
2,12-Diaza[6]helicene: An Efficient Non-Conventional Stereogenic Scaffold for Enantioselective Electrochemical Interphases


 , , and
, , and
Abstract
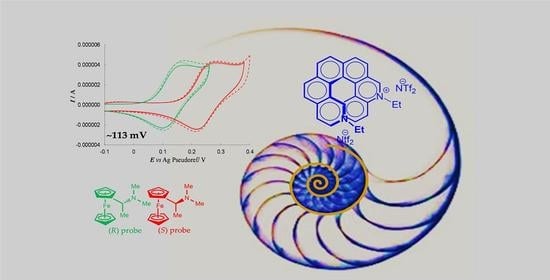
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
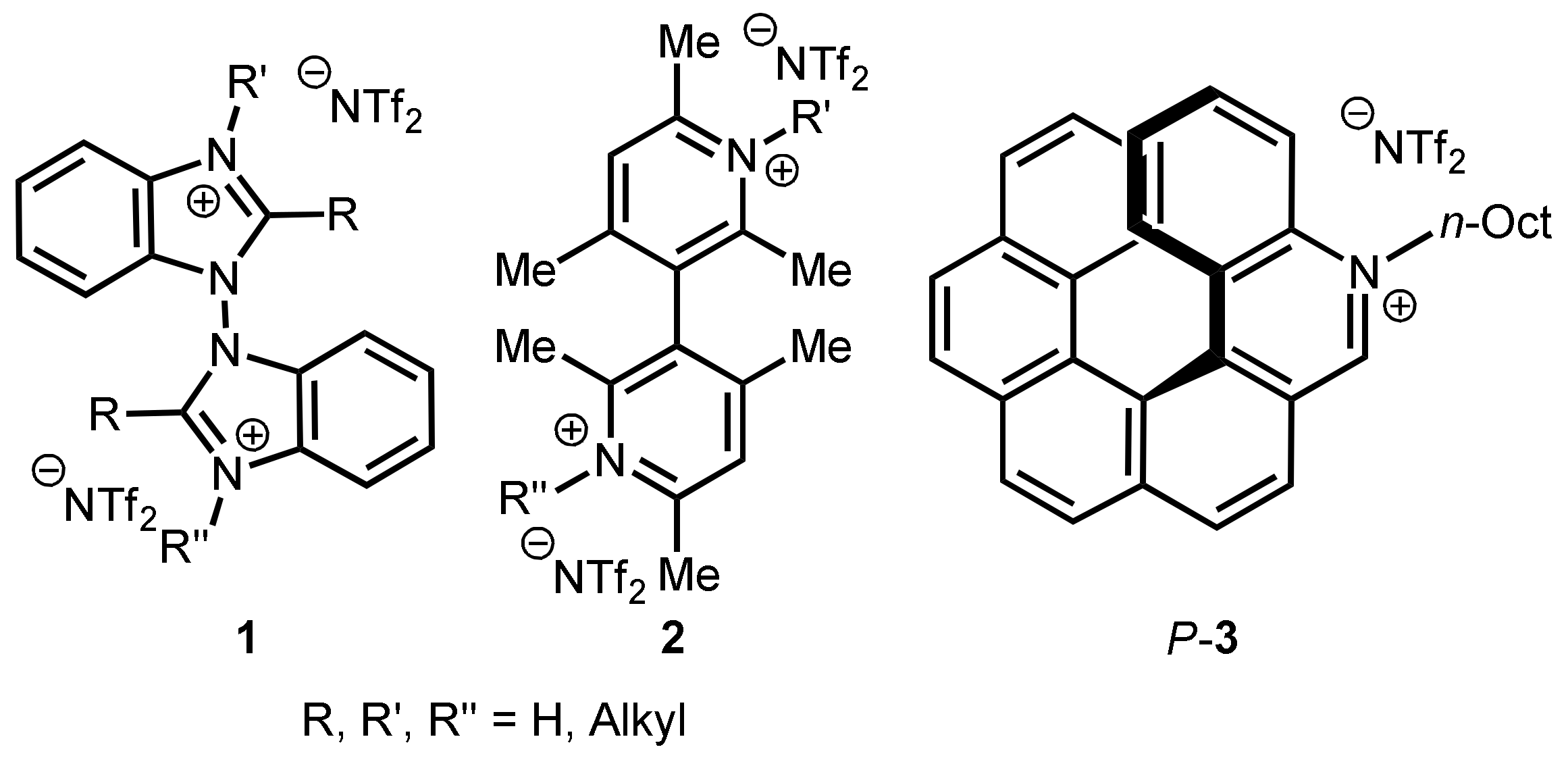
3.1. Synthesis of 2,12-Diaza[6]helicene
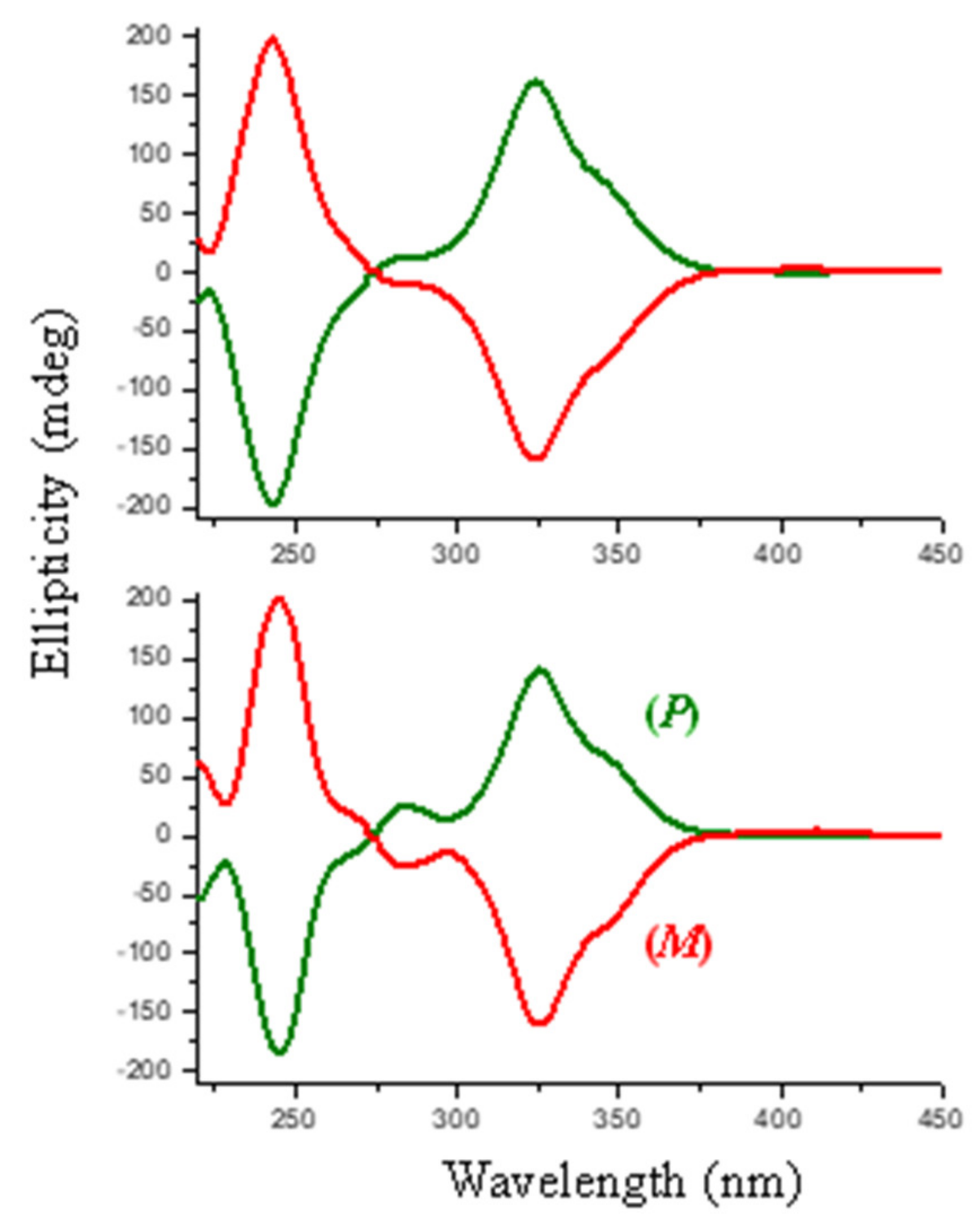
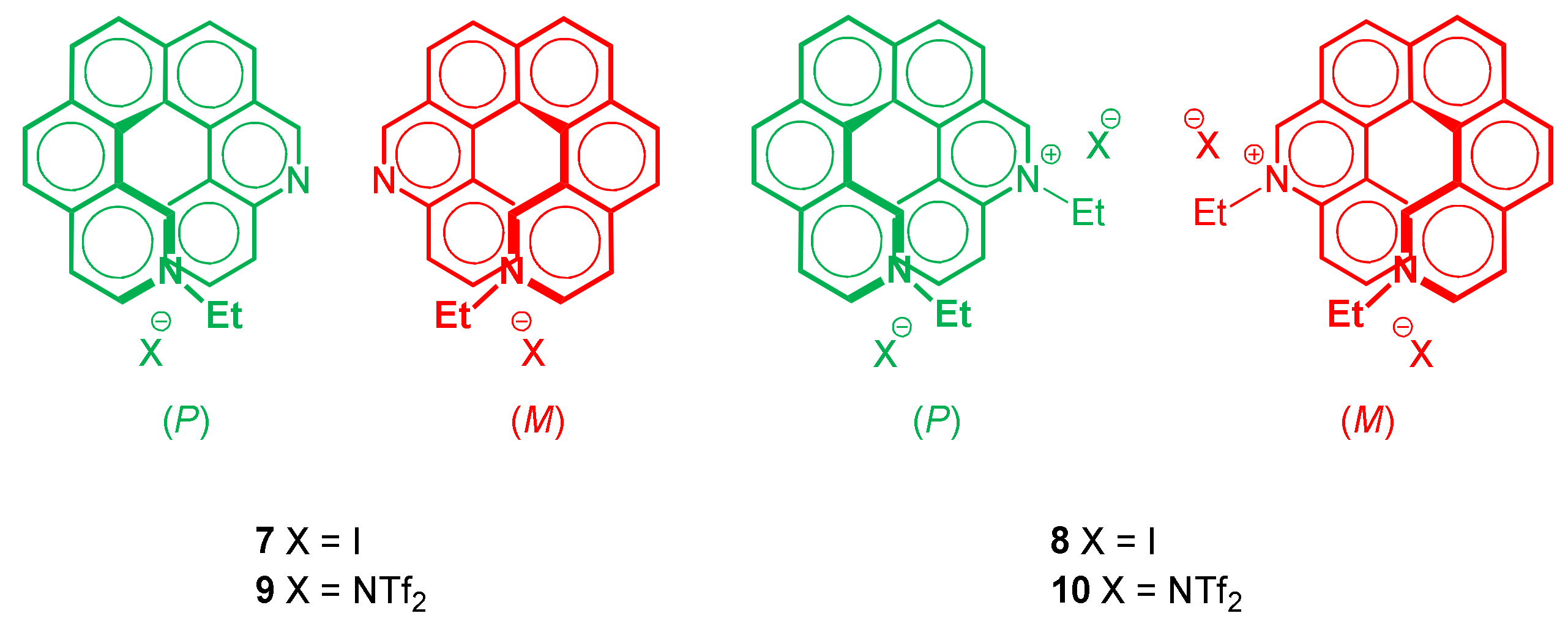
3.2. Resolution of the Enantiomers
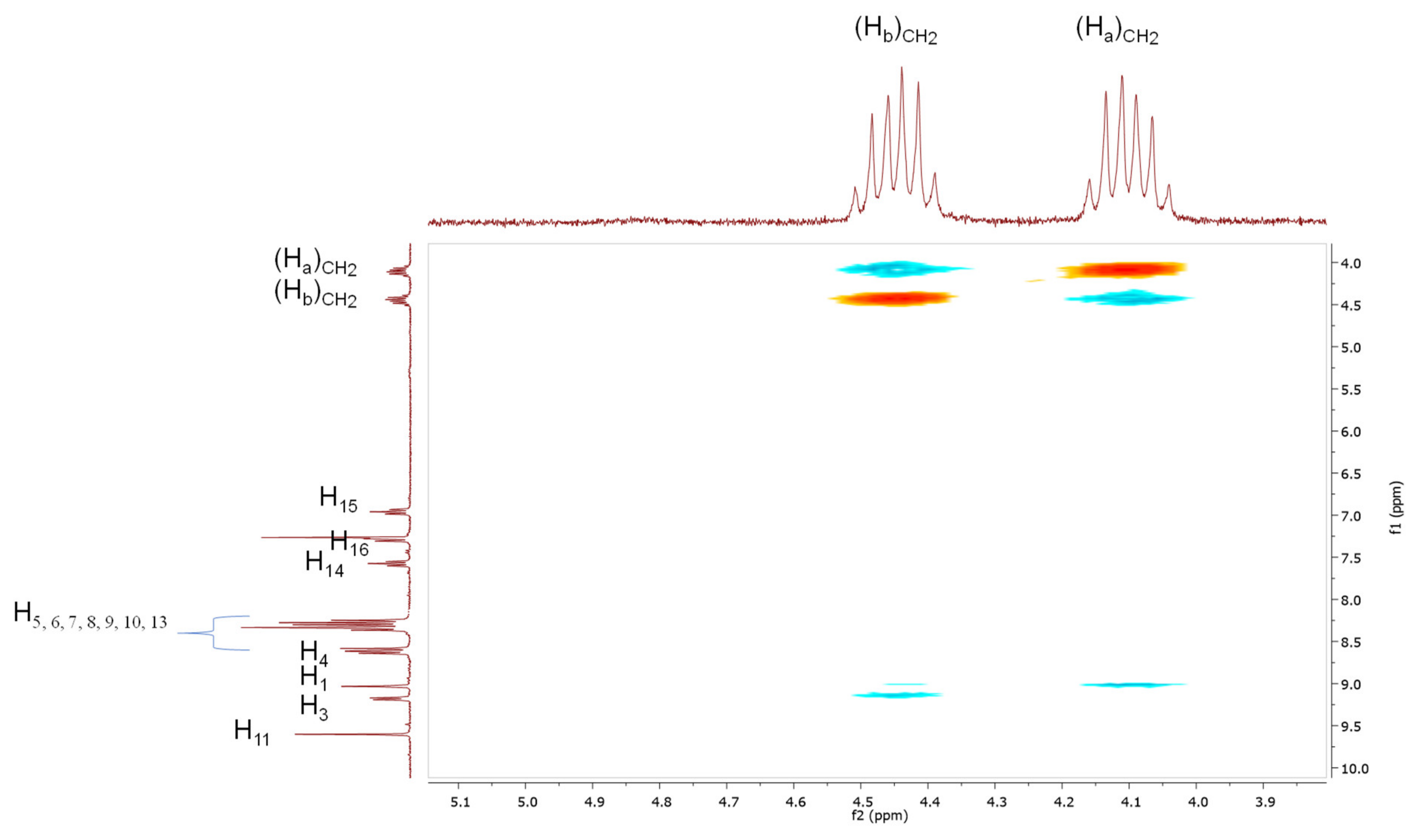
3.3. Alkylation of (P)-4 and (M)-4
3.4. Electrochemical Characterization
- (i)
- only diazahelicene undergoes oxidation within the available electrochemical window. Only one irreversible oxidation peak, followed by a shoulder, is perceivable before the background. It should correspond to oxidation of one of the nitrogen sites, according to the similar value measured for monoazahelicene [6] and significantly less positive than that of carbahelicenes. Given the above-mentioned conjugation between nitrogen sites, the second oxidation should take place at more positive potentials.
- (ii)
- huge positive shift of reduction peak potentials with strong reciprocal interaction between N sites for cations: Parent diazahelicene 4 features a complex reduction peak system, although starting with a monoelectronic chemically and electrochemically reversible peak. This can be assumed to account for reductions on both helicene sides, in the −2.2/−2.8 V range, similar to that formerly observed for the monoazahelicene case. There are, however, differences in peak potentials and multiplicity, consistently with the change in the nature of the two terminals. Instead in the case of 9 a reduction peak is observed at −1.52 V, i.e., much more positive respect to 4, as the nitrogen site, becoming cationic, now provides a preferential site for the first reduction. However, such potential is significantly less positive than the one observed in the case of the monoaza analogue. Since in monoazahelicene salts it has been observed that the first reduction potential is nearly constant, regardless of the position of the nitrogen atom, such a large potential difference could be ascribed, again, to the interaction in space between the aza cation and the free aza terminal, making the first reduction site less electron poor. At the same time, the reduction peak system in the −2.2/−2.8 V range looks much simpler compared to 5-aza[6]helicenium, and should correspond to reduction of the non-alkylated aza site. The azahelicenium double salt 10 features a neat couple of first reduction peaks (spaced about 0.5 V) that, considering again the above remarks about the first reduction potential of an aza cation, should be likely described as the response of two nearly equivalent redox sites, with significant reciprocal interaction along the carbohelicene chain as well as across space. This was also observed in a similar literature case [12], for which two reversible monoelectronic twin peaks are reported at −1.32 and −1.59 V. However, compared with ref. [12], in the case of 10 both reductions occur at less negative potentials, consistently with the overall more efficient conjugation. In our case, the distance between the peaks is higher, which could be ascribed to stronger reciprocal interactions. It is also worth mentioning that in cases 9 and 10, where cation reduction results in radical formation, a possible follow-up is dimerization [12,13].
3.5. Chiral Discrimination Experiments
- (i)
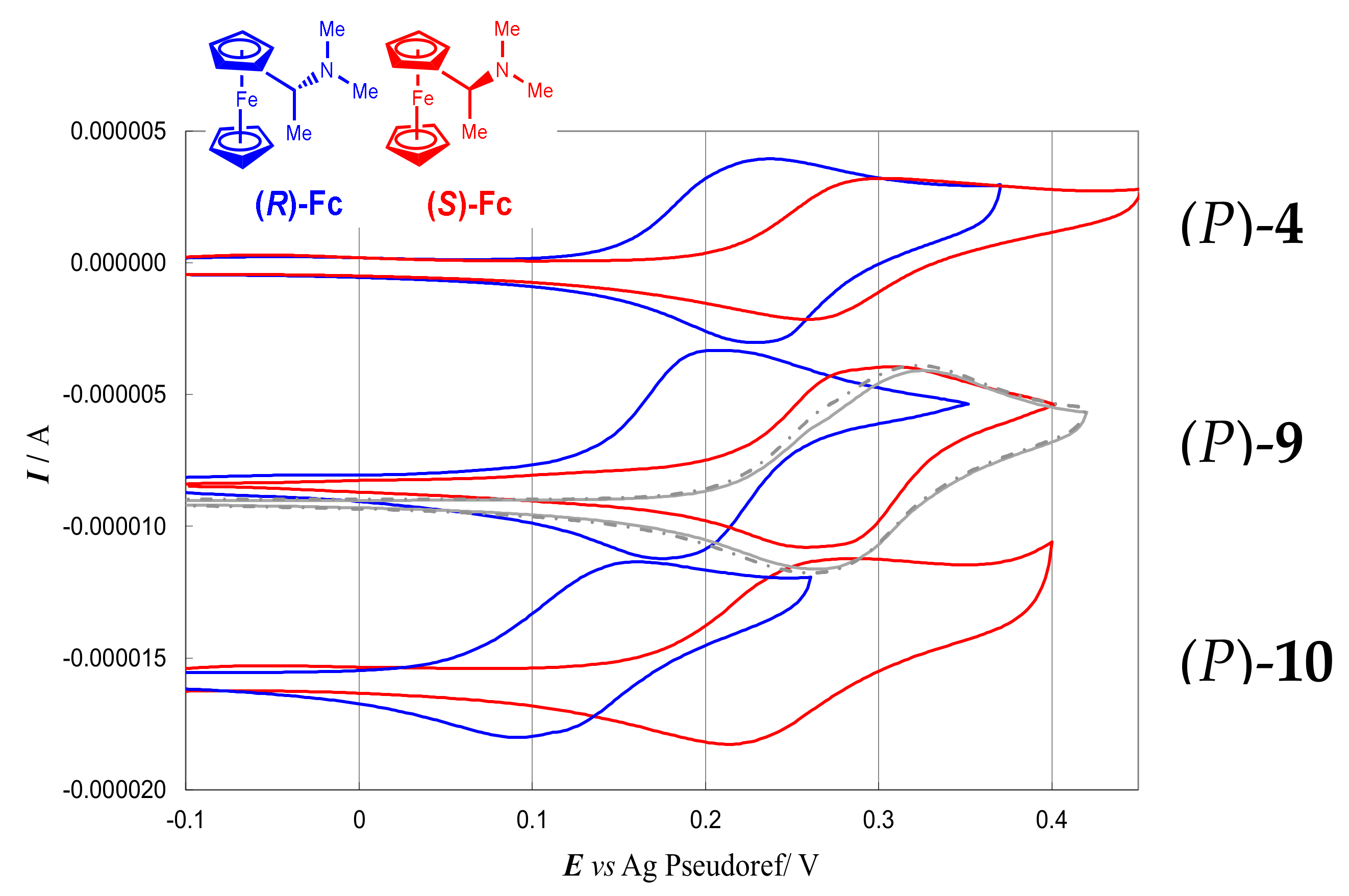
- for enantioselective cyclic voltammetry experiments (CV) with the benchmark electroactive enantiomer probes (R)-(+)- or (S)-(−)- N,N′-dimethyl- 1-ferrocenylethylamine ((R)-Fc and (S)-Fc, Figure 10),
- (ii)
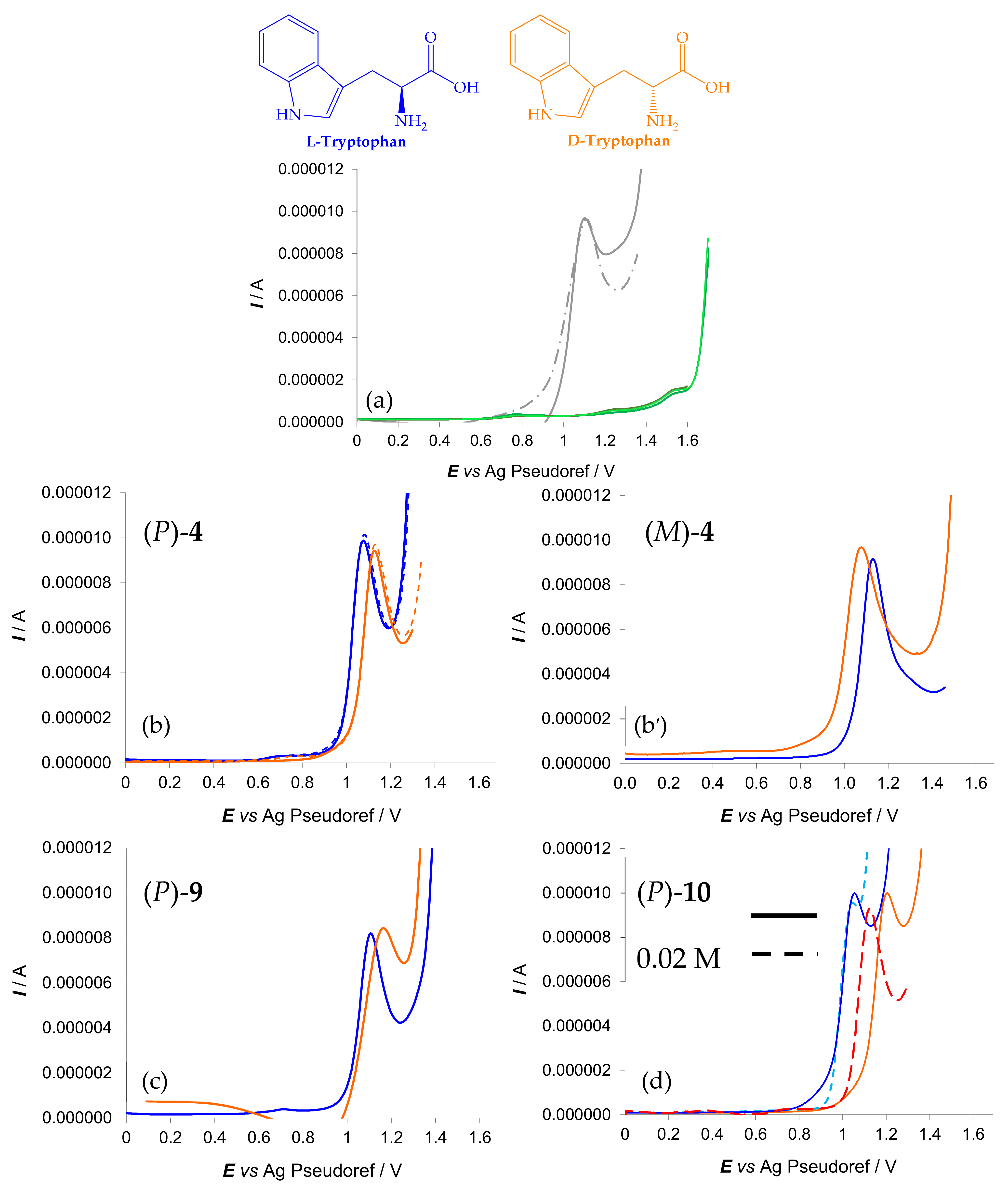
- for enantioselective differential pulse voltammetry (DPV) experiments with the enantiomers of an electroactive probe of biological interest, i.e., L- or D-tryptophan (Trp), an α-amino acid (Figure 11),
- Figure 11a–c provide a comparison of the enantiodiscrimination ability of 4, 9 and 10 as 0.02 M chiral additives in achiral commercial (BMIM)NTf2 with L- and D-Trp (0.03 M). As in the former study case, once more the same probe enantiomer sequence (in this case, L- before D-) is obtained with all the inherently chiral additives, but the peak potential differences significantly increase from 1 to 3 (4 ~60 mV < 9 ~100 mV < 10 ~180 mV) that is, with increasing number of alkylated nitrogen sites.
- Figure 11d highlights the selector concentration effect in the case of inherently chiral salt 10, the most effective selector in the series. In particular, the observed peak potential difference for the probe enantiomers is significantly larger when working with 10 at 0.02 M concentration (solid lines) with respect to 0.01 M concentration (dashed lines). A similar beneficial concentration effect has already been observed by employing as additives in achiral ionic liquids an inherently chiral molecular salt based on either an atropisomeric bipyridinium scaffold [1] or on a chiral bile acid scaffold with many stereocentres as stereogenic elements [14]. Altogether, by considering such three cases, we can assume the above concentration effect to be general.
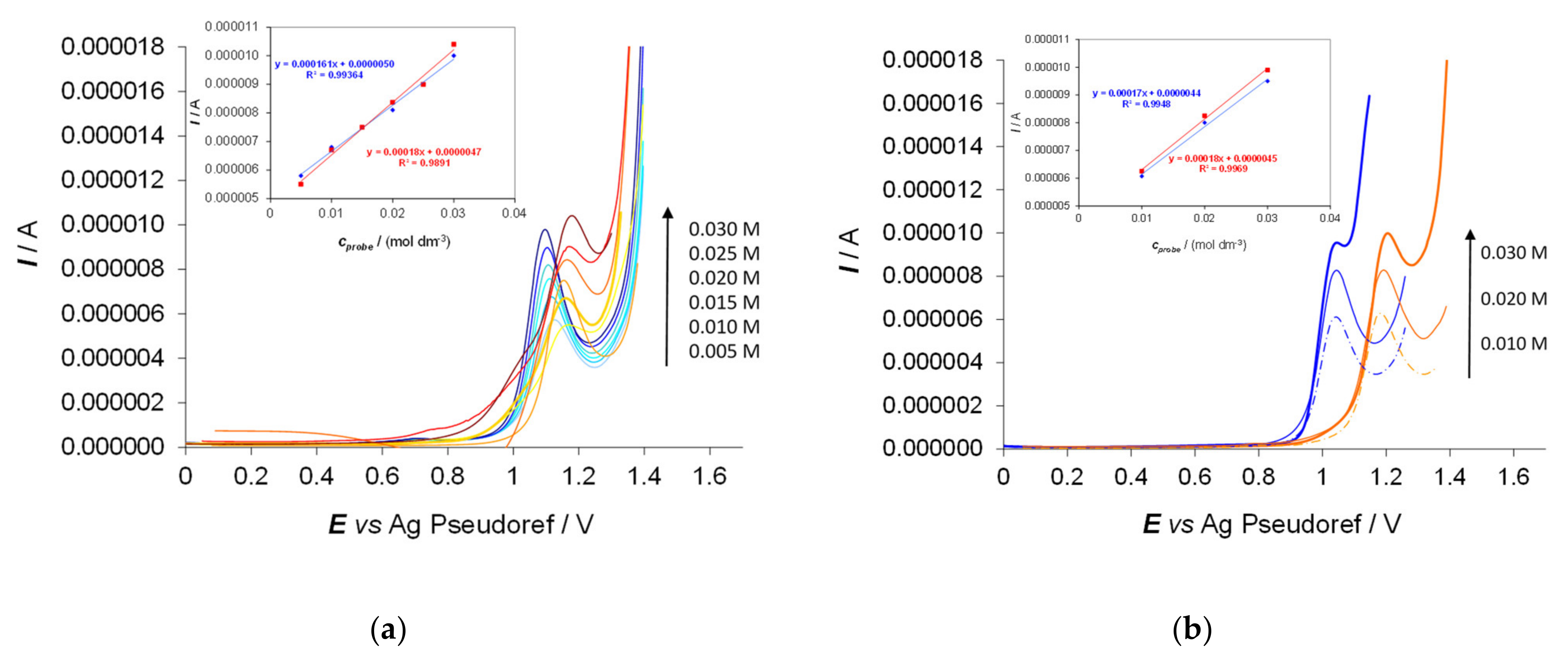
- Testing 9 and 10 with L- and D-Trp in the 0.005–0.03 M concentration range, a linear dynamic range with very good correlation coefficients is observed for the peak currents as a function of the concentration for both enantiomers (Figure 12). Thus, quantification through peak current analysis can be added to configuration assignment based on the peak potential.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rizzo, S.; Arnaboldi, S.; Cirilli, R.; Gennaro, A.; Isse, A.A.; Sannicolò, F.; Mussini, P.R. An “inherently chiral” 1,1′-bibenzimidazolium additive for enantioselective voltammetry in ionic liquid media. Electrochem. Commun. 2018, 89, 57–61. [Google Scholar] [CrossRef]
- Rizzo, S.; Arnaboldi, S.; Mihali, V.; Cirilli, R.; Forni, A.; Gennaro, A.; Isse, A.A.; Pierini, M.; Mussini, P.R.; Sannicolò, F. “Inherently Chiral” Ionic-Liquid Media: Effective Chiral Electroanalysis on Achiral Electrodes. Angew. Chem. Int. Ed. 2017, 56, 2079–2082. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.; Arnaboldi, S.; Husanu, E.; Grecchi, S.; Buzzi, I.F.; Cirilli, R.; Rizzo, S.; Chiappe, C.; Mussini, P.R.; Guazzelli, L. A family of chiral ionic liquids from the natural pool: Relationships between structure and functional properties and electrochemical enantiodiscrimination tests. Electrochim. Acta 2019, 298, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Bazzini, C.; Brovelli, S.; Caronna, T.; Gambarotti, C.; Giannone, M.; Macchi, P.; Meinardi, P.; Mele, A.; Panzeri, W.; Recupero, F.; et al. Synthesis and Characterization of Some Aza [5] helicenes. Eur. J. Org. Chem. 2005, 2005, 1247–1257. [Google Scholar] [CrossRef]
- Janke, R.H.; Haufe, G.; Würthwein, E.-U.; Borkent, J.H. Racemization Barriers of Helicenes: A Computational Study. J. Am. Chem. Soc. 1996, 118, 6031–6035. [Google Scholar] [CrossRef] [Green Version]
- Fontana, F.; Carminati, G.; Bertolotti, B.; Mussini, P.; Arnaboldi, S.; Grecchi, S.; Cirilli, R.; Micheli, L.; Rizzo, S. Helicity: A Non-Conventional Stereogenic Element for Designing Inherently Chiral Ionic Liquids for Electrochemical Enantiodifferentiation. Molecules 2021, 26, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chem. Acc. 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Batesky, D.C.; Goldfogel, M.J.; Weix, D.J. Removal of Triphenylphosphine Oxide by Precipitation with Zinc Chloride in Polar Solvents. J. Org. Chem. 2017, 82, 9931–9936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbate, S.; Longhi, G.; Lebon, F.; Castiglioni, E.; Superchi, S.; Pisani, L.; Fontana, F.; Torricelli, F.; Caronna, T.; Villani, C.; et al. Helical Sense-Responsive and Substituent-Sensitive Features in Vibrational and Electronic Circular Dichroism, in Circularly Polarized Luminescence, and in Raman Spectra of Some Simple Optically Active Hexahelicenes. J. Phys. Chem. C 2014, 118, 1682–1695. [Google Scholar] [CrossRef]
- Caronna, T.; Castiglione, F.; Famulari, A.; Fontana, F.; Malpezzi, L.; Mele, A.; Mendola, D.; Natali Sora, I. Quantum Mechanics Calculations, Basicity and Crystal Structure: The Route to Transition Metal Complexes of Azahelicenes. Molecules 2012, 17, 463–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, J.R.; Pospisil, L.; Bednarova, L.; Correa da Costa, R.; White, A.J.P.; Mori, T.; Teply, F.; Fuchter, M.J. Intense redox-driven chiroptical switching with a 580 mV hysteresis actuated through reversible dimerization of an azoniahelicene. Chem. Commun. 2017, 53, 9059–9062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacek, J.; Zadny, J.; Storch, J.; Hrbac, J. Chiral Electrochemistry: Anodic Deposition of Enantiopure Helical Molecules. ChemPlusChem 2020, 85, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Grecchi, S.; Federghini, C.; Longhi, M.; Mezzetta, A.; Guazzelli, L.; Khawthong, S.; Arduini, F.; Chiappe, C.; Iuliano, A.; Mussini, P.R. Chiral biobased ionic liquids with cations or anions including bile acid building blocks as chiral selectors in voltammetry. ChemElectroChem 2021, 8, 1377–1387. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B3LYP/6–31g(d,p) | B3LYP/Jun-cc-pVTZ | M06–2X/6–31g(d,p) | M06–2X/Jun-cc-pVTZ | |
|---|---|---|---|---|
| Energy (ha) | −1032.54128065 | −1032.85778413 | −1032.12555221 | −1032.46026159 |
| Q[N(2)] | −0.196 | −0.190 | −0.194 | −0.190 |
| Q[N(12)] | −0.194 | −0.188 | −0.192 | −0.188 |
| Q[N(2)] − Q[N(12)] | −0.002 | −0.002 | −0.002 | −0.002 |
| B3LYP/jun-cc-pVTZ | ||
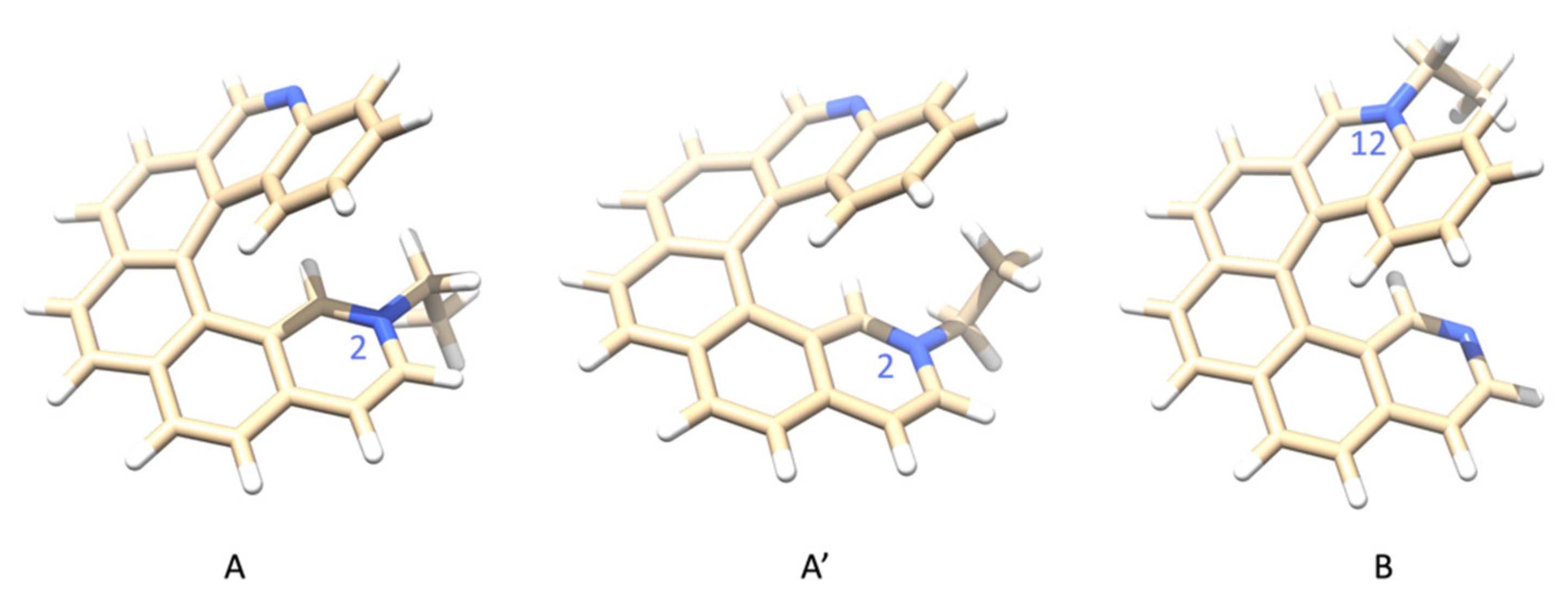
| ID and Ethylation Site | Energy (ha) | ΔE (kcal/mol) |
| A—N(2) | −1111.90878265 | 0.0 |
| A′—N(2) | −1111.90864474 | 0.1 |
| B—N(12) | −1111.90386408 | 3.1 |
| M06–2X/jun-cc-pVTZ | ||
| ID and Ethylation Site | Energy (ha) | ΔE (kcal/mol) |
| A—N(2) | −1111.46570147 | 0.8 |
| A′—N(2) | −1111.46703268 | 0.0 |
| B—N(12) | −1111.46068086 | 4.0 |
| Ep, Ia/V | Ep, Ic/V | |
|---|---|---|
| 5-aza[6]helicene [6] | 1.07, 1.47 | −2.25, −2.52, −2.66 |
| N-octyl-5-aza[6]helicenium bistriflimidate [6] | 1.46 | −1.19 (nearly splitting), −2.58 |
| 4 | 1.26 | −2.16, 2.41,−2.48, −2.73 |
| 9 | no peaks | −1.52 (with prepeak); −2.47 |
| 10 | no peaks | −1.00, −1.52, −2.45, −2.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, F.; Bertolotti, B.; Grecchi, S.; Mussini, P.R.; Micheli, L.; Cirilli, R.; Tommasini, M.; Rizzo, S. 2,12-Diaza[6]helicene: An Efficient Non-Conventional Stereogenic Scaffold for Enantioselective Electrochemical Interphases. Chemosensors 2021, 9, 216. https://doi.org/10.3390/chemosensors9080216
Fontana F, Bertolotti B, Grecchi S, Mussini PR, Micheli L, Cirilli R, Tommasini M, Rizzo S. 2,12-Diaza[6]helicene: An Efficient Non-Conventional Stereogenic Scaffold for Enantioselective Electrochemical Interphases. Chemosensors. 2021; 9(8):216. https://doi.org/10.3390/chemosensors9080216
Chicago/Turabian StyleFontana, Francesca, Benedetta Bertolotti, Sara Grecchi, Patrizia Romana Mussini, Laura Micheli, Roberto Cirilli, Matteo Tommasini, and Simona Rizzo. 2021. "2,12-Diaza[6]helicene: An Efficient Non-Conventional Stereogenic Scaffold for Enantioselective Electrochemical Interphases" Chemosensors 9, no. 8: 216. https://doi.org/10.3390/chemosensors9080216
APA StyleFontana, F., Bertolotti, B., Grecchi, S., Mussini, P. R., Micheli, L., Cirilli, R., Tommasini, M., & Rizzo, S. (2021). 2,12-Diaza[6]helicene: An Efficient Non-Conventional Stereogenic Scaffold for Enantioselective Electrochemical Interphases. Chemosensors, 9(8), 216. https://doi.org/10.3390/chemosensors9080216