3.1. Materials Composition, Crystalline Structure, and Morphology
At present, many solution-based synthesis techniques including precipitation, sol-gel, pyrosol, pyrolysis, sonochemical, hydrothermal, and solvothermal processes have been developed for ZrO
2 nanoparticles during the past few decades [
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
31,
32,
33], which are favorable for controlling the size, morphology, and surface chemistry of the resulting product. Herein, a facile solvothermal method was adopted to prepare the pristine, Eu
3+ singly doped and Ti
4+/Eu
3+-co-doped ZrO
2 NCs using alkoxide precursors as the Zr source and n-butoxide as solvents.
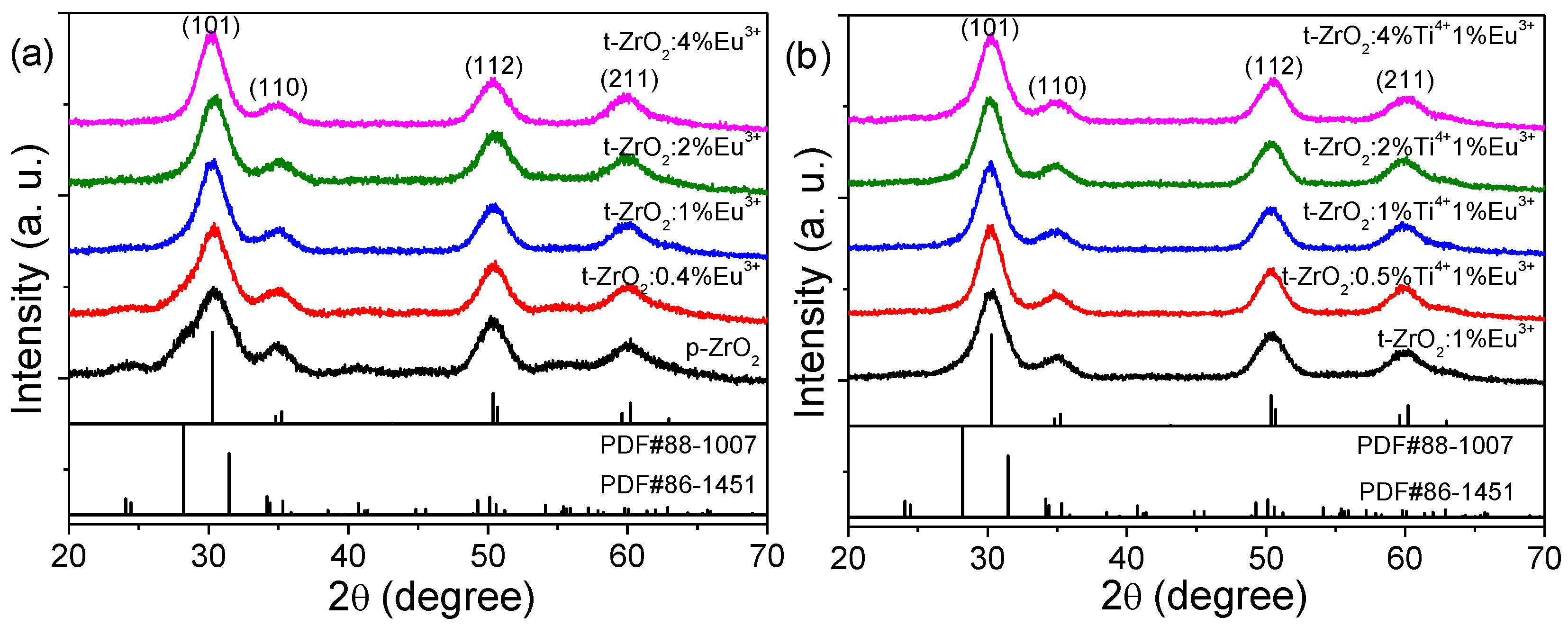
Figure 1 shows the powder XRD patterns of the as-obtained nanoparticles. Broad reflection peaks were observed in all of the samples, indicating a smaller crystallite size. Phase purity was examined upon indexing the reflection peaks to the standard patterns; a mixed phase of the predominant t-ZrO
2 and a relatively small amount of m-ZrO
2 (<10 vol%, additional weaker peaks at ~24.5°, 40.7°, and 55°) could be distinguished in the p-ZrO
2 and the samples doped with a low concentration of Eu
3+ (<2 mol%), while additional Ti
4+ doping had no significant effect on the crystalline-phase purity. A high purity of t-ZrO
2 NCs was obtained for the Eu
3+ content above 2 mol% due to the production of more compensation defects of oxygen vacancy, which agrees well with a previous report [
38]. Due to the same valency (4+) and slightly smaller ionic radius (CN8, 0.74 Å@Ti
4+, 0.84 Å @Zr
4+), the Ti
4+ ions were expected to substitute for Zr sites in the cation network, and the obtained samples were a true solid solution of two oxides.
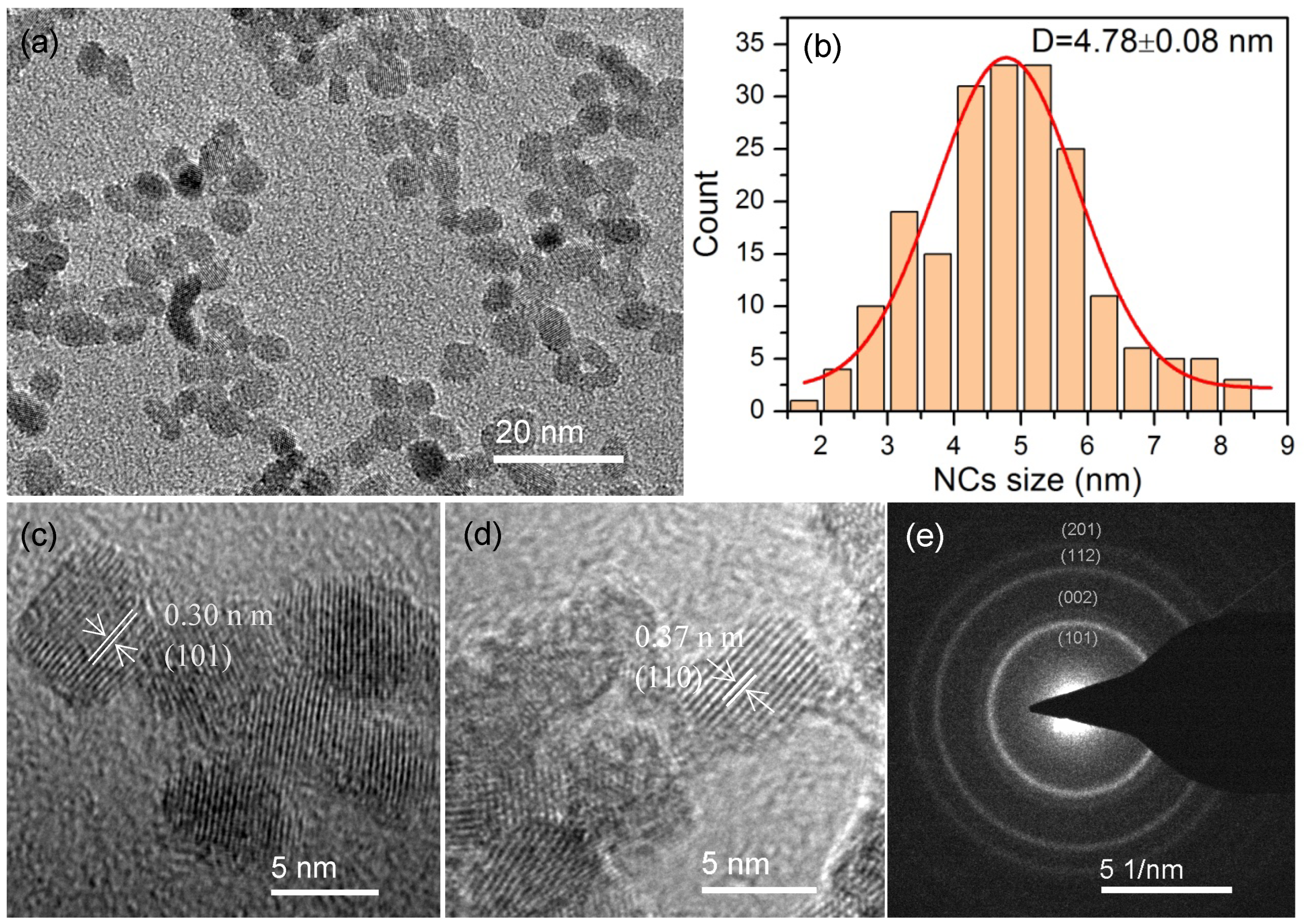
Figure 2a exhibits the TEM images of the as-synthesized p-ZrO
2 NCs. The NCs were mostly round-shaped with similar sizes, despite the presence of a few oval-shaped or interconnected NCs. A histogram of more than 200 NCs is shown in
Figure 2b, and a mean size of ~4.78 nm with a standard deviation of ~1.7% (0.08 nm) was derived, indicating a narrow-size distribution of the NCs. The high-resolution (HRTEM) image (
Figure 2c) clearly demonstrates the single crystallinity of individual NCs. Lattice fringes were very clear with an observed d-spacing of ~0.30 nm, which is consistent with the lattice spacing for the (101) plane of tetragonal ZrO
2 (PDF#88-1007). In addition, monoclinic ZrO
2 NCs (PDF#86-1451) were also observed with the lattice spacing of ~0.37 nm for the (110) plane after a careful examination (
Figure 2d). The selective area electron diffraction (SAED,
Figure 2e) on an aggregation of NCs displayed a ring pattern that could be easily matched with the XRD pattern of t-ZrO
2, indicating the presence of small crystallites within the aggregate and that the tetragonal crystalline phase is the principal polymorph. It is noteworthy that additional Eu
3+ and Ti
4+ doping did not induce any significant changes in the size and morphology of NCs upon extensive TEM observations on the other samples (see
Figure S1).
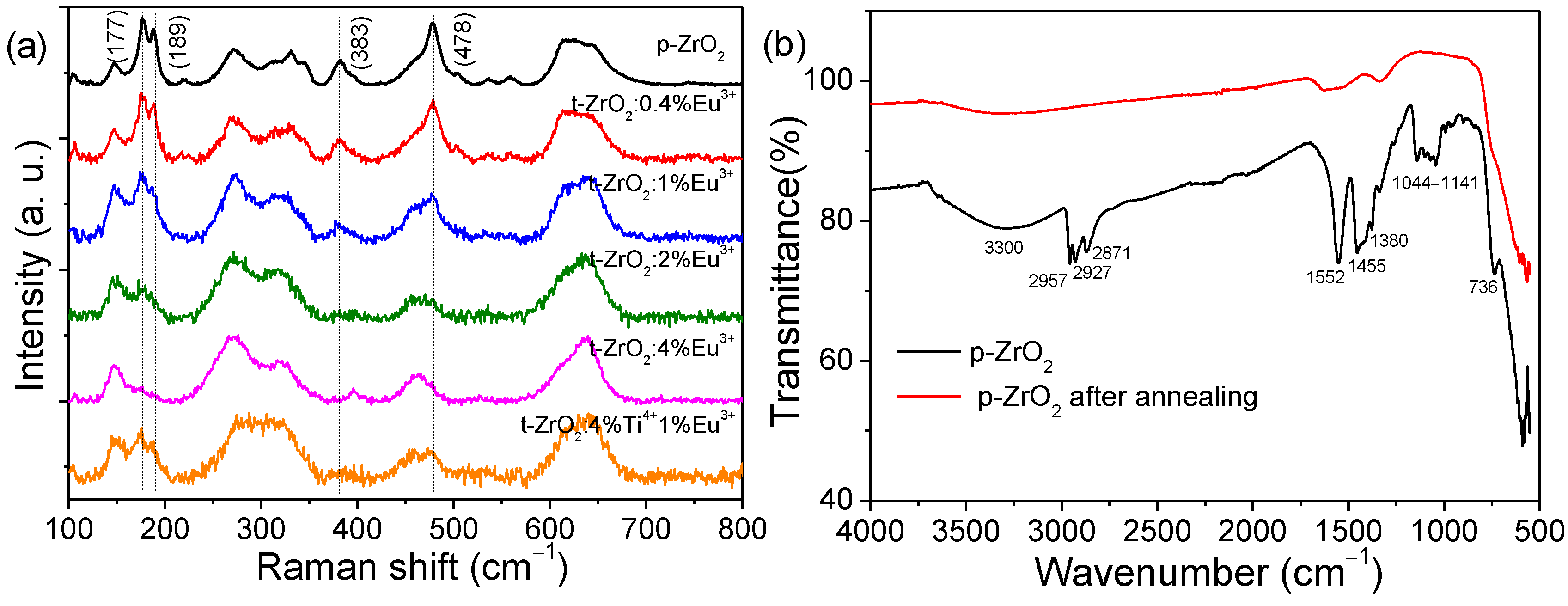
Raman spectroscopy is a powerful tool to determine phase compositions in the case of many transition metal oxides [
31,
39], which was further employed to verify the phase purity of the NCs prepared in this study. According to the group theory, six Raman active vibrational modes (A1
g + 2B1
g + 3E
g) of 18 normal modes were allowed for t-ZrO
2 in the 100–800 cm
−1 range while 18 of the 36 normal modes were allowed for Raman modes in the lower symmetry of m-ZrO
2, among which the peaks at ~145, 270, and 650 cm
−1 could be assumed as being characteristic of t-ZrO
2, while a sharp doublet centered at ~180 cm
−1 and a peak at ~478 cm
−1 could be considered as the most sensitive analytical marks of the monoclinic phase [
31,
39]. Note that there was intense background luminescence when all as-synthesized particles were stimulated with a sub-bandgap laser at 325, 488, and 532 nm. Raman analysis was only possible with excitation at a longer wavelength of 830 nm by an argon-ion laser, but it showed much weaker Raman scattering. The background emissions were nearly quenched after thermal treatment at 400 °C in air. The Raman spectra of some typical samples (p-ZrO
2, t-ZrO
2:
xEu
3+, and t-ZrO
2:4%Ti
4+, 1%Eu
3+) under investigation are reported in
Figure 3a and
Figure S2b. The characteristic Raman modes of m-ZrO
2 appeared in p-ZrO
2, but the peak intensity decreased greatly upon Eu
3+ doping and almost disappeared in the 4 mol% Eu
3+-doped sample. These observations of the evolution of phase purity closely followed the XRD analysis. By comparing the spectrum of t-ZrO
2:1%Eu
3+ with that of t-ZrO
2:4%Ti
4+, 1%Eu
3+, it was found that the modes of m-ZrO
2 were significantly weakened; this indicates that the extra Ti
4+ doping suppressed the growth of m-ZrO
2 NCs and favored the stabilization of t-ZrO
2 NCs. Without creating oxygen vacancies, the stabilization of tetragonal ZrO
2 bulk materials by undersized tetravalent dopants (Ti, Si, Ge, Sn, Ce, Th, and U) has been demonstrated previously [
30].
The surface composition was characterized by FTIR spectroscopy. All of the as-synthesized ZrO
2 NCs had similar spectral features. The typical FTIR spectra of p-ZrO
2 before and after thermal treatment are shown in
Figure 3b. For the as-obtained NCs, the presence of vibrational modes associated with hydrocarbon (~2957 (ν
as(CH
3) C-CH
3), 2927 (ν
as (CH
2) C-CH
2), 2871 (ν
s(CH
3) C-CH
3), and 1380 cm
−1 (δ
as(CH
3)) and C-O (~1141 and 1044 cm
−1)) species indicated that butoxyl groups were bound to the surface of NCs, which were most likely from the Zr source or the solvents [
40]; meanwhile, the indicative absorption bands of carboxylate groups centered at ~1552 (COO
− antisymmetric stretch), 1455, and 1415 cm
−1 (COO
− symmetric stretches) show that the acetate ligands (CH
3COO
−) were definitely bonded to the NCs’ surface, since the only organic carboxylic acid involved in the synthetic procedure was acetic acid. These carboxyl groups were suggested to be coordinated to the zirconium atoms in a chelating mode given the small separation between asymmetric and symmetric stretches (Δν = ν
a(COO
−) − ν
s(COO
−) = 100 cm
−1) [
40]. In addition, the surface hydroxyl groups (O-H) characterized by a broad band in the 3000–3700 cm
−1 range appeared in the samples. In the lower-frequency range, a peak centered at 736 cm
−1 was assigned to Zr–O vibration. As shown in
Figure 3b and
Figure S2a, thermal treatment at 400 °C significantly removed these surface organic groups but slightly promoted the phase transformation to m-ZrO
2.
The XPS analysis (
Figure S3) was performed on a typical sample of t-ZrO
2:4%Ti
4+, 1%Eu
3+ to further determine the surface composition of the as-grown NCs. From the XPS survey spectra (
Figure S3a), peaks from Zr (3s, 3p, 3d, 4s and 4p), O 1s, and C 1s can be clearly seen. The doped metal elements of Ti and Eu were detected by the high-resolution XPS but with much weaker signals. As shown in
Figure S3c,d, only one of the paired peaks could be clearly resolved for Ti and Eu, respectively. The peak-fitting was processed to analyze the chemical state and environment. The Ti 2p
3/2 spectrum exhibited only one component at 458.7 eV, which could be assigned to Ti
4+ in the oxides. No obvious extra components appeared at the lower-binding-energy side, commonly ascribed to Ti
3+. This indicates that most of the Ti ions were doped in the form of a 4+ valency. As for the europium spectrum, the observed signals were mainly from Eu
3+; the full width at half maximum (FWHM) of the 3d
5/2 peak was approximately 6 eV, which was much greater than that of the host element of Zr (~1.5 eV,
Figure S3b) and another dopant of Ti (~1.75 eV). Such a broadening is indicative of the appearance of a complex-site environment surrounding the Eu
3+ immediately. The 3d
5/2 peak can be further decomposed into two sub-peaks at 1133.0 and 1136.0 eV, implying that at least two types of Eu
3+ ions occurred in the nanocrystals under investigation.
3.2. Optical Absorption
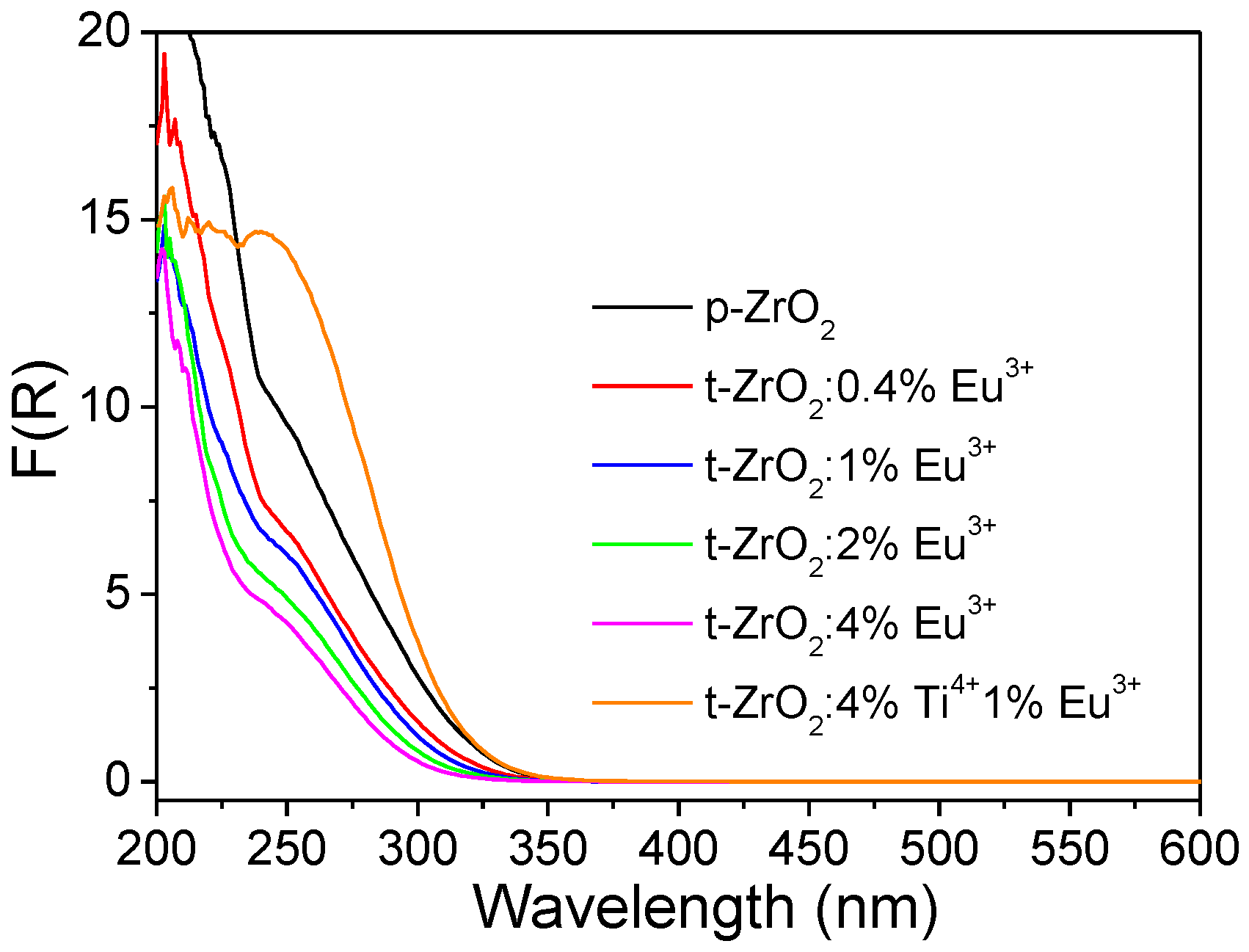
The absorption characteristics of the as-obtained NCs from 200 to 600 nm were studied by measuring the reflection of a plaque of the powders ≥1 mm thick. In the ultraviolet region, this sample thickness is sufficient to guarantee the measurement of R
∞, i.e., the reflection at an “infinite” sample thickness [
9,
41]. The experimental spectra were replotted and are presented in the form of the Kubelka–Munk function F(R
∞) vs. wavelength (
Figure 4). F(R
∞) is given by F(R
∞)∝ (1−R)
2/2R = K/S, where K and S are the phenomenological absorption and scattering coefficients, respectively [
9,
41].
In p-ZrO
2, a weak absorption commenced in the visible region (~530 nm), followed by a steep edge developing rapidly to the shorter-wave UV region (
Figure S4). Note that such intense absorption had an onset at ~350 nm (3.54 eV), and the corresponding optical absorption edge evaluation yielded a value of ~4.3 eV (this was performed following the case of a directly allowed electron transition which occurred in the tetragonal phase of ZrO
2), which was much smaller than the experimentally determined intrinsic bandgap value (~5.78 eV) of t-ZrO
2 [
10,
11,
12,
13]. Thus, the weaker visible absorption and stronger UV absorption were both located in the optically transparent region of the host matrix of t-ZrO
2 and did not correspond to the electronic transitions from the valence band to the conduction band. These absorptions were then distinguished to correlate with some impurity ions or lattice defects, which commonly exhibit absorption below the optical band gap. The weaker visible absorption should be the main factor leading to the background luminescence encountered during Raman measurements and was suggested to arise from the carbon impurities, which were produced under the environment of a high temperature, a high pressure, and hypoxia in a sealed autoclave containing organic solvents, while the stronger UV absorption was considered to originate from the titanium impurity ions (Ti
4+). We and other researchers have demonstrated previously that Ti impurities may be naturally present as traces in ZrO
2 and unintentionally substitute Zr, since Zr is commonly extracted from Ti ores (e.g., ilmenite or rutile) [
9,
13]. The Ti impurities significantly affected the optical properties of the following experimentally reprocessed materials and induced much puzzle or debate when interpreting the optical behavior observed [
9,
13]. The high-purity ZrO
2 reagent supplied by Aladdin Industrial Inc. (Shanghai, China) was recently evidenced to contain trace amounts of titanium impurities [
9]. A corresponding charge transfer band (Ti
4+ + O
2−→Ti
3+ + O
−, electron transitions from the valence O 2p states to those of the empty Ti
4+ 3d orbitals) well below the fundamental absorption was observed in the pristine m-ZrO
2, showing an onset at ~350 nm as well. Following this line, the Zr source of zirconium butoxide used in this study was also supposed to contain the titanium impurity. This was exactly confirmed by the ICP analysis, as a higher content of 1915.2 ppm Ti was determined in this commercial Zr source (with a Zr content of 211,108.4 ppm). The relative content of Ti to the sum of the metallic element of Zr and Ti was further determined to be ~0.9 wt% in mass percentage or ~1.7mol% in molar percentage, respectively. The above assignments are further discussed hereafter.
The doping of Eu3+ ions into inorganic oxides commonly involves additionally allowed transitions of CT (O2−→Eu3+, an electron of O 2p states transitioning to the partially filled Eu3+ 4f orbitals) in the short-wave UV region. An increased absorption can then be expected, however, by comparison with the p-ZrO2; the addition of Eu3+ ions anomalously decreased the UV absorption continuously. We suggest that the degradation of the CT (O2−→Ti4+) band was caused by the more competitive absorption from the CT (O2−→Eu3+) transition but with a lower absorption coefficient, since both processes shared the electrons at the top of the valence band.
As shown in
Figure 4 and
Figure S4, additionally introducing Ti increased the absorption in both the visible and UV regions (t-ZrO
2:4%Ti
4+, 1%Eu
3+). Obviously, the enhanced UV absorption (<350 nm) could be explained as being due to the incorporation of more Ti ions into the host lattice. The enhancement of the near-UV and visible absorption confirmed that the presence of such carbon impurities had more correlation with the titanium element.
3.3. Photoluminescence of p-ZrO2 NCs
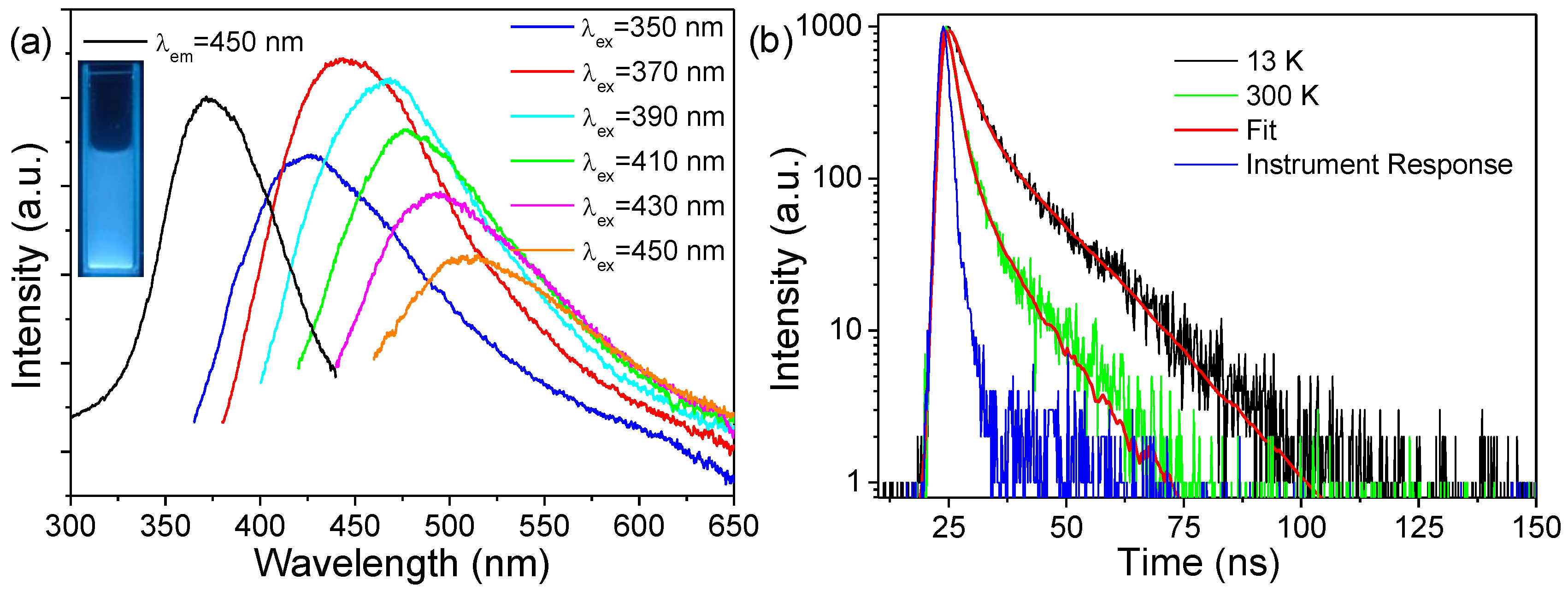
The as-synthesized p-ZrO
2 NCs showed a bluish color under a hand-held UV lamp (inset of
Figure 5a), which could be quenched after lower-temperature annealing at 400 °C for 4 h. In the fluorescence spectra (
Figure 5a), p-ZrO
2 NCs have optimal excitation and emission wavelengths at ~370 nm and 450 nm. The effective PL excitation ranged mainly from the long-wavelength UV-to-visible spectral region and did not display pronounced emissions under shorter-wave UV excitation. An excitation-dependent PL behavior was observed; as λ
ex increased, the emission spectra were systematically displaced toward longer wavelengths and the intensity decreased with λ
ex > 380 nm. The dependence of emission wavelength and intensity on λ
ex has been identified as a generic feature of various types of carbon nanomaterials and silica xerogels [
42,
43]. These emissions feature a very small Stokes shift and lead to background luminescence during Raman measurements. The fluorescence decays (
Figure 5b, λ
ex = 370 nm, λ
em = 450 nm) displayed a non-exponential behavior and had a lifetime of a nanoseconds magnitude even at a lower temperature of 13 K. The faster fluorescence decays indicated that the electronic transitions in the present luminescent species had a full parity/spin-allowed character. The traces could be fitted into bi-exponential function as I(t) = A
1 exp(−t/
τ1) + A
2 exp(−t/
τ2), where
τ1 and
τ2 are the time constants corresponding to slow and fast components, and A
1 and A
2 represent the amplitudes of each component at t = 0, respectively. The yielded time constants had the values of
τ1~12.1 (A
1~0.024) and
τ2~2.9 ns (A
2~0.099) at 13 K, and
τ1~6.8 (A
1~0.016) and
τ2~1.4 ns (A
2~0.175) at 300 K. The prolonged average decay times at a lower temperature were attributed to the reduced nonradiative transitions via electron–phonon coupling or thermally activated energy migration or transfer to quenching centers. By considering the PL characteristics including long-wave UV excitation, excitation-dependent emission, smaller Stokes shift, and shorter fluorescence lifetimes, we assigned the luminescence to the trace amounts of carbon dopant impurities, as performed previously for the PL of sol–gel-derived pristine ZrO
2 and Y
2O
3 nanoparticles [
43]. The carbon dopant impurities could be readily formed during the solvothermal processes, where the high temperature and the high pressure acted upon the organic ligands and solvents and favored the incorporation of carbon impurities.
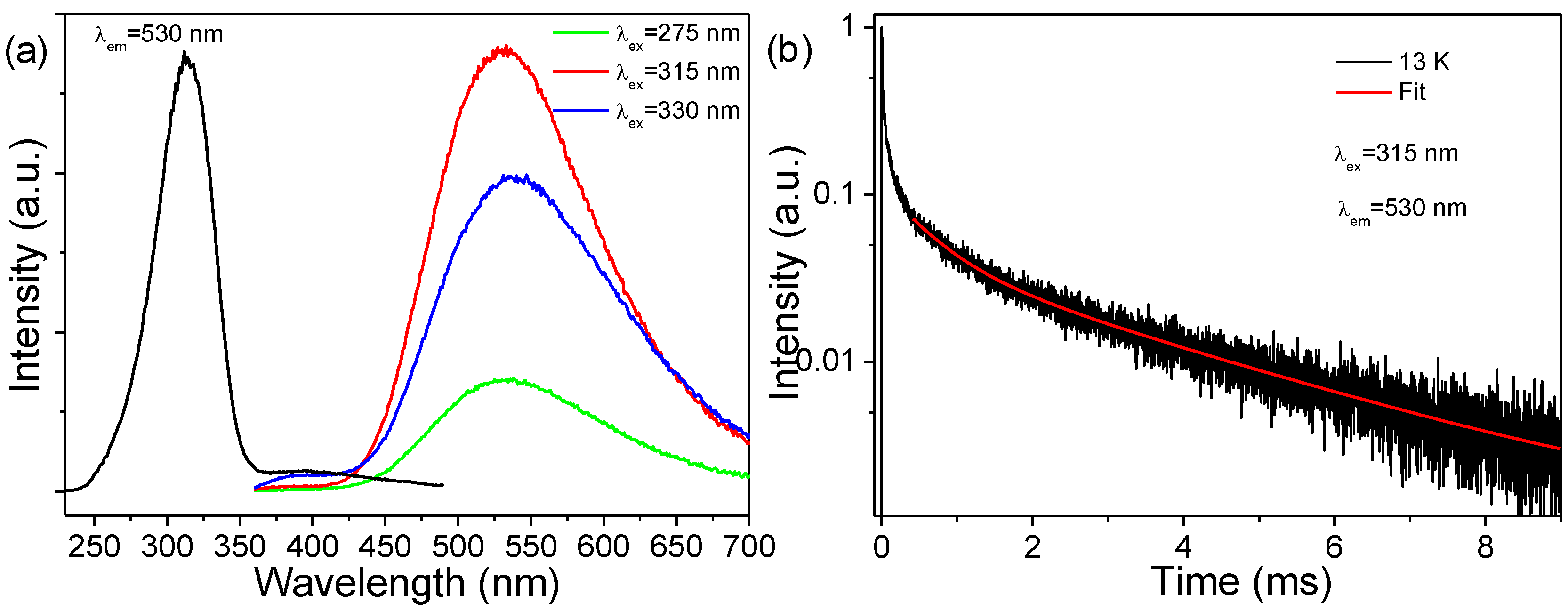
Figure 6a shows the PL excitation and emission spectra of the p-ZrO
2 NCs at 13 K. In sharp contrast to the room-temperature PL, there existed an intense shorter-wave UV excitation band covering the 230–360 nm range (centered at 315 nm), which yielded bright broadband white luminescence in the 400–700 nm range (centered at 530 nm) with a large Stokes shift reaching ~13,000 cm
−1 and an insignificant excitation-dependent emission. Such an excitation band coincides well with the observed strong absorption in
Figure 4, which is, thus, ascribed to CT (O
2−→Ti
4+) transitions in the titanate groups [
9]. Note that the carbon impurities-related excitation was also observed in the range of 360–490 nm by reason of the spectral overlapping of PL emissions [
43], but it was much weaker in intensity. In
Figure 6b, the fluorescence decay shows a much longer lifetime in the scale of milliseconds. There exists an initial rapid non-exponential component, followed by a much slower component at later times. The rapid component corresponds to the luminescence of carbon impurities discussed above, which was slightly excited by the short-wave UV irradiation at a low temperature, while the slower one arose from the CT (Ti
3+→O
−) emissions in the titanate groups, of which the long decay times at lower temperatures have been previously observed for Ti
4+ in monoclinic hafnia (HfO
2) [
8], indicating that the emission results from a strongly forbidden transition, presumably a triplet state of a strong spin-forbidden character. The slow component in the trace is clearly two-exponential with the two resolved decay time constants having values of
τ1~0.56 and
τ2~2.98 ms, which are comparable with previous reports [
8].
3.4. Photoluminescence of t-ZrO2:Eu3+ NCs
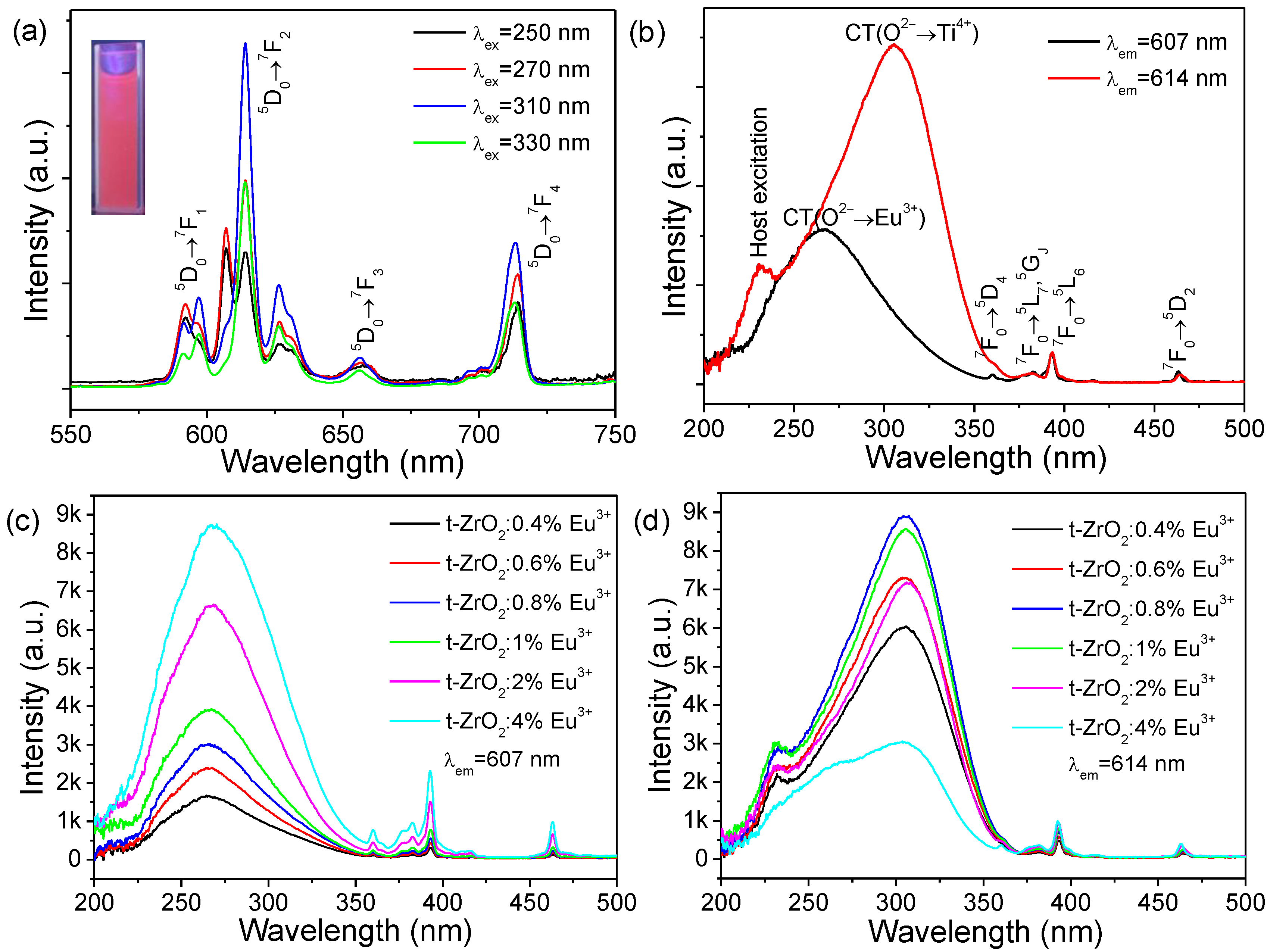
Since the general PL characteristic features of the sample series of t-ZrO
2:
xEu
3+ (
x = 0.4–4%) were experimentally found to be analogous at room temperature, the typical PL excitation and emission spectra from t-ZrO
2:1 mol%Eu
3+ were selected and are shown in
Figure 7a,b. The emission of these nanophosphors exhibits the well-known Eu
3+ emission lines from the
5D
0 level to the
7F manifold; however, a closer inspection of the PL spectra recorded with different irradiations in the short-wave UV region (
Figure 7a, λ
ex = 250, 270, 310 and 330 nm) suggests that at least two sets of emissions, arising from two types of Eu
3+ (designated as Eu(I) and Eu(II)), could be distinguished to be present in the samples, the assignment of which is tabulated in
Table S1. The main emission lines for Eu(I) and Eu(II) are both from the
5D
0→
7F
2 transitions and are located at ~607 and 614 nm, respectively. Clearly, the asymmetry ratio (R) of the integrated intensities of the
5D
0→
7F
2 and
5D
0→
7F
1 transitions (R = I(
5D
0→
7F
2)/I(
5D
0→
7F
1)) in Eu(II) is much larger than that of Eu(I) [
15]. Due to the different intensity mechanisms, it is known that the R values can be considered indicative of the asymmetry of the coordination polyhedron of the Eu
3+ ion. In particular, the lower the R value is, the higher the site symmetry of the Eu
3+ ion [
15]. It follows that Eu(II) occupies a noncentrosymmetric site with a much lower symmetry while Eu(I) experiences a much higher symmetry of the local surrounding. Noteworthy is the spectral features of the Eu(II) luminescence (number, relative intensities, and energetical positions of lines) which closely resemble those of bulky and nanocrystalline m-ZrO
2 reported previously [
9,
15].
Figure 7b presents the excitation spectra of the emissions monitored at their relative maxima of Eu(I) (λ
em = 607 nm) and Eu(II) (λ
em = 614 nm). Both excitation spectra consist of a broad band covering the 200−370 nm range of wavelengths and some sharp lines of the intraconfigurational transitions of the 4f
6 configuration of the Eu
3+ ion. Obviously, the broad excitation bands both show an asymmetric profile, but they peak at different energetical positions, which are located at ~265 nm and 305 nm for Eu(I) and Eu(II), respectively. An additional shoulder (λ
ex = 230 nm) could be clearly observed for Eu(II) on the high-energy side of the main band. This observation confirms the presence of two types of Eu
3+. Upon a closer examination, the asymmetric broadband in the range of 220–360 nm has at least two components for Eu(I) and three components for Eu(II). If we fit such spectral curves to the Gauss function roughly, we could obtain two sub-bands peaking at 260 and 283 nm for Eu(I) (denoted as Eu(I)-G260 and Eu(I)-G283, respectively) and three sub-bands peaking at 232, 279, and 313 nm (denoted as Eu(II)-G232, Eu(II)-G279, and Eu(II)-G313, respectively), as shown in
Figure S5. The small energy difference between Eu(I)-G283 and Eu(II)-G279 points to the same origin for both bands.
Figure 7c,d show a comparison of the excitation spectra of samples with different doping levels by monitoring the relative maxima of Eu(I) (λ
em = 607 nm) and Eu(II) (λ
em = 614 nm), respectively. Clearly, the broad excitation band of the λ
em = 607 nm emission intensified successively with the increasing Eu
3+ concentration, while those of the λ
em = 614 nm emission reach a maximum intensity at
x =0.8 mol % and then decrease. For Eu(I), the normalized spectra, as shown in
Figure S6, indicate a slightly increased relative ratio of the Eu(I)-G283 sub-peak with increasing Eu
3+ concentration.
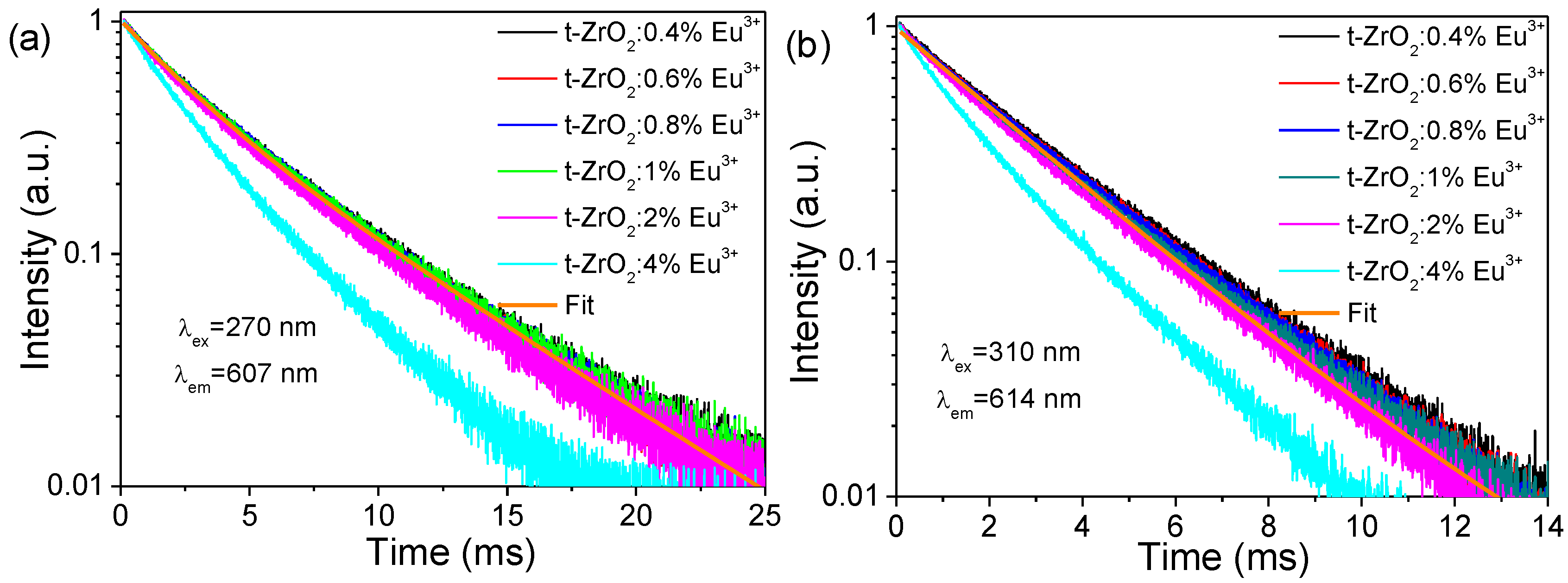
Figure 8a,b show the fluorescence decays of Eu(I) and Eu(II) in the t-ZrO
2:
xEu
3+ (
x = 0.4–4 mol%) samples. It can be seen that, regardless of Eu(I) or Eu(II), the traces almost superimpose with each other in the content range of 0.4–1 mol% and then decay much faster with increasing Eu
3+ concentration. All of the traces are rarely pure single-exponential in character even at lower doping levels. For the t-ZrO
2:
xEu
3+ (
x = 0.4–1 mol%) samples, the experimental decays could be well approximated by the fit using the bi-exponential function, yielding the time constants of
τ1~6.23 and
τ2~2.54 ms, where the only differences are the relative contribution of the fast and slow components to the whole decay traces. For Eu(I), the slow component dominates the decays (Rel. 72.6%), while for Eu(II), the fast component dominates the decays (Rel. 91.8%). It is, thus, considered that Eu(I) and Eu(II) have a lifetime constant of ~6.23 and 2.54 ms, respectively; the non-exponential behaviors are due to spectral overlapping between the emissions of Eu(I) and Eu(II). Faster decays at the ≥2 mol% Eu
3+ contents were caused by the concentration quenching, which occurred via energy migration through the lattice, or they were caused by the clustering of activators. Note that the lifetime constant of Eu(II) (≤2 mol%) was approximately 3.7-fold larger than that of Eu
3+ in m-ZrO
2 with a determined value of~0.68 ms [
9].
As demonstrated in
Section 3.2, there is little doubt that the absorption in the short-wave UV region was mainly from the host lattice, CT (O
2−→Eu
3+), and CT (O
2−→Ti
4+) transitions in titanate groups. The question we now face is the assignments of these sub-bands. Such a Eu(II)-G232 band could be ascribed to the excitation in the host lattice followed by energy transfer to the Eu(II) ion upon considering the higher-energy nature (~5.3 eV), which approach the bandgap value of ~5.78 eV [
10]. The dependence of the Eu(I)-G260 sub-band on the Eu
3+ concentration coincides with the analysis of the optical absorption and suggests that it is due to CT (O
2−→Eu
3+) transitions (direct excitation of Eu
3+ centers). The sum of the sub-bands of Eu(II)-G279 and Eu(II)-G313, thus, arose from the titanate groups’ excitation, as was further confirmed by the bright CT (Ti
3+→O
−) emissions under such excitations at 13 K (
Figure S7a, t-ZrO
2:0.4%Eu
3+). In addition, the spectral profile of such a room-temperature broad excitation band of the Eu
3+ emissions closely followed the low-temperature excitation spectra of the CT (Ti
3+→O
−) broadband emissions in p-ZrO
2 and t-ZrO
2:0.4%Eu
3+ (
Figure S7b; note that for the FLS 920 spectrofluorometer, the excitation radiation below about 250 nm is too weak to obtain reliable excitation spectra in the deep UV [
8]). The indirect excitation of the Eu
3+ centers by the naturally present Ti
4+ indicated that an effective energy transfer from CT (O
2−→Ti
4+) transitions to Eu
3+ centers occurred, which was previously observed in m-ZrO
2:Ti
4+,Eu
3+ bulk phosphors [
9]. Alternatively, some previous studies on the solution-synthesized ZrO
2: Eu
3+ NCs have attributed the broad UV excitation band to host excitation, such as the study by X. Chen et al. [
16], and to the only CT (O
2−→Eu
3+), such as the study by A. Speghini et al. [
15],and their spectroscopic analyses neglected the effects of naturally occurring Ti
4+ ions on the PL of the Eu
3+-doped on purpose. In this study, we present more evidence to perform the assignment of the above excitation bands. It follows that the excitation of Eu(I) was dominated by the CT (O
2−→Eu
3+) transitions, with a minor but increasing contribution from CT (O
2−→Ti
4+) transitions at a higher Eu
3+ concentration, while the inverse case occurred for Eu(II), where the excitation by CT (O
2−→Eu
3+) transitions were negligibly small. This indicates that Eu(II) is particularly closely correlated with the Ti impurities in the host lattices, while Eu(I) is not.
We now turn to the distribution of Eu(I) and Eu(II), and some questions arise. Were Eu(I) and Eu(II) both built into the lattice structure of t-ZrO
2 NCs together, that is, was most of the red luminescence emitted by the dominant phase of t-ZrO
2? Or did Eu(I) and Eu(II) enter into two segregated crystalline phases, that is, Eu(I) in the dominant t-ZrO
2 while Eu(II) in the m-ZrO
2, given the presence of the m-ZrO
2 NCs’ impurities in the lower-doping systems and the high similarity of PL emission in the case of the m-ZrO
2 phase? Herein, we ruled out the second question upon the following consideration. First, the m-ZrO
2 NCs impurities were of a small amount (<10 vol%), even in the less-doped samples (≤2 mol%), but a substantial intensity was invariably observed, which was more intense than that of Eu(I) up to 2 mol% under the peak excitation of the UV broadband (see
Figure 7 and
Figure 8). Even in the high purity of t-ZrO
2:4%Eu
3+ NCs, the Eu(II) emissions remained to be observed. Then, the color of the PL emissions was homogeneous across the whole body of the powders upon a careful observation under UV irradiation, which is unexpected if the resulting samples are a mechanical mixture based on a tiny amount of m-ZrO
2:Eu(II) and a large fraction of t-ZrO
2:Eu(I) NCs. One may argue that XRD is not sensitive enough to detect the many impurity phases of m-ZrO
2 NCs and that Raman spectra (see
Figure 3a) are more powerful and precise, where an intense Raman peak of a sharp doublet appeared in the samples (
x = 0.4 and 1 mol%). This could be explained by the fact that monoclinic zirconia had stronger Raman signals than tetragonal zirconia under equivalent situations, as claimed by Kim et al. [
44]. A third and an effective host excitation observed when monitoring Eu(II) emissions also indicated that the volume amount of the host materials should be large. The last is the much longer PL decay by a factor of ~3.7 for Eu(II) than that of Eu
3+ in bulk monoclinic zirconia. This is exactly contrary to our common expectation, that is, its fluorescence lifetime constant is suggested to be much shorter than that of the bulk ones (~0.68 ms) because of the enhanced nonradiative transfer to the many quenching centers, if the Eu(II) is actually located in m-ZrO
2 NCs. Fluorescence decay commonly has much more fingerprint characteristics for PL identification. Alternatively, the presence of two distinct sites in the tetragonal Ce
0.
2Zr
0.
8O
2 bulk materials was observed in the literature using EXAFS and Eu
3+ as a structural probe, respectively [
45,
46]. It is, thus, rational to reach the conclusion that most of the photoluminescent Eu
3+ ions were incorporated into the t-ZrO
2 NCs but located at different symmetries of sites.
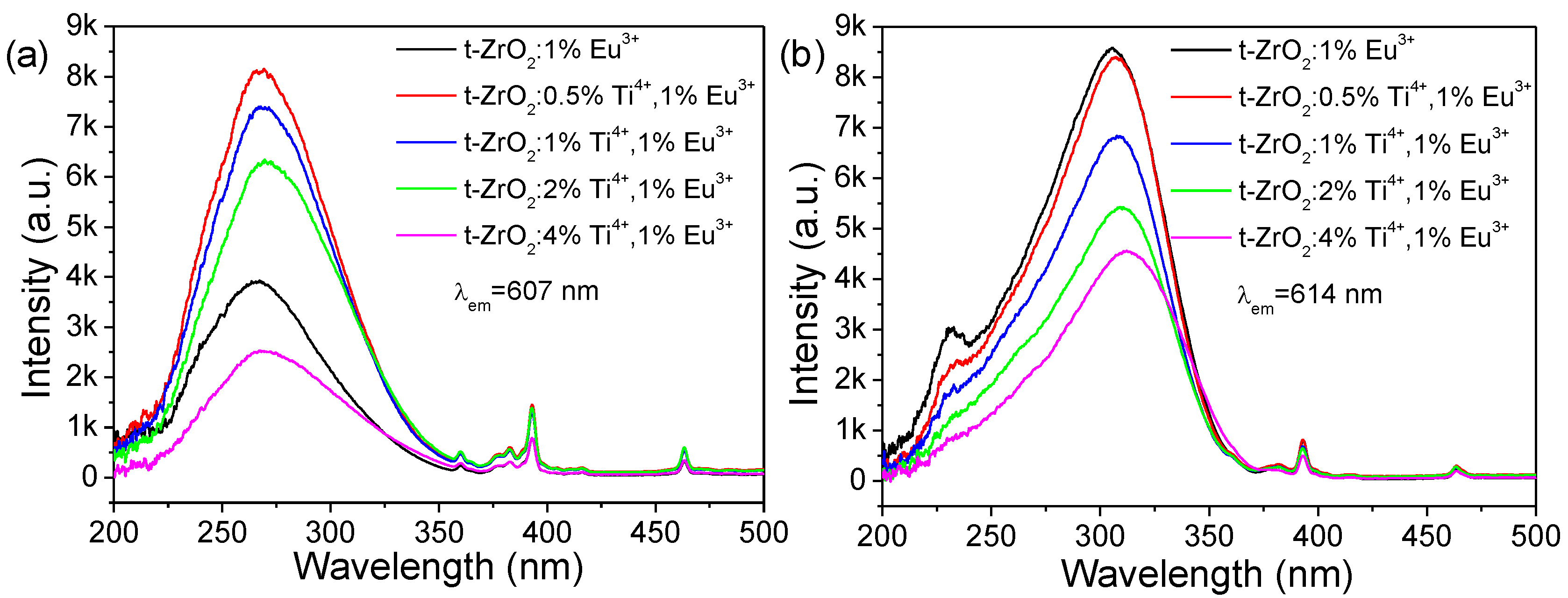
Following the above elucidation on the Eu3+ distribution, we could further conclude that the Ti4+ impurities are distributed somewhat non-randomly over the sites normally occupied by Zr in the nanocrystalline matrix but approach Eu(II) ions, where the CT (O2−→Ti4+) absorption in the titanate groups is more competitive to that of the direct CT (O2−→Eu3+) ones. As a consequence, the indirect sensitization luminescence of Eu3+ by the titanate groups is more effective than that of the direct excitation of the Eu3+ centers. In contrast, the Ti4+ and Eu(I) ions are proposed to be a bit far apart since the energy transfer is distance-dependent; the following sensitization process is not more competitive, and then the CT (O2−→Eu3+) transitions dominate the excitation of the Eu(I) centers. It is reasonable that more Eu(I) approached the Ti4+ ions upon increasing the Eu3+ concentration, and a much greater luminescence contribution from the process of the sensitization of CT (O2−→Ti4+) was then observed. At present, the underlying mechanism responsible for the dopant distribution, however, remains unclear.
The regular tetragonal structure cannot explain the presence of more than one site. The following question is why there exists two types of Eu
3+ in the predominant t-ZrO
2 NCs. In the crystalline lattice of t-ZrO
2, the Zr
4+ ion is situated in octo-coordinated sites (ZrO
8) of a D
2d symmetry [
15,
47] where the Eu
3+ could find itself, and for this symmetry, the
7F
1 and
7F
2 multiplets are split in two and four levels [
15], respectively, and then two peaks in the 590–600 nm region assigned to the
5D
0→
7F
1 transition and four peaks in the 600–630 nm region assigned to the
5D
0→
7F
2 transition should be observed commonly. The luminescence spectra of Eu(I) and Eu(II) observed (see
Table S1), however, are not fully compatible with an emitting Eu
3+ ion in the D
2d symmetry. It seems, therefore, that in the t-ZrO
2 NCs, the incorporation of the Eu
3+ on a Zr
4+ site distorts the surroundings of the cations. This fact could be readily understandable by considering the effect of O
2− vacancies near the Eu
3+ ion, which might be produced during the nucleation of NCs as mentioned in the Introduction section, and is yielded by the need for charge compensation. Obviously, one isolated oxygen vacancy would significantly affect its nearest and next nearest neighbors directly by their relative positions acting on cationic sites and would create four sevenfold coordination (CN7) sites in its first cationic coordination shell and 28 cationic sites of an eightfold coordination (CN8) in the second cationic shell. In addition, the O
2− vacancies would further modify the cationic site symmetry indirectly by means of ion relaxation [
47]. The removal of one O
2− ion would cause the displacement of the nearest-neighbor oxygens and the secondary relaxation of oxygens in the different directions. These displacements of atoms from their position in the “ideal” position, in turn, change the symmetry of cationic sites. Moreover, the longer-range interactions such as vacancy–vacancy, cation–vacancy, or cation–cation may also occur [
47]. Finally, the common nature of nanocrystals including a larger surface stress and disorder also favors the formation of at least two types of Eu
3+ in t-ZrO
2 NCs. Herein, we cannot find a more precise description of the lattice distortions with our experimental data and simply suggest that the high site symmetry of Eu(I) ions are of an eightfold coordination, while the low site symmetry of Eu(I) ions are of a sevenfold coordination, like in the m-ZrO
2 (
Figure S8). The predominated PL from Eu(II) shown in the lower doping level of the samples suggests a preferential occupancy of vacancy first-neighbor sites (CN7) by Eu
3+ in these samples. The oxygen vacancy played an important role in the growth and stabilization of tetragonal ZrO
2 NCs [
48,
49]; for the p-ZrO
2 and t-ZrO
2:Eu
3+ NCs with a low content of Eu
3+, the oxygen vacancies could be considered as the“pristine” nucleated ones (referred to as pristine vacancy), while for the highly doped samples, most of the vacancies are produced by the need for a charge compensation (referred to as compensated vacancy). On the basis of the above observation, we come to the reasoning that the two types of oxygen vacancies differ in the association with the dopants of Eu
3+; the pristine vacancy prefers to be located in the close vicinity of Eu
3+, while the compensated vacancy and Eu
3+ ions tend to be far apart. The latter was demonstrated previously by EXAFS [
50]: it was found that vacancies introduced by oversized dopants of lanthanides (Y and Gd) are located as the nearest neighbors to Zr atoms, leaving an eightfold oxygen coordination to dopant cations. The above discussion is consistent with the XPS analysis of the Eu
3+ spectrum. It is known that positively charged oxygen vacancies are able to strongly localize/trap one or two extra electrons near the vacant site, producing a singly charged or neutral vacancy. Upon simply considering the additional influence of localized electrons on the core levels of the neighboring Eu
3+ (CN7), it was suggested that the lower binding energy component (1133.0 eV) of the 3d
5/2 peak of Eu
3+ corresponds to Eu
3+ (CN7), while the higher binding energy component (1136.0 eV) corresponds to Eu
3+ (CN8).
3.6. Temperature Dependence of PL and Optical Thermometry
As demonstrated in the preceding section, at low temperatures, energy transfer-derived dual emissions were observed in t-ZrO2:Eu3+ NCs upon excitation into the naturally occurring titanate groups: the white broadband CT (Ti3+→O−) emissions and red sharp Eu(II) emissions. The former was temperature-sensitive and was almost quenched nonradiatively at room temperature, while the latter was somewhat stable against temperature; the efficient sensitization and the following intense characteristic emission of Eu3+ centers were observed even at room temperature. This fact showed that Eu(II) ions were much more effective to nonradiatively trap the excitation energy than the other radiationless processes occurring at the titanate groups after optical absorption. This agrees well with the preceding illustration of the specific association of Eu(II) with Ti4+ ions in the crystalline lattice of NCs.
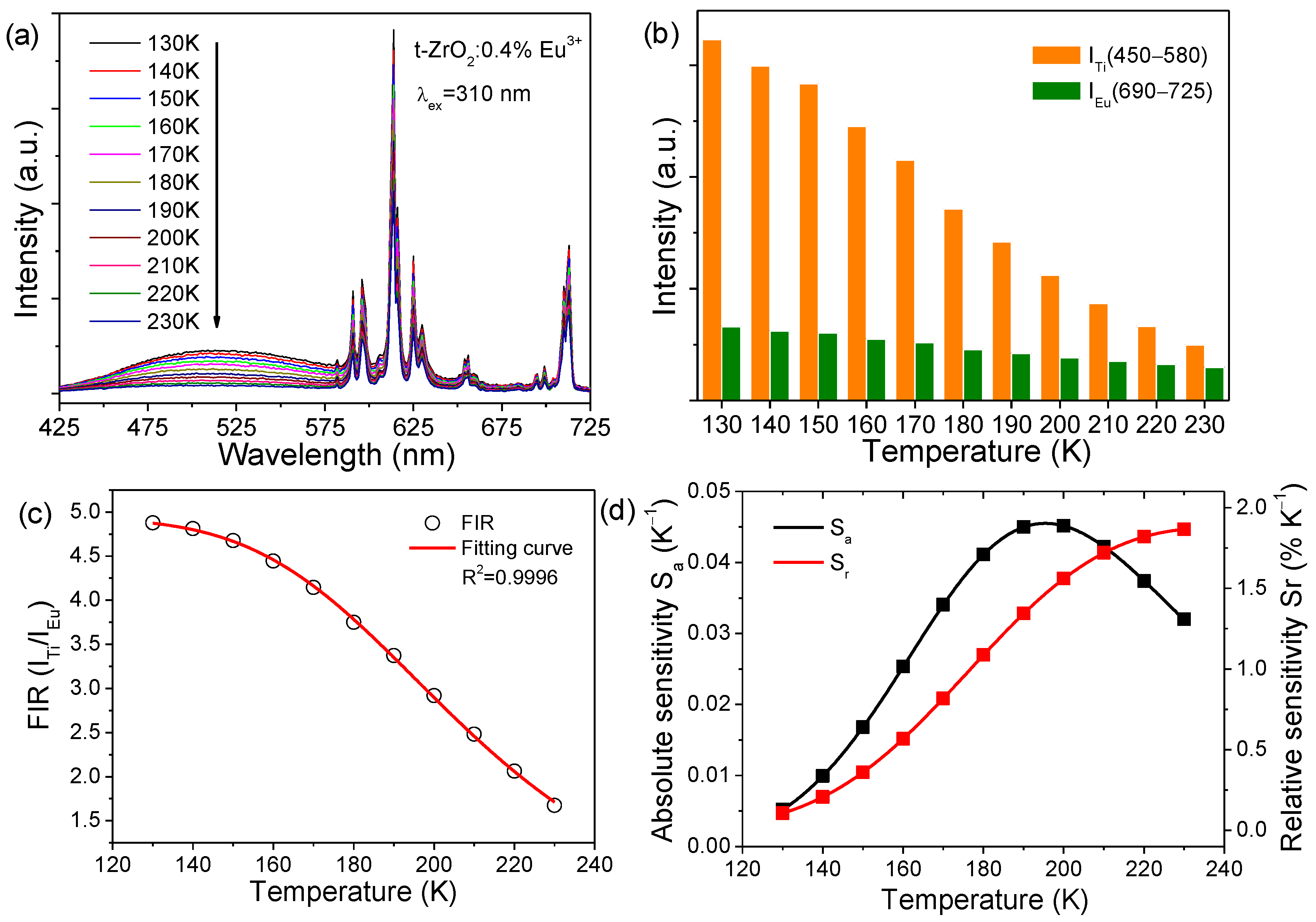
The different temperature sensitivities of PL enables t-ZrO
2:Eu
3+ nanophosphors to have promising potential for ratiometric self-referencing optical thermometry upon single-wavelength excitation, with titanate luminescence as temperature signals and Eu
3+ luminescence as an internal reference. The temperature dependence (130–230 K) of the luminescence of typical t-ZrO
2:0.4%Eu
3+ NCs was examined upon 310 nm excitation for temperature sensing, as shown in
Figure 10a. Note that the peak intensity of the broadband luminescence of titanate groups was much weaker than that of Eu
3+ emissions at each temperature, which was caused by the efficient energy transfer from the titanate groups to Eu
3+ and the higher content of the naturally occurring Ti
4+ ions in the NCs. It was anticipated that such peak intensity would be greatly enhanced if the concentration quenching was suppressed by decreasing the corresponding Ti
4+ content via the use of a high-purity reagent of a Zr source and the stoichiometric addition of a Ti source, which would be favorable for the temperature sensing. The luminescence in a spectral region of 450–580 nm was extracted as signals for the titanate groups (I
Ti), while the luminescence in the range of 690–725 nm was detected for Eu
3+ (I
Eu). The temperature-dependent integrated intensity in such spectral regions is calculated according to the following:
where λ is the wavelength, and T is the corresponding temperature.
Figure 10b shows a histogram of the above calculated integrated intensity. Clearly, the signals from the titanate groups weakened quickly with increasing temperature, whereas those from Eu
3+ decreased much more slowly. The FIR (I
Ti/I
Eu) was found to closely follow the Arrhenius-type Mott equation [
37]:
where I
0 is the initial intensity, A is a constant, E
0 is the activation energy for thermal quenching, and k is the Boltzmann constant. As shown in
Figure 10c, the calculated FIR (I
Ti/I
Eu) values could be well approximated by the fitting using Equation (3), which could be specifically written as
. The absolute sensitivity (S
a) and relative sensitivity (S
r) of optical thermometric materials can be expressed as below [
9,
37]:
The S
a and S
r parameters were determined using Equations (4) and (5) and are plotted in
Figure 10d. The maximal S
a and S
r of the sensing materials of t-ZrO
2:0.4%Eu
3+ NCs were ~0.045 K
−1 (at 190 K) and ~1.9% K
−1 (at 230 K), respectively.
The temperature uncertainty (or resolution) δ
T of the present thermometer can be calculated by the following Equation (6) [
9,
37]:
As shown in
Figure S11, the present optical thermometric material of t-ZrO
2:0.4%Eu
3+ NCs has a high resolution within ~0.2 K in the temperature range of 160–230 K, indicating a high precision towards temperature sensing. The repeatability and reversibility of this thermometry was checked through seven cycling experiments.
Figure S12 shows the plot of FIR at specific temperatures. It is found that the data only show a small deviation from the corresponding average values (dashed lines) for the schemes, indicating a high durability and reproducibility for optical thermometry.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









