
Distinctive Labeling of Live Monocytes and Neutrophils with a Single Fluorescent Molecule

 ,
,  ,
, {kind=link}
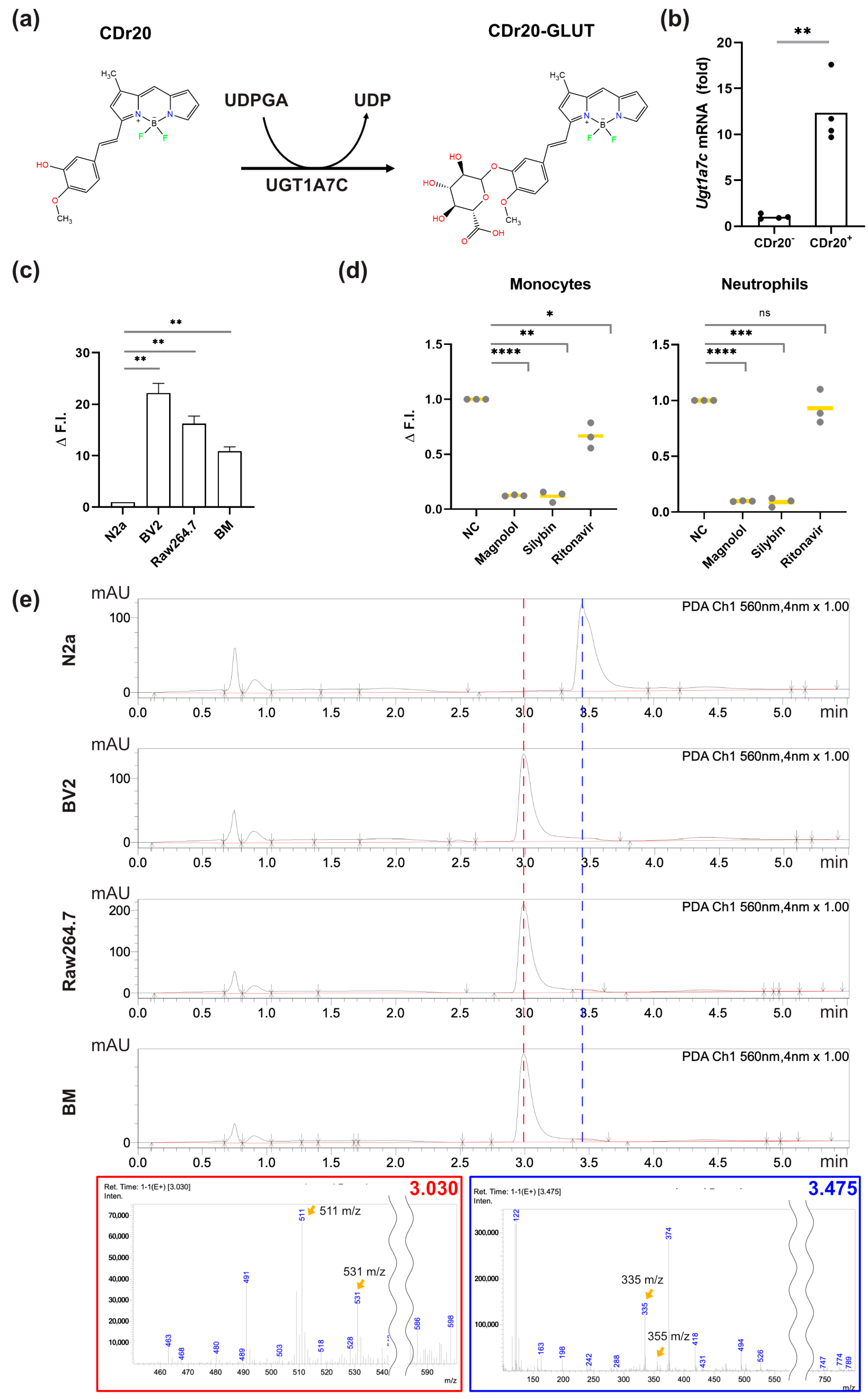{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Model and Cell Culture
2.2. Murine Immune Cell Isolation from Bone Marrow, Spleen, and Peripheral Blood
2.3. Surgical Procedures for In Vivo Imaging
2.4. Multiphoton In Vivo Imaging
2.5. Flow Cytometry Analysis and Fluorescence-Activated Cell Sorting (FACS)
2.6. Fluorimetric Cell Counting Using CDr20
2.7. Microsome Isolation
2.8. In Vitro UGT1A7C Conversion Test
2.9. Inhibition of UGT1 Isoforms and ABC Transporters
2.10. HPLC-MS to Detect CDr20-Gluc
2.11. Statistical Analysis
3. Results and Discussion
3.1. CDr20 Labels Circulating Phagocytic Myeloid Cells In Vivo
3.2. UGT1A7C Activity Is Responsible for the CDr20-Derived Fluorescence in Sub-Populations of Murine WBCs
3.3. CDr20 Preferentially Targets UGT1A7C-Expressed WBCs: Monocytes/Macrophages and Neutrophils
3.4. CDr20-Gluc Gets Exported out of Intracellular Space via ABCC Transporter
4. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Kantari, C.; Pederzoli-Ribeil, M.; Witko-Sarsat, V. The role of neutrophils and monocytes in innate immunity. Contrib. Microbiol. 2008, 15, 118–146. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.A. Chronic neutrophilic leukemia and chronic myelomonocytic leukemia: WHO defined. Best Pract. Res. Clin. Haematol. 2006, 19, 571–593. [Google Scholar] [CrossRef] [PubMed]
- Buoro, S.; Moioli, V.; Seghezzi, M.; Previtali, G.; Alessio, M.G.; Simon Lopez, R.; Ortolani, C.; Ottomano, C.; Lippi, G. Evaluation and comparison of automated hematology analyzer, flow cytometry, and digital morphology analyzer for monocyte counting. Int. J. Lab. Hematol. 2018, 40, 577–585. [Google Scholar] [CrossRef]
- Alamudi, S.H.; Chang, Y.T. Advances in the design of cell-permeable fluorescent probes for applications in live cell imaging. Chem. Commun. 2018, 54, 13641–13653. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.W.; Leong, C.; Zhai, D.; Tan, Y.L.; Lim, L.; Bi, X.; Lee, J.J.; Kim, H.J.; Kang, N.Y.; Ng, S.H.; et al. Neural stem cell specific fluorescent chemical probe binding to FABP7. Proc. Natl. Acad. Sci. USA 2012, 109, 10214–10217. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Lee, S.H.; Park, S.H.; Ciaramicoli, L.M.; Kwon, H.Y.; Cho, H.; Jeong, J.; Chang, Y.T. Neutrophil-Selective Fluorescent Probe Development through Metabolism-Oriented Live-Cell Distinction. Angew. Chem. Int. Ed. Engl. 2021, 60, 23743–23749. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, B.; Choi, S.; Balasubramaniam, S.; Lee, S.C.; Lee, J.Y.; Kim, H.S.; Kim, J.Y.; Kim, J.J.; Lee, Y.A.; et al. Imaging inflammation using an activated macrophage probe with Slc18b1 as the activation-selective gating target. Nat. Commun. 2019, 10, 1111. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Kwon, H.Y.; Sharma, A.; Lee, S.H.; Liu, X.; Miyamoto, N.; Kim, J.J.; Im, S.H.; Kang, N.Y.; Chang, Y.T. Visualizing inflammation with an M1 macrophage selective probe via GLUT1 as the gating target. Nat. Commun. 2022, 13, 5974. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Fukuda, M.; Lee, J.Y.; Su, D.; Sanu, S.; Silvin, A.; Khoo, A.T.T.; Kwon, T.; Liu, X.; Chi, W.; et al. Visualizing Microglia with a Fluorescence Turn-On Ugt1a7c Substrate. Angew. Chem. Int. Ed. Engl. 2019, 58, 7972–7976. [Google Scholar] [CrossRef]
- Wu, B.; Xu, B.; Hu, M. Regioselective glucuronidation of flavonols by six human UGT1A isoforms. Pharm. Res. 2011, 28, 1905–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaassen, C.D. Xenobiotic transporters: Another protective mechanism for chemicals. Int. J. Toxicol. 2002, 21, 7–12. [Google Scholar] [CrossRef]
- Doring, B.; Petzinger, E. Phase 0 and phase III transport in various organs: Combined concept of phases in xenobiotic transport and metabolism. Drug Metab. Rev. 2014, 46, 261–282. [Google Scholar] [CrossRef]
- Liu, X.; Quan, N. Immune Cell Isolation from Mouse Femur Bone Marrow. Bio-Protocol 2015, 5, e1631. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.F.; Berger, H.; Su, I.H. Isolation and Activation of Murine Lymphocytes. J. Vis. Exp. 2016, 116, e54596. [Google Scholar] [CrossRef]
- Walsky, R.L.; Bauman, J.N.; Bourcier, K.; Giddens, G.; Lapham, K.; Negahban, A.; Ryder, T.F.; Obach, R.S.; Hyland, R.; Goosen, T.C. Optimized assays for human UDP-glucuronosyltransferase (UGT) activities: Altered alamethicin concentration and utility to screen for UGT inhibitors. Drug Metab. Dispos. 2012, 40, 1051–1065. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, C.A.; Watt, F.; Murray, J.; Pajic, M.; Prokvolit, A.; Xue, C.; Flemming, C.; Smith, J.; Purmal, A.; Isachenko, N.; et al. Small-molecule multidrug resistance-associated protein 1 inhibitor reversan increases the therapeutic index of chemotherapy in mouse models of neuroblastoma. Cancer Res. 2009, 69, 6573–6580. [Google Scholar] [CrossRef] [Green Version]
- Gekeler, V.; Ise, W.; Sanders, K.H.; Ulrich, W.R.; Beck, J. The leukotriene LTD4 receptor antagonist MK571 specifically modulates MRP associated multidrug resistance. Biochem. Biophys. Res. Commun. 1995, 208, 345–352. [Google Scholar] [CrossRef]
- Maguire, A.R.; Plunkett, S.J.; Papot, S.; Clynes, M.; O’Connor, R.; Touhey, S. Synthesis of indomethacin analogues for evaluation as modulators of MRP activity. Bioorg. Med. Chem. 2001, 9, 745–762. [Google Scholar] [CrossRef]
- Barraud de Lagerie, S.; Comets, E.; Gautrand, C.; Fernandez, C.; Auchere, D.; Singlas, E.; Mentre, F.; Gimenez, F. Cerebral uptake of mefloquine enantiomers with and without the P-gp inhibitor elacridar (GF1210918) in mice. Br. J. Pharmacol. 2004, 141, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Weidner, L.D.; Zoghbi, S.S.; Lu, S.; Shukla, S.; Ambudkar, S.V.; Pike, V.W.; Mulder, J.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. The Inhibitor Ko143 Is Not Specific for ABCG2. J. Pharmacol. Exp. Ther. 2015, 354, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Koike, K.; Ohga, T.; Kubo, T.; Wada, M.; Kohno, K.; Mori, T.; Hidaka, K.; Kuwano, M. Chemosensitisation of spontaneous multidrug resistance by a 1,4-dihydropyridine analogue and verapamil in human glioma cell lines overexpressing MRP or MDR1. Br. J. Cancer 1995, 72, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alliance of Genome Resources, C. Harmonizing model organism data in the Alliance of Genome Resources. Genetics 2022, 220, iyac022. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Shi, R.; Wang, T.; Tan, H.; Xu, H.; Ma, Y. Interaction between oblongifolin C and UDP-glucuronosyltransferase isoforms in human liver and intestine microsomes. Xenobiotica 2015, 45, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ge, G.; Zhang, H.; Liu, H.; He, G.; Liang, S.; Zhang, Y.; Fang, Z.; Dong, P.; Finel, M.; et al. Characterization of hepatic and intestinal glucuronidation of magnolol: Application of the relative activity factor approach to decipher the contributions of multiple UDP-glucuronosyltransferase isoforms. Drug Metab. Dispos. 2012, 40, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Gufford, B.T.; Chen, G.; Vergara, A.G.; Lazarus, P.; Oberlies, N.H.; Paine, M.F. Milk Thistle Constituents Inhibit Raloxifene Intestinal Glucuronidation: A Potential Clinically Relevant Natural Product-Drug Interaction. Drug Metab. Dispos. 2015, 43, 1353–1359. [Google Scholar] [CrossRef] [Green Version]
- Algeelani, S.; Alam, N.; Hossain, M.A.; Mikus, G.; Greenblatt, D.J. In vitro inhibition of human UGT isoforms by ritonavir and cobicistat. Xenobiotica 2018, 48, 764–769. [Google Scholar] [CrossRef]
- Heng, T.S.; Painter, M.W.; Immunological Genome Project, C. The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef]
- Petzinger, E.; Geyer, J. Drug transporters in pharmacokinetics. Naunyn Schmiedebergs Arch. Pharmacol. 2006, 372, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Lu, Y.; Wu, T. The impact of ATP-binding cassette transporters on metabolic diseases. Nutr. Metab. 2020, 17, 61. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Fukuda, M.; Lee, J.Y.; Chang, Y.-T.; Je, H.S.; Kim, B. Distinctive Labeling of Live Monocytes and Neutrophils with a Single Fluorescent Molecule. Chemosensors 2023, 11, 265. https://doi.org/10.3390/chemosensors11050265
Kim S, Fukuda M, Lee JY, Chang Y-T, Je HS, Kim B. Distinctive Labeling of Live Monocytes and Neutrophils with a Single Fluorescent Molecule. Chemosensors. 2023; 11(5):265. https://doi.org/10.3390/chemosensors11050265
Chicago/Turabian StyleKim, Songhui, Masahiro Fukuda, Jung Yeol Lee, Young-Tae Chang, H. Shawn Je, and Beomsue Kim. 2023. "Distinctive Labeling of Live Monocytes and Neutrophils with a Single Fluorescent Molecule" Chemosensors 11, no. 5: 265. https://doi.org/10.3390/chemosensors11050265
APA StyleKim, S., Fukuda, M., Lee, J. Y., Chang, Y. -T., Je, H. S., & Kim, B. (2023). Distinctive Labeling of Live Monocytes and Neutrophils with a Single Fluorescent Molecule. Chemosensors, 11(5), 265. https://doi.org/10.3390/chemosensors11050265