-J.; Lin, J.-J. Study of the preferred modification sites of the quinone methide intermediate resulting from the latent trapping device of the activity probes for hydrolases. Biochem. Biophys. Res. Commun. 2005, 326, 30–35. [Google Scholar] [CrossRef]
- Lo, L.-C.; Wang, H.-Y.; Wang, Z.-J. Design and Synthesis of an Activity Probe for Protein Tyrosine Phosphatases. J. Chin. Chem. Soc. 1999, 46, 715–718. [Google Scholar] [CrossRef]
- Lo, L.-C.; Pang, T.-L.; Kuo, C.-H.; Chiang, Y.-L.; Wang, H.-Y.; Lin, J.-J. Design and Synthesis of Class-Selective Activity Probes for Protein Tyrosine Phosphatases. J. Proteome Res. 2002, 1, 35–40. [Google Scholar] [CrossRef]
- Zhu, Q.; Huang, X.; Chen, G.Y.J.; Yao, S.Q. Activity-based fluorescent probes that target phosphatases. Tetrahedron Lett. 2003, 44, 2669–2672. [Google Scholar] [CrossRef]
- Shen, K.; Qi, L.; Ravula, M.; Klimaszewski, K. Synthesis and peptide incorporation of an unnatural amino acid containing activity-based probe for protein tyrosine phosphatases. Bioorg. Med. Chem. Lett. 2009, 19, 3264–3267. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Qi, L.; Ravula, M. Facile Incorporation of a Phosphatase Activity-Dependent Quinone Methide Generating Motif into Phosphotyrosine. Synthesis 2009, 22, 3765–3768. [Google Scholar] [CrossRef]
- Kalesh, K.A.; Pheng, T.L.; Lu, K.; Gao, L.; Wang, J.; Yao, S.Q. Peptide-based activity-based probes (ABPs) for target-specific profiling of proteintyrosine phosphatases (PTPs). Chem. Commun. 2010, 46, 589–591. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; Kuo, C.-C.; Chu, C.-Y.; Huang, Y.-H.; Hu, Y.-L.; Lin, J.-J.; Lo, L.-C. Development of activity-based probes with tunable specificity for protein tyrosine phosphatase subfamilies. Tetrahedron 2010, 66, 4521–4529. [Google Scholar] [CrossRef]
- Polaske, N.W.; Kelly, B.D.; Ashworth-Sharpe, J.; Bieniarz, C. Quinone Methide Signal Amplification: Covalent Reporter Labeling of Cancer Epitopes using Alkaline Phosphatase Substrates. Bioconjug. Chem. 2016, 27, 660–666. [Google Scholar] [CrossRef]
- Song, H.; Li, Y.; Xue, C.; Xie, H. Highly Efficient Multiple-Labeling Probes for the Visualization of Enzyme Activities. Eur. J. Chem. 2019, 25, 13994–14002. [Google Scholar] [CrossRef]
- Li, X.; Gao, X.; Shi, W.; Ma, H. Design Strategies for Water-Soluble Small Molecular Chromogenic and Fluorogenic Probes. Chem. Rev. 2014, 114, 590–659. [Google Scholar] [CrossRef]
- Ge, J.; Li, L.; Yao, S.Q. A self-immobilizing and fluorogenic unnatural amino acid that mimics phosphotyrosine. Chem. Commun. 2011, 47, 10939–10941. [Google Scholar] [CrossRef]
- Li, Y.; Song, H.; Xue, C.; Fang, Z.; Xiong, L.; Xie, H. A self-immobilizing near-infrared fluorogenic probe for sensitive imaging of extracellular enzyme activity in vivo. Chem. Sci. 2020, 11, 5889–5894. [Google Scholar] [CrossRef]
- Chen, Y.; Xue, C.; Wang, J.; Xu, M.; Li, Y.; Ding, Y.; Song, H.; Xu, W.; Xie, H. High-contrast and real-time visualization of membrane proteins in live cells with malachite green-based fluorogenic probes. Chin. Chem. Lett. 2022, 33, 1637–1642. [Google Scholar] [CrossRef]
- Ichikawa, M.; Ichikawa, Y. A Mechanism-Based Affinity-Labeling Agent for Possible Use in Isolating N-Acetylglucosaminidase. Bioorg. Med. Chem. Lett. 2001, 11, 1769–1773. [Google Scholar] [CrossRef]
- Tsai, C.-S.; Li, Y.-K.; Lo, L.-C. Design and Synthesis of Activity Probes for Glycosidases. Org. Lett. 2002, 4, 3607–3610. [Google Scholar] [CrossRef]
- Kurogochi, M.; Nishimura, S.-I.; Chuan Lee, Y. Mechanism-based Fluorescent Labeling of β-Galactosidases. J. Biol. Chem. 2004, 279, 44704–44712. [Google Scholar] [CrossRef] [Green Version]
- Shie, T.-H.; Chiang, Y.-L.; Lin, J.-J.; Li, Y.-K.; Lo, L.-C. Facile synthesis toward the construction of an activity probe library for glycosidases. Carbohydr. Res. 2006, 341, 443–456. [Google Scholar] [CrossRef]
- Lo, L.-C.; Chu, C.-Y.; Pan, Y.-R.; Wan, C.-F.; Li, Y.-K.; Lin, J.-J. Rapid and selective isolation of β-xylosidase through an activity-based chemical approach. Biotechnol. J. 2006, 1, 197–202. [Google Scholar] [CrossRef]
- Komatsu, T.; Kikuchi, K.; Takakusa, H.; Hanaoka, K.; Ueno, T.; Kamiya, M.; Urano, Y.; Nagano, T. Design and Synthesis of an Enzyme Activity-Based Labeling Molecule with Fluorescence Spectral Change. J. Am. Chem. Soc. 2006, 128, 15946–15947. [Google Scholar] [CrossRef]
- Kalidasan, K.; Su, Y.; Wu, X.; Yao, S.Q.; Uttamchandani, M. Fluorescence-activated cell sorting and directed evolution of α-N-acetylgalactosaminidases using a quenched activity-based probe (qABP). Chem. Commun. 2013, 49, 7237–7239. [Google Scholar] [CrossRef] [Green Version]
- Kwan, D.H.; Chen, H.-M.; Ratananikom, K.; Hancock, S.M.; Watanabe, Y.; Kongsaeree, P.T.; Samuels, A.L.; Withers, S.G. Self-Immobilizing Fluorogenic Imaging Agents of Enzyme Activity. Angew. Chem. Int. Ed. 2011, 50, 300–303. [Google Scholar] [CrossRef]
- Cheng, T.-C.; Roffler, S.R.; Tzou, S.-C.; Chuang, K.-H.; Su, Y.-C.; Chuang, C.-H.; Kao, C.-H.; Chen, C.-S.; Harn, I.-H.; Liu, K.-Y.; et al. An Activity-Based Near-Infrared Glucuronide Trapping Probe for Imaging β-Glucuronidase Expression in Deep Tissues. J. Am. Chem. Soc. 2012, 134, 3103–3110. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Nandakumar, M.; Lai, H.-Y.; Chou, T.-C.; Chu, C.-Y.; Lin, C.-H.; Lo, L.-C. Development of Activity-Based Probes for Imaging Human α-l-Fucosidases in Cells. J. Org. Chem. 2015, 80, 8458–8463. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, M.; Hsu, Y.-L.; Lin, J.C.-Y.; Lo, C.; Lo, L.-C. Detection of Human α-L-Fucosidases by a Quinone Methide-Generating Probe: Enhanced Activities in Response to Helicobacter pylori Infection. ChemBioChem 2015, 16, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
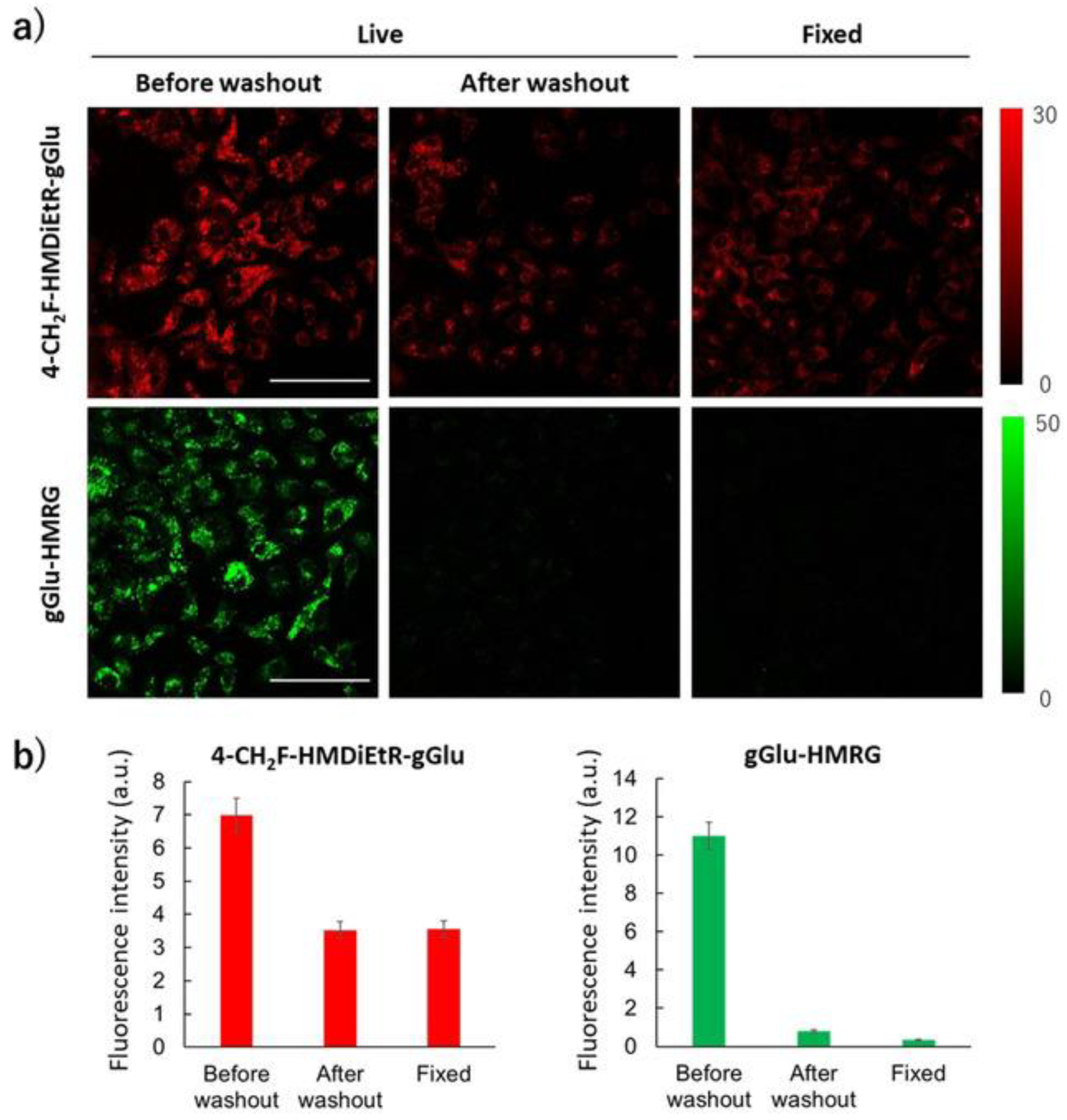
- Doura, T.; Kamiya, M.; Obata, F.; Yamaguchi, Y.; Hiyama, T.Y.; Matsuda, T.; Fukamizu, A.; Noda, M.; Miura, M.; Urano, Y. Detection of LacZ-Positive Cells in Living Tissue with Single-Cell Resolution. Angew. Chem. Int. Ed. 2016, 55, 9620–9624. [Google Scholar] [CrossRef]
- Kamiya, M.; Asanuma, D.; Kuranaga, E.; Takeishi, A.; Sakabe, M.; Miura, M.; Nagano, T.; Urano, Y. β-Galactosidase Fluorescence Probe with Improved Cellular Accumulation Based on a Spirocyclized Rhodol Scaffold. J. Am. Chem. Soc. 2011, 133, 12960–12963. [Google Scholar] [CrossRef]
- Ito, H.; Kawamata, Y.; Kamiya, M.; Tsuda-Sakurai, K.; Tanaka, S.; Ueno, T.; Komatsu, T.; Hanaoka, K.; Okabe, S.; Miura, M.; et al. Red-Shifted Fluorogenic Substrate for Detection of lacZ-Positive Cells in Living Tissue with Single-Cell Resolution. Angew. Chem. Int. Ed. 2018, 57, 15702–15706. [Google Scholar] [CrossRef]
- Chiba, M.; Kamiya, M.; Tsuda-Sakurai, K.; Fujisawa, Y.; Kosakamoto, H.; Kojima, R.; Miura, M.; Urano, Y. Activatable Photosensitizer for Targeted Ablation of lacZ-Positive Cells with Single-Cell Resolution. ACS Cent. Sci. 2019, 5, 1676–1681. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Kamiya, M.; Obata, F.; Miura, M.; Terai, T.; Komatsu, T.; Ueno, T.; Hanaoka, K.; Nagano, T.; Urano, Y. Selective Ablation of β-Galactosidase-Expressing Cells with a Rationally Designed Activatable Photosensitizer. Angew. Chem. Int. Ed. 2014, 53, 6772–6775. [Google Scholar] [CrossRef]
- Jiang, J.; Tan, Q.; Zhao, S.; Song, H.; Hu, L.; Xie, H. Late-stage difluoromethylation leading to a self-immobilizing fluorogenic probe for the visualization of enzyme activities in live cells. Chem. Commun. 2019, 55, 15000–15003. [Google Scholar] [CrossRef]
- Noguchi, K.; Shimomura, T.; Ohuchi, Y.; Ishiyama, M.; Shiga, M.; Mori, T.; Katayama, Y.; Ueno, Y. β-Galactosidase-Catalyzed Fluorescent Reporter Labeling of Living Cells for Sensitive Detection of Cell Surface Antigens. Bioconjug. Chem. 2020, 31, 1740–1744. [Google Scholar] [CrossRef]
- Hirata, M.; Kogame, T.; Adachi, S.; Haga, H. Galactosidase-catalyzed fluorescence amplification method (GAFAM): Sensitive fluorescent immunohistochemistry using novel fluorogenic β-galactosidase substrates and its application in multiplex immunostaining. Histochem. Cell Biol. 2022, 1–14. [Google Scholar] [CrossRef]
- Hyun, J.Y.; Park, S.-H.; Park, C.W.; Kim, H.B.; Cho, J.W.; Shin, I. Trifunctional Fluorogenic Probes for Fluorescence Imaging and Isolation of Glycosidases in Cells. Org. Lett. 2019, 21, 4439–4442. [Google Scholar] [CrossRef] [PubMed]
- Whidbey, C.; Sadler, N.C.; Nair, R.N.; Volk, R.F.; DeLeon, A.J.; Bramer, L.M.; Fansler, S.J.; Hansen, J.R.; Shukla, A.K.; Jansson, J.K.; et al. A Probe-Enabled Approach for the Selective Isolation and Characterization of functionally Active Subpopulations in the Gut Microbiome. J. Am. Chem. Soc. 2019, 141, 42–47. [Google Scholar] [CrossRef]
- Luijkx, Y.M.C.A.; Henselijn, A.; Bosman, G.P.; Cramer, D.A.T.; Giesbers, K.C.A.P.; van ’t Veld, E.M.; Boons, G.-J.; Heck, A.J.R.; Reiding, K.R.; Strijbis, K.; et al. Detection of Bacterial α-l-Fucosidases with an Ortho-Quinone Methide-Based Probe and Mapping of the Probe-Protein Adducts. Molecules 2022, 27, 1615. [Google Scholar] [CrossRef]
- Liu, J.; Ma, X.; Cui, C.; Chen, Z.; Wang, Y.; Deenik, P.R.; Cui, L. Noninvasive NIR Imaging of Senescence via In Situ Labeling. J. Med. Chem. 2021, 64, 17969–17978. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-P.; Ren, C.-T.; Lai, Y.-N.; Wu, S.-H.; Wang, W.-M.; Chen, J.-Y.; Lo, L.-C. Design of a Mechanism-Based Probe for Neuraminidase To Capture Influenza Viruses. Angew. Chem. Int. Ed. 2005, 44, 6888–6892. [Google Scholar] [CrossRef]
- Hinou, H.; Kurogochi, M.; Shimizu, H.; Nishimura, S.-I. Characterization of Vibrio cholerae Neuraminidase by a Novel Mechanism-Based Fluorescent Labeling Reagent. Biochemistry 2005, 44, 11669–11675. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, S.; Xue, Z.; Han, J.; Han, S. Senescence-associated sialidase revealed by an activatable fluorescence-on labeling probe. Chem. Commun. 2018, 54, 11566–11569. [Google Scholar] [CrossRef]
- Gao, Z.; Thompson, A.J.; Paulson, J.C.; Withers, S.G. Proximity Ligation-Based Fluorogenic Imaging Agents for Neuraminidases. Angew. Chem. Int. Ed. 2018, 57, 13538–13541. [Google Scholar] [CrossRef]
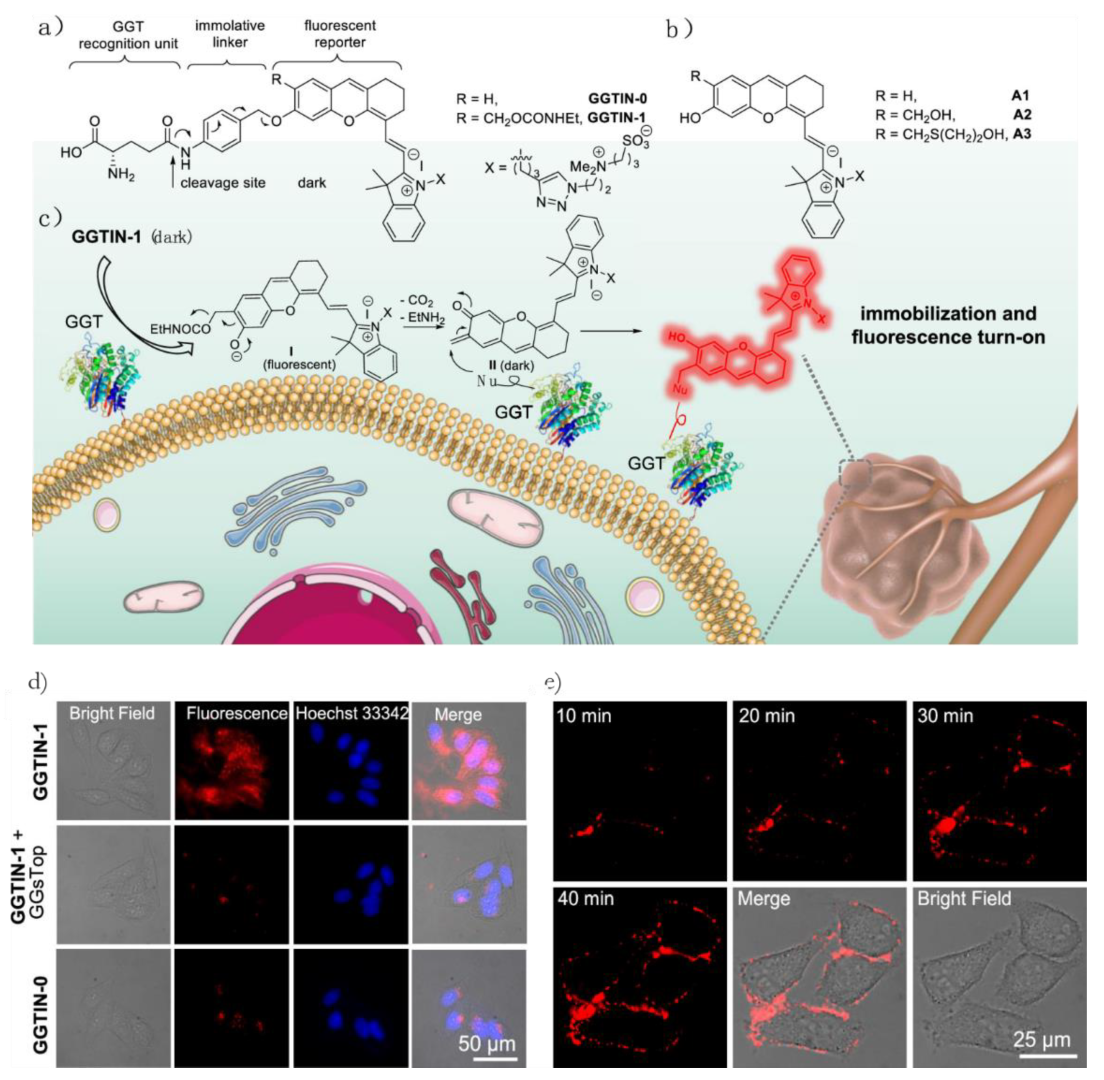
- Li, Y.; Xue, C.; Fang, Z.; Xu, W.; Xie, H. In Vivo Visualization of γ-Glutamyl Transpeptidase Activity with an Activatable Self-Immobilizing Near-Infrared Probe. Anal. Chem. 2020, 92, 15017–15024. [Google Scholar] [CrossRef]
- Obara, R.; Kamiya, M.; Tanaka, Y.; Abe, A.; Kojima, R.; Kawaguchi, T.; Sugawara, M.; Takahashi, A.; Noda, T.; Urano, Y. γ-Glutamyltranspeptidase (GGT)-Activatable Fluorescence Probe for Durable Tumor Imaging. Angew. Chem. Int. Ed. 2021, 60, 2125–2129. [Google Scholar] [CrossRef]
- Lu, C.-P.; Ren, C.-T.; Wu, S.-H.; Chu, C.-Y.; Lo, L.-C. Development of an Activity-Based Probe for Steroid Sulfatases. ChemBioChem 2007, 8, 2187–2190. [Google Scholar] [CrossRef]
- Lenger, J.; Schröder, M.; Ennemann, E.C.; Müller, B.; Wong, C.-H.; Noll, T.; Dierks, T.; Hanson, S.R.; Sewald, N. Evaluation of sulfatase-directed quinone methide traps for proteomics. Bioorg. Med. Chem. 2012, 20, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, C.-H.; Lu, C.-P.; Wu, S.-H.; Lo, L.-C. Synthesis and evaluation of turn-on fluorescent probes for imaging steroid sulfatase activities in cells. Chem. Commun. 2014, 50, 6116–6119. [Google Scholar] [CrossRef]
- Park, H.-J.; Rhee, H.-W.; Hong, J.-I. Activity-based fluorescent probes for monitoring sulfatase activity. Bioorg. Med. Chem. Lett. 2012, 22, 4939–4941. [Google Scholar] [CrossRef]
- Shao, Q.; Zheng, Y.; Dong, X.; Tang, K.; Yan, X.; Xing, B. A Covalent Reporter of β-Lactamase Activity for Fluorescent Imaging and Rapid Screening of Antibiotic-Resistant Bacteria. Chem. Eur. J. 2013, 19, 10903–10910. [Google Scholar] [CrossRef]
- Mao, W.; Xia, L.; Wang, Y.; Xie, H. A Self-Immobilizing and Fluorogenic Probe for β-Lactamase Detection. Chem. Asian J. 2016, 11, 3493–3497. [Google Scholar] [CrossRef]
- Sellars, J.-D.; Landrum, M.; Congreve, A.; Dixon, D.P.; Mosely, J.A.; Beeby, A.; Edwards, R.; Steel, P.G. Fluorescence quenched quinone methide based activity probes–a cautionary tale. Org. Biomol. Chem. 2010, 8, 1610–1618. [Google Scholar] [CrossRef]
- Shi, Y.; Zhu, R.; Xue, Z.; Han, J.; Han, S. An in cellulo-activated multicolor cell labeling approach used to image dying cell clearance. Analyst 2019, 144, 4687–4693. [Google Scholar] [CrossRef]
- Wang, S.; Tan, W.; Lang, W.; Qian, H.; Guo, S.; Zhu, L.; Ge, J. Fluorogenic and Mitochondria-Localizable Probe Enables Selective Labeling and Imaging of Nitroreductase. Anal. Chem. 2022, 94, 7272–7277. [Google Scholar] [CrossRef]
- Cao, S.; Wang, Y.; Peng, X. ROS-Inducible DNA Cross-Linking Agent as a New Anticancer Prodrug Building Block. Chem. Eur. J. 2012, 18, 3850–3854. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Wang, Y.; Peng, X. The Leaving Group Strongly Affects H2O2-Induced DNA Cross-Linking by Arylboronates. J. Org. Chem. 2014, 79, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fan, H.; Balakrishnan, K.; Lin, Z.; Cao, S.; Chen, W.; Fan, Y.; Guthrie, Q.A.; Sun, H.; Teske, K.A.; et al. Hydrogen peroxide activated quinone methide precursors with enhanced DNA cross-linking capability and cytotoxicity towards cancer cells. Eur. J. Med. Chem. 2017, 133, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Tamura, T.; Fujisawa, A.; Nishikawa, Y.; Cheng, R.; Takato, M.; Hamachi, I. Imaging and Profiling of Proteins under Oxidative Conditions in Cells and Tissues by Hydrogen-Peroxide-Responsive Labeling. J. Am. Chem. Soc. 2020, 142, 15711–15721. [Google Scholar] [CrossRef]
- Iwashita, H.; Castillo, E.; Messina, M.S.; Swanson, R.A.; Chang, C.J. A tandem activity-based sensing and labeling strategy enables imaging of transcellular hydrogen peroxide signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2018513118. [Google Scholar] [CrossRef]
- Jung, Y.L.; Sarkar, S.; Ha, J.; Park, S.B.; Ahn, K.H. Fluorophore Labeling of Proteins: A Versatile Trigger–Release–Conjugation Platform Based on the Quinone Methide Chemistry. Bioconjug. Chem. 2022, 33, 1543–1551. [Google Scholar] [CrossRef]
- Basarić, N.; Mlinarić-Majerski, K.; Kralj, M. Quinone Methides: Photochemical Generation and its Application in Biomedicine. Curr. Org. Chem. 2014, 18, 3–18. [Google Scholar] [CrossRef]
- Zlatić, K.; Antol, I.; Uzelac, L.; Mikecin Dražić, A.-M.; Kralj, M.; Bohne, C.; Basarić, N. Labeling of Proteins by BODIPY-Quinone Methides Utilizing Anti-Kasha Photochemistry. ACS. Appl. Mater. Interfaces 2020, 12, 347–351. [Google Scholar] [CrossRef]
- Zlatić, K.; Cindrić, M.; Antol, I.; Uzelac, L.; Mihaljević, B.; Kralj, M.; Basarić, N. Wavelength dependent photochemistry of BODIPY–phenols and their applications in the fluorescent labeling of proteins. Org. Biomol. Chem. 2021, 19, 4891–4903. [Google Scholar] [CrossRef]
- Kashima, H.; Kamiya, M.; Obata, F.; Kojima, R.; Nakano, S.; Miura, M.; Urano, Y. Photoactivatable fluorophores for durable labelling of individual cells. Chem. Commun. 2021, 57, 5802–5805. [Google Scholar] [CrossRef]
- Zlatić, K.; Bogomolec, M.; Cindrić, M.; Uzelac, L.; Basarić, N. Synthesis, photophysical properties, anti-Kasha photochemical reactivity and biological activity of vinyl- and alkynyl-BODIPY derivatives. Tetrahedron 2022, 124, 132995. [Google Scholar] [CrossRef]
- Wang, P.; Song, Y.; Zhang, L.; He, H.; Zhou, X. Quinone Methide Derivatives: Important Intermediates to DNA Alkylating and DNA Cross-linking Actions. Curr. Med. Chem. 2005, 12, 2893–2913. [Google Scholar] [CrossRef] [PubMed]
- Percivalle, C.; Doria, F.; Freccero, M. Quinone Methides as DNA Alkylating Agents: An Overview on Efficient Activation Protocols for Enhanced Target Selectivity. Curr. Org. Chem. 2014, 18, 19–43. [Google Scholar] [CrossRef]
- Huang, C.; Rokita, S.E. DNA alkylation promoted by an electron-rich quinone methide intermediate. Front. Chem. Sci. Eng. 2016, 10, 213–221. [Google Scholar] [CrossRef]
- Hutchinson, M.A.; Deeyaa, B.D.; Byrne, S.R.; Williams, S.J.; Rokita, S.E. Directing Quinone Methide-Dependent Alkylation and Cross-Linking of Nucleic Acids with Quaternary Amines. Bioconjug. Chem. 2020, 31, 1486–1496. [Google Scholar] [CrossRef]
- Deeyaa, B.D.; Rokita, S.E. Migratory ability of quinone methide-generating acridine conjugates in DNA. Org. Biomol. Chem. 2020, 18, 1671–1678. [Google Scholar] [CrossRef]
- Byrne, S.R.; Rokita, S.E. Unraveling Reversible DNA Cross-Links with a Biological Machine. Chem. Res. Toxicol. 2020, 33, 2903–2913. [Google Scholar] [CrossRef]
- Qian, L.; Li, L.; Yao, S.Q. Two-Photon Small Molecule Enzymatic Probes. Acc. Chem. Res. 2016, 49, 626–634. [Google Scholar] [CrossRef]
- Wang, S.; Li, B.; Zhang, F. Molecular Fluorophores for Deep-Tissue Bioimaging. ACS Cent. Sci. 2020, 6, 1302–1316. [Google Scholar] [CrossRef]
- Zhang, X.; Li, S.; Ma, H.; Wang, H.; Zhang, R.; Zhang, X.-D. Activatable NIR-II organic fluorescent probes for bioimaging. Theranostics 2022, 12, 3345–3371. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Target Enzyme | ki (min−1) | Ki (mM) | Selectivity between PTPs | |
|---|---|---|---|---|---|
 | GGDEGIHXSELI | SHP1 | 0.78 | 2.81 | moderate selectivity |
| GGGSAAPXLKTK | PTPB1/TCPTP | 0.68 | 1.00 | moderate selectivity | |
| GGKAVDGXVKPQ | PTP1B | 0.71 | 0.97 | high PTPB1 selectivity | |
| GGLNSDGXTPEP | LMWPTP | 0.80 | 2.19 | moderate selectivity | |
| GGLPPEGXVVVV | SHP2 | 0.74 | 3.89 | moderate selectivity | |
| GGNSDVQXTEVQ | SHP1/SHP2 | 0.73 | 1.93 | moderate selectivity | |
| GGPEGHEXpYRVR | PTP1B | 0.75 | 0.94 | moderate selectivity | |
| GGPQDKEXYKVK | SHP2/PTP1B | 0.75 | 2.45 | moderate selectivity | |
| GGVDADEXLIPQ | SHP2/PTP1B | 0.71 | 1.48 | moderate selectivity | |
| GGELEFXMDE | PTP1B | 0.61 | 1.00 | moderate selectivity | |
| Structure a | Target Enzyme | λ (nm) abs/em b | Notes | Studied System | Ref. |
|---|---|---|---|---|---|
 | PTP1B | 330/520 | no cross-labeling in a mixture of proteins | in vitro | [30] |
 | PTP (yeast), ALPs (calf, shrimp, human) | 555/565 | selective against other enzyme classes, no selectivity among phosphatases | in vitro | [31] |
 | PTP (yeast), ALPs (calf, shrimp, human) | 555/565 | selective against other enzyme classes, no selectivity among phosphatases | in vitro | [31] |
 | PTP1B | 540/565 | high PTPB1 selectivity against other PTPs, no cross-labeling in cell lysates | in vitro | [34] |
 | ALP | 490/520 | low labeling efficiency | in vitro | [37] c |
 | ALP | 490/520 | low labeling efficiency | in cellulo (HeLa) | [37] c |
 | ALP | 490/520 | cross-labeling with other protein | in vitro | [37] c |
 | ALP | 490/520 | cross-labeling with other protein | in vitro | [37] c |
 | ALP | 490/520 | cross-labeling with other protein, most efficient labeling d in vitro | in cellulo (HeLa) | [37] c |
 | ALP | 490/520 | cross-labeling with other protein, most efficient labeling d in cellulo | in cellulo (HeLa) | [37] c |
 | ALP | 620/670 | cross-labeling with other protein, most efficient labeling e in vitro | in cellulo (HeLa), in vivo (mice) | [37] c |
 | ALP | 620/670 | cross-labeling with other protein, most efficient labeling e in cellulo, low cytotoxicity | in cellulo (HeLa), in vivo (mice) | [37] c |
| Structure a | Target Enzyme | Turn-On Ratio | λ (nm) abs/em b | Notes | Studied System | Ref. |
|---|---|---|---|---|---|---|
 | PTP | ND | 360/460 | in vitro model compound for the other probes in [34], low efficiency but specific labeling, no loss of enzyme activity, kcat = 8.8 s−1, KM = 1.1 mM | in vitro | [39] |
 | PTP | ND | 360/460 | needs UV irradiation to uncage the active probe; localizes in plasma membrane | in cellulo (HeLa) | [39] |
 | PTP | ND | 360/460 | needs UV irradiation to uncage the active probe; localizes in ER | in cellulo (HeLa) | [39] |
 | PTP | ND | 360/460 | needs UV irradiation to uncage the active probe; localizes in mitochondria | in cellulo (HeLa) | [39] |
 | ALP | ND | 685/720 | unstable, hydrolyzes in PBS pH 7.4 | in vitro | [40] |
 | ALP | ND | 685/720 | stable, but ALP cannot hydrolyze the phosphate moiety | in vitro | [40] |
 | ALP | 40× | 685/720 | cross-labeling with other protein, but in cellulo most of the fluorescence is detected in the plasma membrane, where ALP is located, low cytotoxicity | in cellulo (HeLa) in vivo (mice) | [40] |
 | ALP | 10× c up to 200× d | 633/655–685 | higher photostability compared to the NIR dye from [32]; no cross-labeling studies, but in cellulo most of the fluorescence is detected in the plasma membrane, where ALP is located, low cytotoxicity | in cellulo (HeLa, HepG2, HCT116 cells) | [41] |
| Structure a | Target Enzyme b | λ (nm) abs/em c | Notes | Studied System | Ref. |
|---|---|---|---|---|---|
 | GLB | 360/520 | inactivates GLB: A. oryzae: Ki = 1.44 mM, t1/2 = 10.5 min bovine liver: Ki = 0.22 mM, t1/2 = 4.22 min B. circulans: Ki = 1.38 mM, t1/2 = 11.2 min X. manihotis: Ki = 38.5 mM, t1/2 = 21.1 min E. coli: Ki = 3.5 mM, t1/2 = 4.3 min | in cellulo (B16 cells) | [44] |
 | GLB | 400/515, 400/460 | the ratiometric fluorescence response can be used for accurate determination of enzyme concentration; kcat = 6.1 s−1, KM = 150 µM | in cellulo (LacZ-positive HEK293 cells) | [47] |
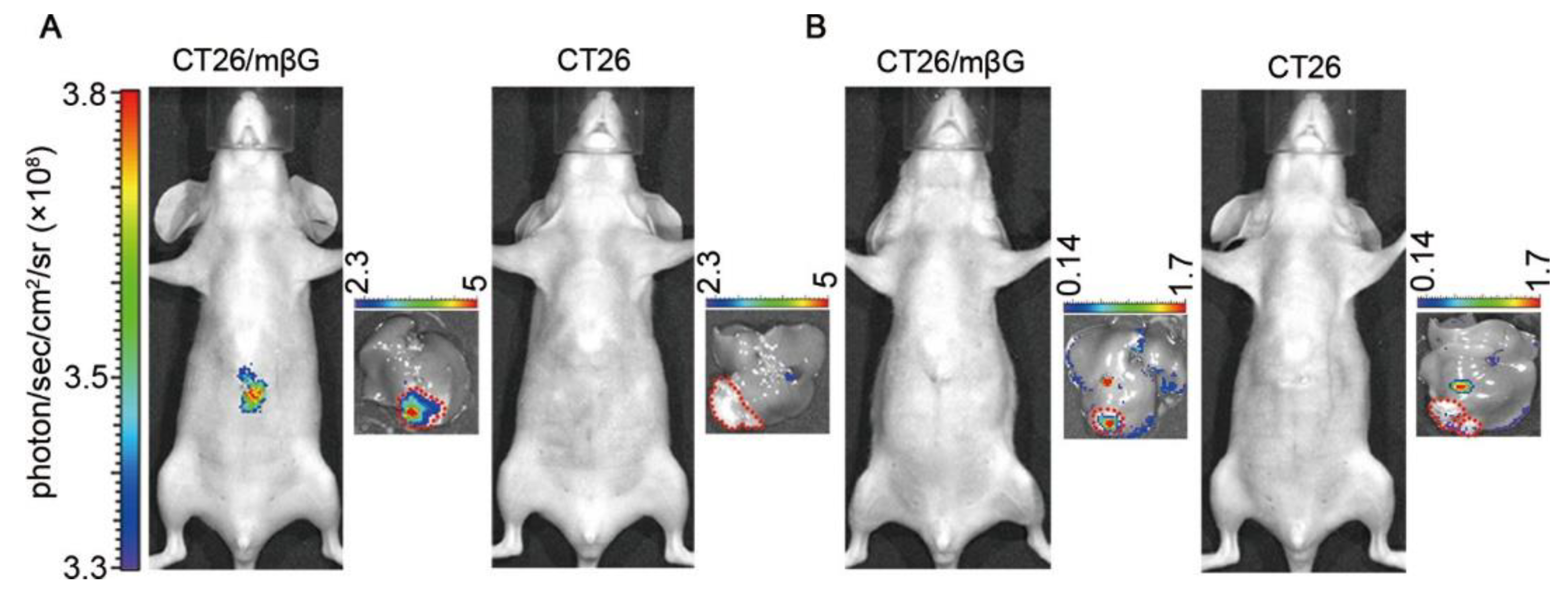
 | GLB | 490/525 | no loss of enzyme activity, cross-labeling with other proteins, can be used for in vivo imaging of subcutaneous tumors | in vitro (E. coli and CT26), in vivo (mice) | [50] |
 | GLB | 710/835 | can be used for in vivo imaging of subcutaneous and deep tissue tumors | in vivo (mice) | [50] |
 | FUCA | 497/520 | efficient labeling, localizes in lysosomes, low cytotoxicity | in cellulo (AGS and HEK293 cells) | [51,52] |
 | FUCA | 497/520 | low labeling efficiency | in vitro | [51] |
| Structure a | Target Enzyme b | Turn-On Ratio | λ (nm) abs/em c | Notes | Studied System | Ref. |
|---|---|---|---|---|---|---|
 | NAGA | ND | 490/520 | used for the directed evolution of NAGAs: wild type: kcat = 384 s−1, KM = 172 µM; variant: kcat = 287 s−1, KM = 62 µM; cross-labeling with other proteins | in vivo (NovaBlue (D3) competent cells) | [48] |
 | GUSB | ND | 330/450 | highly specific fluorescence labeling of the guard cells of stomata | in vivo (Arabidopsis (Col-0) plant) | [49] |
 | Abg/GB | ND | 330/450 | no cytotoxicity in non-Abg encoding E. coli, moderate cytotoxicity in Abg encoding E. coli; both probes can be used for cell sorting | in cellulo (E. coli R1360 cells, P. pastoris cells) | [49] |
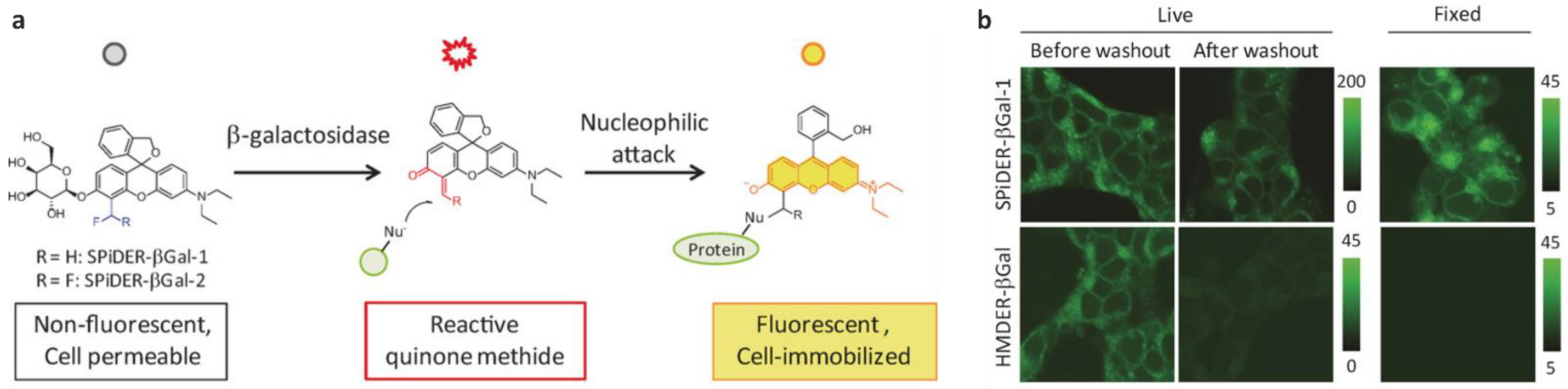
 | GLB | >650× | 525/560 | kcat = 3.38 s−1, KM = 16.9 µM; moderate cytotoxicity, better labeling efficiency than the difluoroderivative, cross-labeling with other proteins | in cellulo (HEK-lacZ(+) cells), ex vivo (Drosophila melanogaster and mice) | [53,60] |
 | GLB | >210× | 525/560 | kcat = 8.0 s−1, KM = 29.6 µM; low labeling efficiency | in vitro | [53] |
 | GLB | >150× | 610/630 | reduced reactivity of the quinone-methide leads to leaking of the probe from the cells | in cellulo (HEK-lacZ(+) cells) | [55] |
 | GLB | >150× | 610/630 | cross-labeling with other proteins | in cellulo (HEK-lacZ(+) cells, ex vivo (Drosophila melanogaster and mice) | [55] |
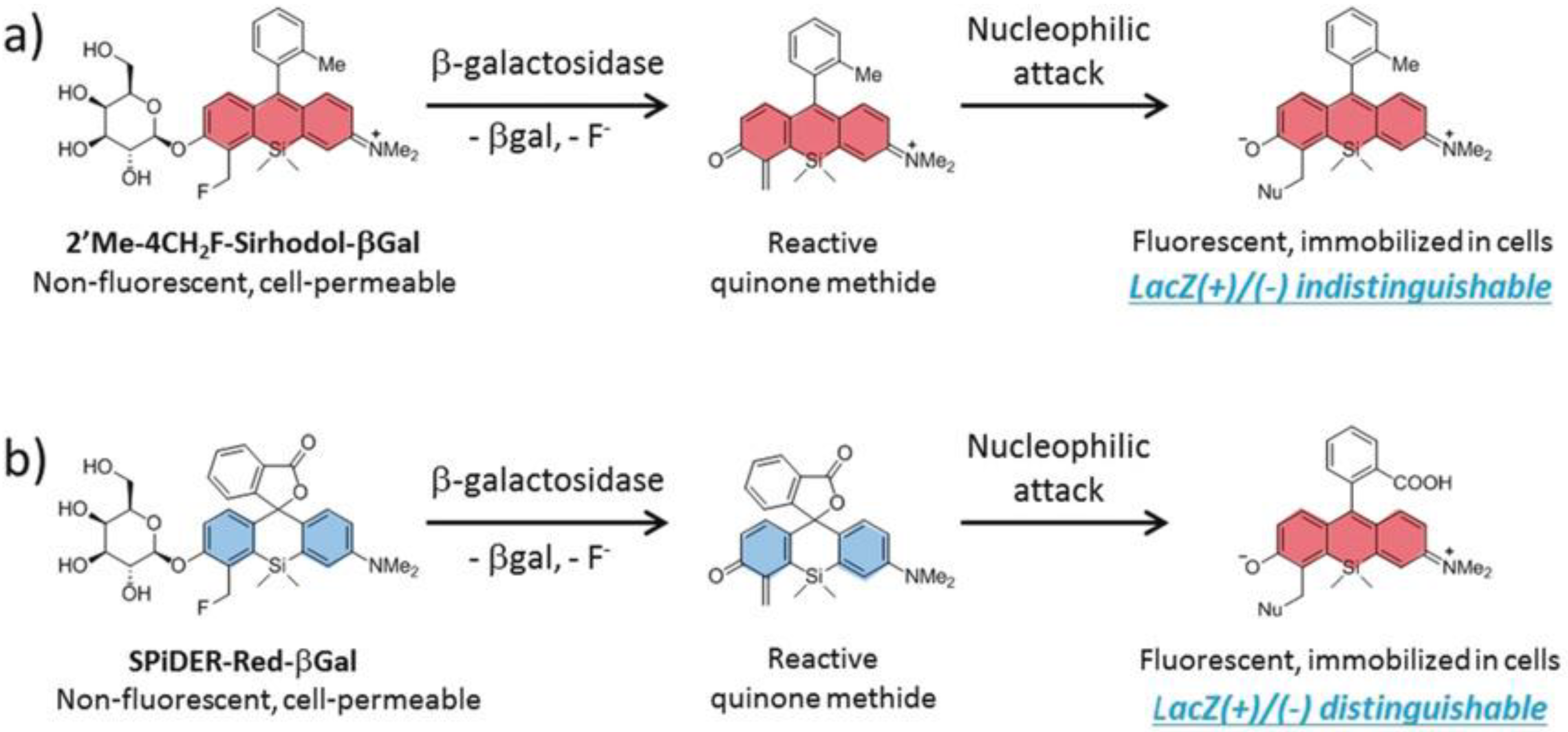
 | GLB | NA d | 525/560 | modulated photosensitizer, selective ablation of lacZ(+) cells, kcat = 3.32 s−1, KM = 10.1 µM | in vitro (HEK-lacZ(+) cells, ex vivo and in vivo (Drosophila melanogaster and mice) | [56] |
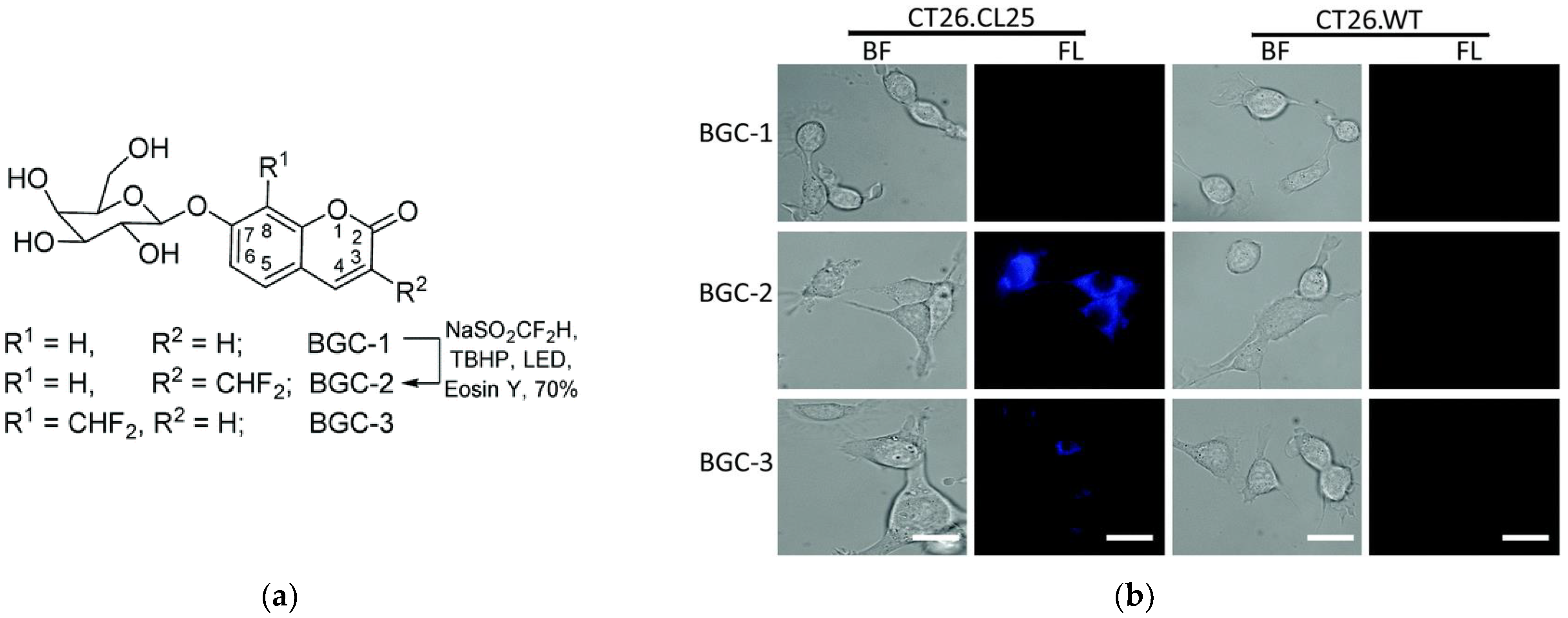
 | GLB | >200× | 416/454 | much higher fluorescence compared to the 8-difluoromethyl analogue, low cytotoxicity, cross-labeling with other proteins | in cellulo (HEK-lacZ(+), HeLa and CT26.CL25 cells) | [58] |
 | GLB | ND | 352/454 | low fluorescence upon hydrolysis by GLB | in cellulo (HEK-lacZ(+) and CT26.CL25 cells) | [58] |
 | GLB | ~200× | 323/460 | kcat = 54 min−1, Km = 0.062 μM fluorescent detection of cell surface antigens using GLB labeled antibody | in cellulo (A549 cells) | [59] |
 | GLB | ND | 330/450 | inactivates the enzyme, no cross-labeling in cell lysate | in vitro | [61] |
 | GB | ND | 330/450 | inactivates the enzyme, no cross-labeling in cell lysate | in vitro | [61] |
 | HEX | ND | 330/450 | inactivates the enzyme, no cross-labeling in cell lysate low cytotoxicity the alkyne enables subsequent attachment of an affinity tag for protein isolation | in cellulo (HT-29 cells) | [61] |
 | GLB | ND | 330/450 | moderate enzyme inactivation, low labeling efficiency | in vitro | [61] |
 | GB | ND | 330/450 | moderate enzyme inactivation, low labeling efficiency | in vitro | [61] |
 | GLB | ND | 650/710 | hydrolyzes in buffer | in vitro | [64] |
 | GLB | 16× | 650/710 | kcat = 14.6 s−1, Km = 9.3 μM more stable in buffer than the monofluoro derivative, low cytotoxicity | in cellulo (CT26.CL25, HeLa, MDA-MB-231, MCF7, IMR-90 cells), in vivo (mice) | [64] |
| Structure a | Turn-On Ratio | λ (nm) abs/em b | Kinetics | Notes | Studied System | Ref. |
|---|---|---|---|---|---|---|
 | NA | ND | Ki = 1.90 mM t1/2 = 2.83 min | inactivates the enzyme | in vitro | [66] |
 | 110× | 580/610 | ND | cross-labeling with other proteins | in cellulo (A549, HeLa, HepG2, MCF-7, U2OS, Huh-7) | [67] |
 | ND | 360/470 | KM = 2.4 μM c | cannot differentiate endogenous MDCK sialidase and viral sialidase | in cellulo (MDCK cells) | [68] |
 | ND | 360/470 | ND | lower fluorescence in cellulo compared to the deacetylated probe | in cellulo (MDCK cells) | [68] |
 | ND | 360/470 | ND | only hydrolyzed by viral sialidase, lower fluorescence in cellulo compared to the deacetylated probe | in cellulo (MDCK cells) | [68] |
 | ND | 360/470 | KM = 159 μM d | only hydrolyzed by viral sialidase | in cellulo (MDCK cells) | [68] |
| Structure a | Turn-On Ratio | λ (nm) abs/em b | Notes | Studied System | Ref. |
|---|---|---|---|---|---|
 | 40× | 690/715 | cross-labeling with other protein, no cytotoxicity | in cellulo (HepG2, U87MG) in vivo (mouse) | [69] |
 | >500× | 530/565 | cross-labeling with other protein, no loss of enzyme activity | in cellulo (SHIN3 A549, MIA PaCa-2, HepG2) in vivo (mouse) | [70] |
| Structure a | Target Enzyme | Turn-On Ratio | λ (nm) abs/em b | Notes | Studied System | Ref. |
|---|---|---|---|---|---|---|
 | PARS KARS STS | NA | 490/520 | inactivates PARS, KARS and STS, ARSG not affected; cross-labeling in cell lysate | in vitro | [72] |
 | STS | 7× | 360/465 | inactivates STS, cross-labeling not studied | in cellulo (CHO/STS) | [73] |
 | STS | NA | 360/465 | hydrolyzed by STS, but non-fluorescent side product both in vitro and in cellulo | in cellulo (CHO/STS) | [73] |
 | Sulfatase from Aerobacter aerogenes | 5× | 330/480 | cross-labeling not studied | in vitro | [74] |
| Structure a | Turn-On Ratio | λ (nm) abs/em b | kcat (min−1) | KM (μM) | Notes | Studied System | Ref. |
|---|---|---|---|---|---|---|---|
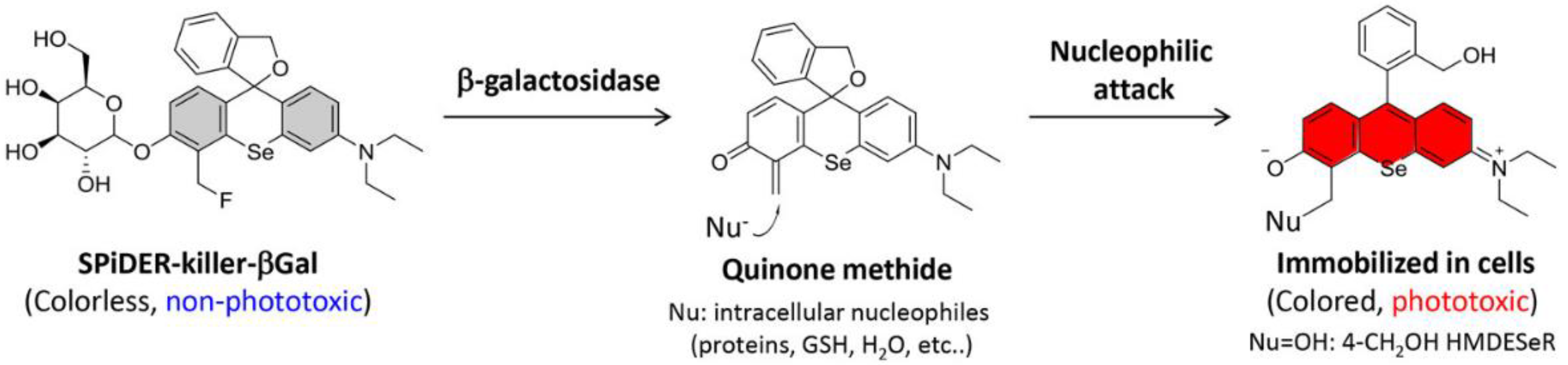
 | 38× | 490/520 | 4.92 | 3.08 | no loss of enzyme activity, cross-labeling not studied | in cellulo (E. coli, B. cereus, MRSA, S. aureus) | [75] c |
 | 110× | 525/570 | 2.16 | 4.28 | no loss of enzyme activity, cross-labeling not studied | in cellulo (E. coli, B. cereus, MRSA, S. aureus) | [75] c |
 | 80× | 650/680 | 1.49 | 5.24 | no loss of enzyme activity, cross-labeling not studied | in cellulo (E. coli, B. cereus, MRSA, S. aureus) | [75] c |
 | 50× | 365/460 | ND | ND | hydrolyzed by β-lactamase but weak fluorescence in cellulo | in cellulo (E. coli) | [76] |
 | <50× | 365/460 | ND | ND | cross-labeling not studied | in cellulo (E. coli, MDR A. baumannii, K. pneumonia) | [76] |
| Structure a | Target Enzyme | Turn-On Ratio | λ (nm) abs/em b | Notes | Studied System | Ref. |
|---|---|---|---|---|---|---|
 | AtCXE12 | 10× | 335/540 | cross-labeling with TaGSTL1 having a reactive Cys residue, no cross-labeling with BSA | in vitro | [77] |
 | esterase | NA | 375/445 | low cytotoxicity, cross-labeling not studied | in cellulo (HeLA, 3T3, A549, B16F10, Huh-7, U2OS and Raw 264.7) | [78] |
 | esterase | NA | 495/520 | low cytotoxicity, cross-labeling not studied | in cellulo (HeLA, 3T3, A549, B16F10, Huh-7, U2OS and Raw 264.7) | [78] |
 | esterase | NA | 550/575 | low cytotoxicity, in cellulo cross-labeling | in cellulo (HeLA, 3T3, A549, B16F10, Huh-7, U2OS and Raw 264.7) | [78] |
 | nitro-reductase | 10× | 520/560 | low cytotoxicity, no cross-labeling | in cellulo (A459) in vivo (zebrafish) | [79] |
| Structure a | ROS | Turn-On Ratio | λ (nm) abs/em b | Notes | Studied System | Ref. |
|---|---|---|---|---|---|---|
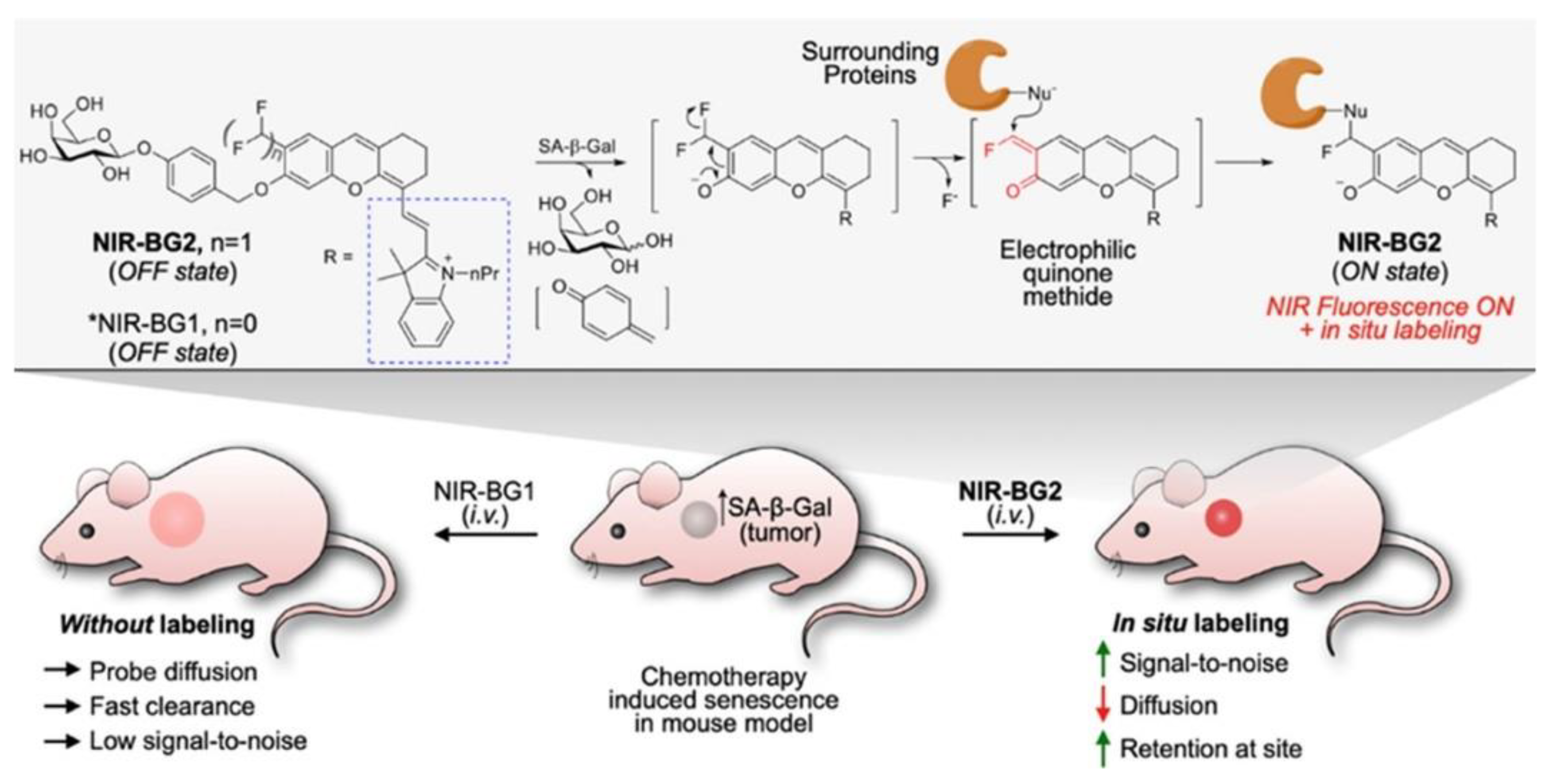
 | H2O2, ONOO− | NA | 360/470 | in vitro model compound | in vitro | [83] |
 | H2O2, ONOO− | ND | 490/520 | minor cellular toxicity, localized in vesicular organelles (phagosomes, endosomes, lysosomes, and autophagosomes) | in cellulo (Raw 264.7) ex-vivo (mouse brain tissue) | [83] |
 | H2O2 | 25× | 490/520 | minor cellular toxicity, no specific localization | in cellulo (HeLA, A431, Raw 264.7, microglia-neuron cocultures) | [84] |
 | H2O2, ONOO−, 1O2, O2−, OH | 30–70× | 350/530 | low cytotoxicity, highly localized in mitochondria and ER, can be used in two-photon microscopy | in cellulo (HeLA, Raw 264.7) ex-vivo (mouse kidney tissue) | [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kern, D.; Kormos, A. Self-Immobilizing Quinone Methides for the Fluorescent Sensing of Enzyme Activity. Chemosensors 2023, 11, 155. https://doi.org/10.3390/chemosensors11030155
Kern D, Kormos A. Self-Immobilizing Quinone Methides for the Fluorescent Sensing of Enzyme Activity. Chemosensors. 2023; 11(3):155. https://doi.org/10.3390/chemosensors11030155
Chicago/Turabian StyleKern, Dóra, and Attila Kormos. 2023. "Self-Immobilizing Quinone Methides for the Fluorescent Sensing of Enzyme Activity" Chemosensors 11, no. 3: 155. https://doi.org/10.3390/chemosensors11030155
APA StyleKern, D., & Kormos, A. (2023). Self-Immobilizing Quinone Methides for the Fluorescent Sensing of Enzyme Activity. Chemosensors, 11(3), 155. https://doi.org/10.3390/chemosensors11030155