
Setting the Stage for Insulin Granule Dysfunction during Type-1-Diabetes: Is ER Stress the Culprit?
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
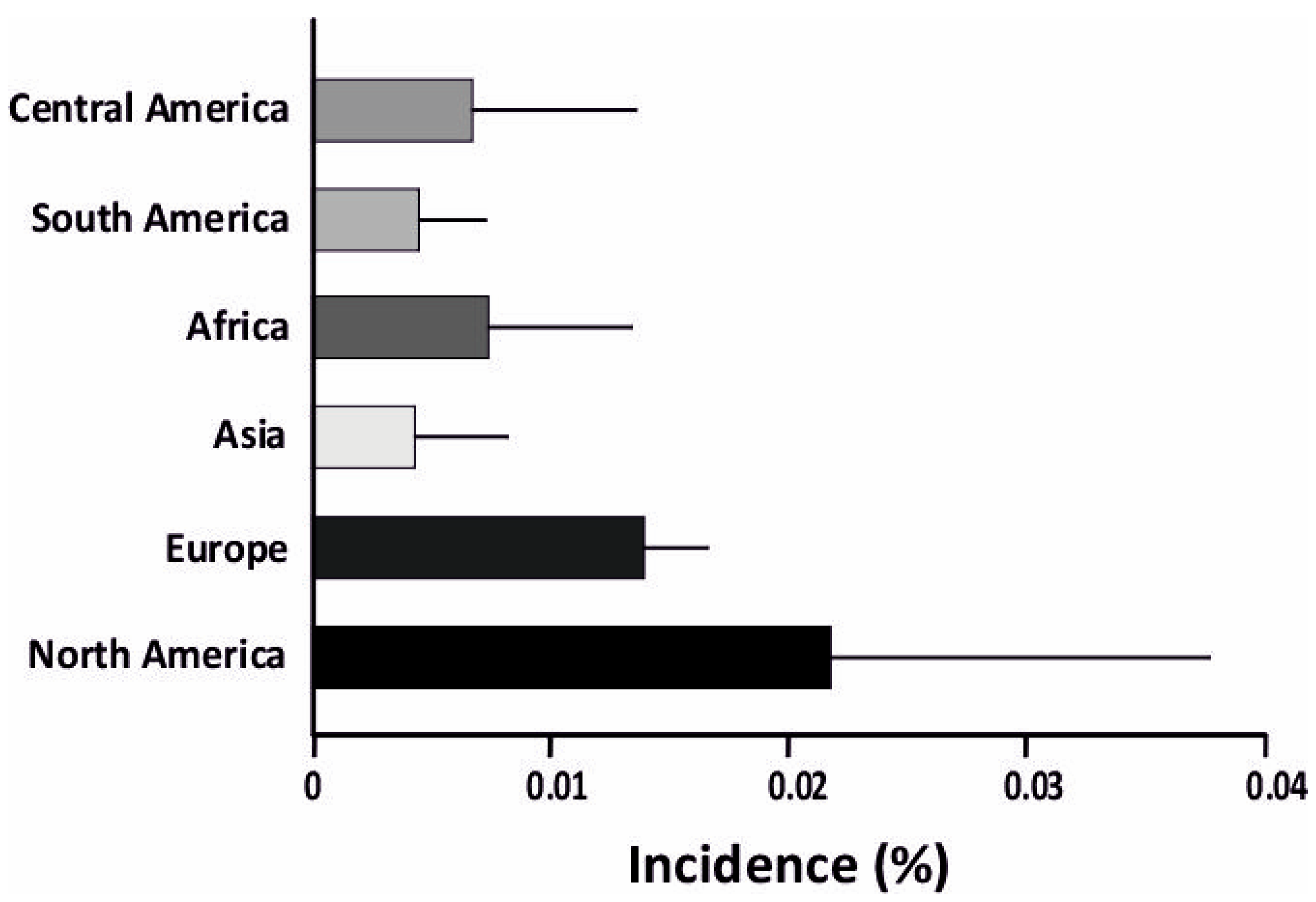
:1. Introduction
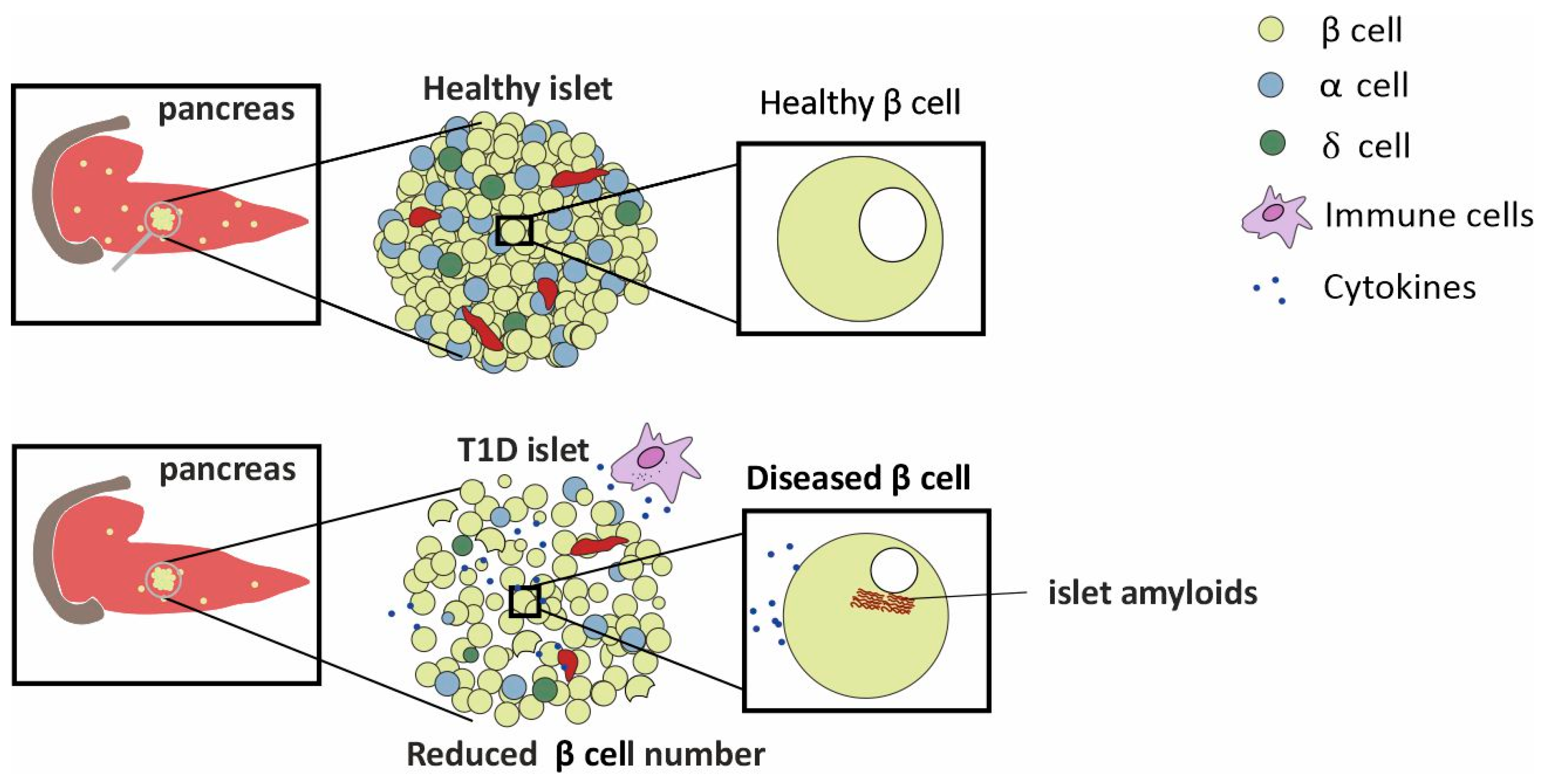
2. A Concise Overview of the Pancreas
3. Factors Influencing T1D Pathogenesis
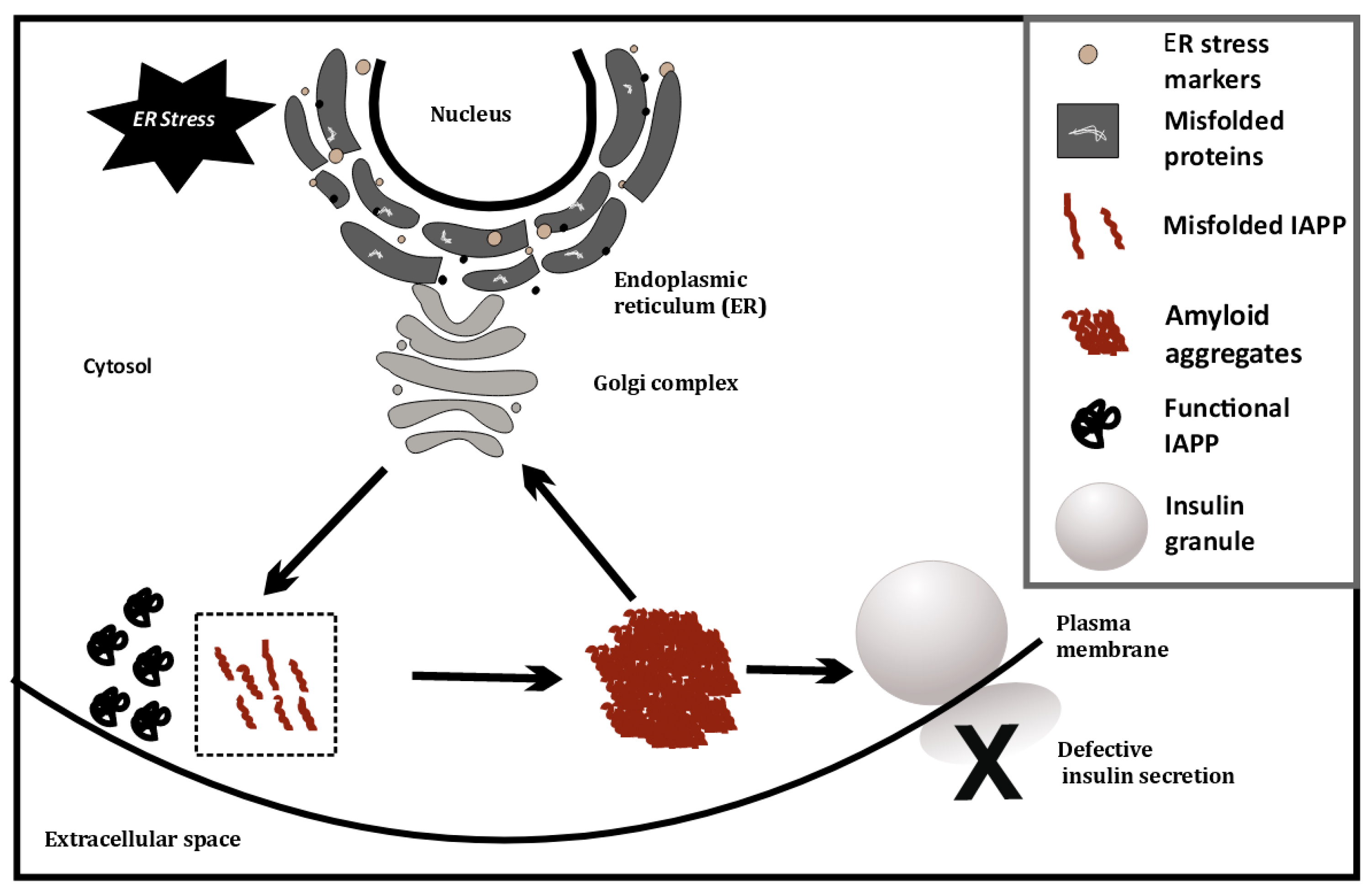
4. ER stress and Its Consequences in T1D
5. IAPP and Amyloids
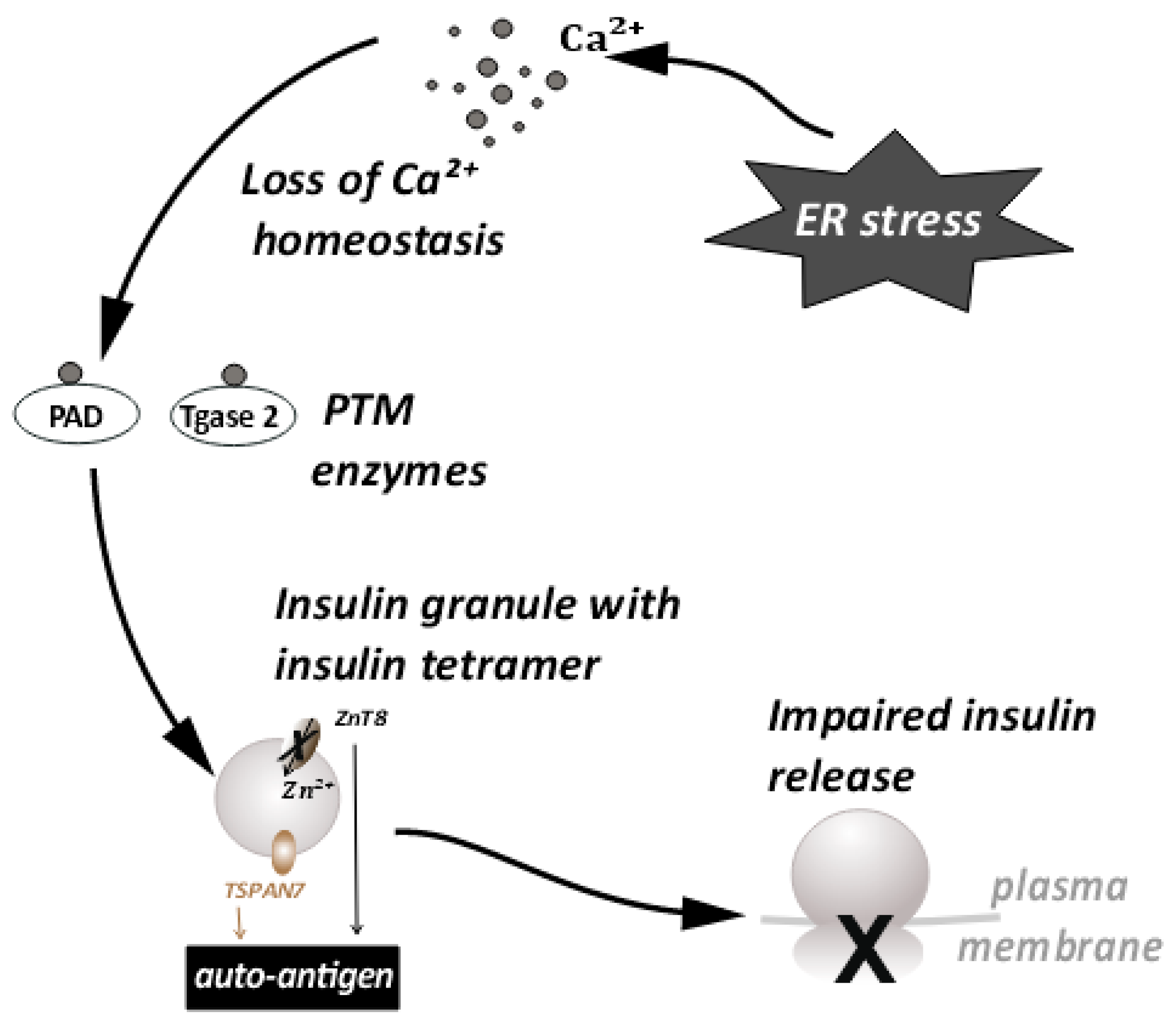
6. Loss of Ca++ Homeostasis as a Mechanism of Autoantigen Generation during ER Stress
7. Effects of ER Stress on Large Dense Core Vesicles—Insulin Secretory Granules (ISGs)
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Syed, F.Z. Type 1 Diabetes Mellitus. Ann. Intern. Med. 2022, 175, ITC34–ITC48. [Google Scholar] [CrossRef] [PubMed]
- Yosten, G.L.C. Alpha Cell Dysfunction in Type 1 Diabetes. Peptides 2018, 100, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Brissova, M.; Haliyur, R.; Saunders, D.; Shrestha, S.; Dai, C.; Blodgett, D.M.; Bottino, R.; Campbell-Thompson, M.; Aramandla, R.; Poffenberger, G.; et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep. 2018, 22, 2667–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komada, H.; Hirota, Y.; Sakaguchi, K.; Okuno, Y.; Ogawa, W.; Seino, S. Impaired Glucagon Secretion in Patients with Fulminant Type 1 Diabetes Mellitus. Endocrine 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.; Begum, M.; Vergari, E.; Briant, L.J.B. Gap Junction Coupling and Islet Delta-Cell Function in Health and Disease. Peptides 2022, 147, 170704. [Google Scholar] [CrossRef]
- Maahs, D.M.; West, N.A.; Lawrence, J.M.; Mayer-Davis, E.J. Epidemiology of Type 1 Diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 481–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IDF Diabetes Atlas 2021|IDF Diabetes Atlas. Available online: https://diabetesatlas.org/atlas/tenth-edition/ (accessed on 14 August 2022).
- Chen, Y.L.; Huang, Y.C.; Qiao, Y.C.; Ling, W.; Pan, Y.H.; Geng, L.J.; Xiao, J.L.; Zhang, X.X.; Zhao, H.L. Climates on Incidence of Childhood Type 1 Diabetes Mellitus in 72 Countries. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Mobasseri, M.; Shirmohammadi, M.; Amiri, T.; Vahed, N.; Fard, H.H.; Ghojazadeh, M. Prevalence and Incidence of Type 1 Diabetes in the World: A Systematic Review and Meta-Analysis. Health Promot. Perspect. 2020, 10, 98–115. [Google Scholar] [CrossRef]
- Harrison, J.W.; Tallapragada, D.S.P.; Baptist, A.; Sharp, S.A.; Bhaskar, S.; Jog, K.S.; Patel, K.A.; Weedon, M.N.; Chandak, G.R.; Yajnik, C.S.; et al. Type 1 Diabetes Genetic Risk Score Is Discriminative of Diabetes in Non-Europeans: Evidence from a Study in India. Sci. Rep. 2020, 10, 9450. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Campbell-Thompson, M.; Kusmartseva, I.; Kaestner, K.H. Organisation of the Human Pancreas in Health and in Diabetes. Diabetologia 2020, 63, 1966–1973. [Google Scholar] [CrossRef]
- Lammert, E.; Thorn, P. The Role of the Islet Niche on Beta Cell Structure and Function. J. Mol. Biol. 2020, 432, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Westermark, G.T.; Krogvold, L.; Dahl-Jørgensen, K.; Ludvigsson, J. Islet Amyloid in Recent-Onset Type 1 Diabetes—the DiViD Study. Ups. J. Med. Sci. 2017, 122, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Todd, J.A. Etiology of Type 1 Diabetes. Immunity 2010, 32, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inshaw, J.R.J.; Cutler, A.J.; Crouch, D.J.M.; Wicker, L.S.; Todd, J.A. Genetic Variants Predisposing Most Strongly to Type 1 Diabetes Diagnosed under Age 7 Years Lie near Candidate Genes That Function in the Immune System and in Pancreatic B-Cells. Diabetes Care 2020, 43, 169–177. [Google Scholar] [CrossRef]
- Noble, J.A.; Valdes, A.M.; Cook, M.; Klitz, W.; Thomson, G.; Erlich, H.A. The Role of HLA Class II Genes in Insulin-Dependent Diabetes Mellitus: Molecular Analysis of 180 Caucasian, Multiplex Families. Am. J. Hum. Genet. 1996, 59, 1134. [Google Scholar] [PubMed]
- Jerram, S.T.; Leslie, R.D. The Genetic Architecture of Type 1 Diabetes. Genes 2017, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Camunas-Soler, J.; Dai, X.-Q.; Hang, Y.; Bautista, A.; Lyon, J.; Suzuki, K.; Kim, S.K.; Quake, S.R.; MacDonald, P.E. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab. 2020, 31, 1017–1031.e4. [Google Scholar] [CrossRef]
- Xia, Y.; Xie, Z.; Huang, G.; Zhou, Z. Incidence and Trend of Type 1 Diabetes and the Underlying Environmental Determinants. Diabetes. Metab. Res. Rev. 2019, 35, e3075. [Google Scholar] [CrossRef] [Green Version]
- Dedrick, S.; Sundaresh, B.; Huang, Q.; Brady, C.; Yoo, T.; Cronin, C.; Rudnicki, C.; Flood, M.; Momeni, B.; Ludvigsson, J.; et al. The Role of Gut Microbiota and Environmental Factors in Type 1 Diabetes Pathogenesis. Front. Endocrinol. (Lausanne) 2020, 11. [Google Scholar] [CrossRef]
- Ylipaasto, P.; Klingel, K.; Lindberg, A.M.; Otonkoski, T.; Kandolf, R.; Hovi, T.; Roivainen, M. Enterovirus Infection in Human Pancreatic Islet Cells, Islet Tropism in Vivo and Receptor Involvement in Cultured Islet Beta Cells. Diabetologia 2004, 47, 225–239. [Google Scholar] [CrossRef]
- Clements, G.B.; Galbraith, D.N.; Taylor, K.W. Coxsackie B Virus Infection and Onset of Childhood Diabetes. Lancet 1995, 346, 221–223. [Google Scholar] [CrossRef]
- André, L.; Hober, D.; Hober-Vandenberghe, C.; Belaich, S.; Vantyghem, M.-C.; Lefebvre, J.; Wattré, P. Detection of Coxsackie B Virus RNA Sequences in Whole Blood Samples From Adult Patients at the Onset of Type I Diabetes Mellitus. J. Med. Virol 1997, 52, 121–127. [Google Scholar] [CrossRef]
- Yap, I.S.; Giddings, G.; Pocock, E.; Chantler, J.K. Lack of Islet Neogenesis Plays a Key Role in Beta-Cell Depletion in Mice Infected with a Diabetogenic Variant of Coxsackievirus B4. J. Gen. Virol. 2003, 84, 3051–3068. [Google Scholar] [CrossRef] [PubMed]
- Colli, M.L.; Paula, F.M.; Marselli, L.; Marchetti, P.; Roivainen, M.; Eizirik, D.L.; Op De Beeck, A. Coxsackievirus B Tailors the Unfolded Protein Response to Favour Viral Amplification in Pancreatic β Cells. J. Innate Immun. 2019, 11, 375–389. [Google Scholar] [CrossRef]
- Christen, U.; Bender, C.; von Herrath, M.G. Infection as a Cause of Type 1 Diabetes? Curr. Opin. Rheumatol. 2012, 24. [Google Scholar] [CrossRef]
- Singh, K. Regulatory T Cells in Type 1 Diabetes: The Role of IL-35 in Counteracting the Disease; Uppsala University, Department of Medical Biochemistry and Microbiology: Uppsala, Sweden, 2017. [Google Scholar]
- Ylipaasto, P.; Smura, T.; Gopalacharyulu, P.; Paananen, A.; Seppänen-Laakso, T.; Kaijalainen, S.; Ahlfors, H.; Korsgren, O.; Lakey, J.R.T.; Lahesmaa, R.; et al. Enterovirus-Induced Gene Expression Profile Is Critical for Human Pancreatic Islet Destruction. Diabetologia 2012, 55, 3273–3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.H.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of Islet-Autoreactive CD8 T Cells in Insulitic Lesions from Recent Onset and Long-Term Type 1 Diabetes Patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Stone, N.L.; Harrison, L.C. T-Cell Epitopes in Type 1 Diabetes Autoantigen Tyrosine Phosphatase IA-2: Potential for Mimicry with Rotavirus and Other Environmental Agents. Mol. Med. 1998, 4, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Shruthi, S.; Mohan, V.; Amutha, A.; Aravindhan, V. Increased Serum Levels of Novel T Cell Cytokines IL-33, IL-9 and IL-17 in Subjects with Type-1 Diabetes. Cytokine 2016, 86, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.S.; Blower, M.D. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Marré, M.L.; James, E.A.; Piganelli, J.D. β Cell ER Stress and the Implications for Immunogenicity in Type 1 Diabetes. Front. cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. 2008, 3, 399. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis Control by the Unfolded Protein Response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 MRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Kaufman, R.J. The Impact of the Unfolded Protein Response on Human Disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Engin, F.; Yermalovich, A.; Ngyuen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the Unfolded Protein Response in Pancreatic β Cells Protects Mice against Type 1 Diabetes. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaça, J.; Caicedo, A.; Landsman, L. Beta Cell Dysfunction in Diabetes: The Islet Microenvironment as an Unusual Suspect. Diabetologia 2020, 63, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.L.; Westermark, G.T.; Westermark, P.; Kahn, S.E. Islet Amyloid: A Critical Entity in the Pathogenesis of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3629–3643. [Google Scholar] [CrossRef] [Green Version]
- Marzban, L.; Trigo-Gonzalez, G.; Verchere, C.B. Processing of Pro-Islet Amyloid Polypeptide in the Constitutive and Regulated Secretory Pathways of Beta Cells. Mol. Endocrinol. 2005, 19, 2154–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskarsson, M.E.; Singh, K.; Wang, J.; Vlodavsky, I.; Li, J.; Westermark, G.T. Heparan Sulfate Proteoglycans Are Important for Islet Amyloid Formation and Islet Amyloid Polypeptide-Induced Apoptosis *. J. Biol. Chem. 2015, 290, 15121–15132. [Google Scholar] [CrossRef] [Green Version]
- Paulsson, J.F.; Ludvigsson, J.; Carlsson, A.; Casas, R.; Forsander, G.; Ivarsson, S.A.; Kockum, I.; Lernmark, Å.; Marcus, C.; Lindblad, B.; et al. High Plasma Levels of Islet Amyloid Polypeptide in Young with New-Onset of Type 1 Diabetes Mellitus. PLoS One 2014, 9, e93053. [Google Scholar] [CrossRef] [Green Version]
- Cadavez, L.; Montane, J.; Alcarraz-Vizán, G.; Visa, M.; Vidal-Fàbrega, L.; Servitja, J.M.; Novials, A. Chaperones Ameliorate Beta Cell Dysfunction Associated with Human Islet Amyloid Polypeptide Overexpression. PLoS One 2014, 9, e101797. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, D.C.; Burnett, L.; Kudaibergenova, Z.; Jeremic, A.M. FoxA2 and RNA Pol II Mediate Human Islet Amyloid Polypeptide Turnover in ER-Stressed Pancreatic β-Cells. Biochem. J. 2021, 478, 1261–1282. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Chang, C.; Zhou, Z. Molecular Mechanisms in Autoimmune Type 1 Diabetes: A Critical Review. Clin. Rev. Allergy Immunol. 2014, 47, 174–192. [Google Scholar] [CrossRef]
- Görlach, A.; Klappa, P.; Kietzmann, T. The Endoplasmic Reticulum: Folding, Calcium Homeostasis, Signaling, and Redox Control. Antioxidants Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Burkhardt, H.; Sehnert, B.; Bockermann, R.; Engström, Å.; Kalden, J.R.; Holmdahl, R. Humoral Immune Response to Citrullinated Collagen Type II Determinants in Early Rheumatoid Arthritis. Eur. J. Immunol. 2005, 35, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Whitaker, J.N.; Rhame, L.; Goodin, R.R.; Mc Farland, H.F. Citrulline-Containing Myelin Basic Protein Is Recognized by T-Cell Lines Derived from Multiple Sclerosis Patients and Healthy Individuals. Neurology 1994, 44, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Marré, M.L.; Profozich, J.L.; Coneybeer, J.T.; Geng, X.; Bertera, S.; Ford, M.J.; Trucco, M.; Piganelli, J.D. Inherent ER Stress in Pancreatic Islet β Cells Causes Self-Recognition by Autoreactive T Cells in Type 1 Diabetes. J. Autoimmun. 2016, 72, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lummel, M.; Duinkerken, G.; Van Veelen, P.A.; De Ru, A.; Cordfunke, R.; Zaldumbide, A.; Gomez-Touriño, I.; Arif, S.; Peakman, M.; Drijfhout, J.W.; et al. Posttranslational Modification of HLA-DQ Binding Islet Autoantigens in Type 1 Diabetes. Diabetes 2014, 63, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delong, T.; Baker, R.L.; He, J.; Barbour, G.; Bradley, B.; Haskins, K. Diabetogenic T-Cell Clones Recognize an Altered Peptide of Chromogranin A. Diabetes 2012, 61, 3239–3246. [Google Scholar] [CrossRef] [Green Version]
- Taupenot, L.; Harper, K.L.; Mahapatra, N.R.; Parmer, R.J.; Mahata, S.K.; O’Connor, D.T. Identification of a Novel Sorting Determinant for the Regulated Pathway in the Secretory Protein Chromogranin A. J. Cell Sci. 2002, 115, 4827–4841. [Google Scholar] [CrossRef] [Green Version]
- Strollo, R.; Vinci, C.; Arshad, M.H.; Perrett, D.; Tiberti, C.; Chiarelli, F.; Napoli, N.; Pozzilli, P.; Nissim, A. Antibodies to Post-Translationally Modified Insulin in Type 1 Diabetes. Diabetologia 2015, 58, 2851–2860. [Google Scholar] [CrossRef] [Green Version]
- Walther, D.; Eugster, A.; Jergens, S.; Gavrisan, A.; Weinzierl, C.; Telieps, T.; Winkler, C.; Ziegler, A.G.; Bonifacio, E. Tetraspanin 7 Autoantibodies in Type 1 Diabetes. Diabetologia 2016, 59, 1973–1976. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, K.A.; Tombs, M.A.; Christie, M.R. Autoimmunity to Tetraspanin-7 in Type 1 Diabetes. Med. Microbiol. Immunol. 2020, 209, 437–445. [Google Scholar] [CrossRef] [Green Version]
- McGinty, J.W.; Chow, I.T.; Greenbaum, C.; Odegard, J.; Kwok, W.W.; James, E.A. Recognition of Posttranslationally Modified GAD65 Epitopes in Subjects with Type 1 Diabetes. Diabetes 2014, 63, 3033–3040. [Google Scholar] [CrossRef]
- Hill, H.; Elksnis, A.; Lundkvist, P.; Ubhayasekera, K.; Bergquist, J.; Birnir, B.; Carlsson, P.-O.; Espes, D. Endogenous Levels of Gamma Amino-Butyric Acid Are Correlated to Glutamic-Acid Decarboxylase Antibody Levels in Type 1 Diabetes. Biomedicines 2021, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Wenzlau, J.M.; Juhl, K.; Yu, L.; Moua, O.; Sarkar, S.A.; Gottlieb, P.; Rewers, M.; Eisenbarth, G.S.; Jensen, J.; Davidson, H.W.; et al. The Cation Efflux Transporter ZnT8 (Slc30A8) Is a Major Autoantigen in Human Type 1 Diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 17040–17045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omar-Hmeadi, M.; Idevall-Hagren, O. Insulin Granule Biogenesis and Exocytosis. Cell. Mol. Life Sci. 2021, 78, 1957–1970. [Google Scholar] [CrossRef] [PubMed]
- Barghouth, M.; Jiang, X.; Nagao, M.; Chen, N.; Yang, D.; Ye, Y.; Luan, C.; Gomez, M.F.; Blom, A.M.; Wollheim, C.B.; et al. The Structure of Insulin Granule Core Determines Secretory Capacity Being Reduced in Type-2 Diabetes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Taoufiq, Z.; Ninov, M.; Villar-Briones, A.; Wang, H.-Y.; Sasaki, T.; Roy, M.C.; Beauchain, F.; Mori, Y.; Yoshida, T.; Takamori, S.; et al. Hidden Proteome of Synaptic Vesicles in the Mammalian Brain. Proc. Natl. Acad. Sci. USA 2020, 117, 33586–33596. [Google Scholar] [CrossRef]
- Gandasi, N.R.; Yin, P.; Omar-Hmeadi, M.; Ottosson Laakso, E.; Vikman, P.; Barg, S. Glucose-Dependent Granule Docking Limits Insulin Secretion and Is Decreased in Human Type 2 Diabetes. Cell Metab. 2018, 27, 470–478.e4. [Google Scholar] [CrossRef] [Green Version]
- Gandasi, N.R.; Barg, S. Contact-Induced Clustering of Syntaxin and Munc18 Docks Secretory Granules at the Exocytosis Site. Nat. Commun. 2014, 5, 3914. [Google Scholar] [CrossRef] [Green Version]
- Gandasi, N.R.; Yin, P.; Riz, M.; Chibalina, M.V.; Cortese, G.; Lund, P.-E.; Matveev, V.; Rorsman, P.; Sherman, A.; Pedersen, M.G.; et al. Ca2+ Channel Clustering with Insulin-Containing Granules Is Disturbed in Type 2 Diabetes. J. Clin. Investig. 2017, 127, 2353–2364. [Google Scholar] [CrossRef] [Green Version]
- Tengholm, A.; Gylfe, E. Oscillatory Control of Insulin Secretion. Mol. Cell. Endocrinol. 2009, 297, 58–72. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. The Presynaptic Active Zone. Neuron 2012, 75, 11–25. [Google Scholar] [CrossRef]
- Aslamy, A.; Oh, E.; Ahn, M.; Moin, A.S.M.; Chang, M.; Duncan, M.; Hacker-Stratton, J.; El-Shahawy, M.; Kandeel, F.; DiMeglio, L.A.; et al. Exocytosis Protein DOC2B as a Biomarker of Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Chabosseau, P.; Bellomo, E.A.; Maret, W.; Mitchell, R.K.; Hodson, D.J.; Solomou, A.; Hu, M. Intracellular Zinc in Insulin Secretion and Action: A Determinant of Diabetes Risk? Proc. Nutr. Soc. 2016, 75, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, E. ZnT8 and Type 1 Diabetes. Endocr. J. 2012, 59, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolson, T.J.; Bellomo, E.A.; Wijesekara, N.; Loder, M.K.; Baldwin, J.M.; Gyulkhandanyan, A.V.; Koshkin, V.; Tarasov, A.I.; Carzaniga, R.; Kronenberger, K.; et al. Insulin Storage and Glucose Homeostasis in Mice Null for the Granule Zinc Transporter ZnT8 and Studies of the Type 2 Diabetes-Associated Variants. Diabetes 2009, 58, 2070–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hald, J.; Galbo, T.; Rescan, C.; Radzikowski, L.; Sprinkel, A.e.; Heimberg, H.; Ahnfelt-Rønne, J.; Jensen, J.; Scharfmann, R.; Gradwohl, G.; et al. Pancreatic Islet and Progenitor Cell Surface Markers with Cell Sorting Potential. Diabetologia 2012, 55, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trexler, A.J.; Sochacki, K.A.; Taraska, J.W. Imaging the Recruitment and Loss of Proteins and Lipids at Single Sites of Calcium-Triggered Exocytosis. Mol. Biol. Cell 2016, 27, 2423–2434. [Google Scholar] [CrossRef]
- Dickerson, M.T.; Dadi, P.K.; Butterworth, R.B.; Nakhe, A.Y.; Graff, S.M.; Zaborska, K.E.; Schaub, C.M.; Jacobson, D.A. Tetraspanin-7 Regulation of L-Type Voltage-Dependent Calcium Channels Controls Pancreatic β-Cell Insulin Secretion. J. Physiol. 2020, 598, 4887–4905. [Google Scholar] [CrossRef]
- Rorsman, P.; Braun, M.; Zhang, Q. Regulation of Calcium in Pancreatic α- and β-Cells in Health and Disease. Cell Calcium 2012, 51, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Wicksteed, B.; Uchizono, Y.; Alarcon, C.; McCuaig, J.F.; Shalev, A.; Rhodes, C.J.J. A Cis-Element in the 5’ Untranslated Region of the Preproinsulin MRNA (PpIGE) Is Required for Glucose Regulation of Proinsulin Translation. Cell Metab. 2007, 5, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Ghiasi, S.M.; Dahlby, T.; Hede Andersen, C.; Haataja, L.; Petersen, S.; Omar-Hmeadi, M.; Yang, M.; Pihl, C.; Bresson, S.E.; Khilji, M.S.; et al. Endoplasmic Reticulum Chaperone Glucose-Regulated Protein 94 Is Essential for Proinsulin Handling. Diabetes 2019, 68, 747–760. [Google Scholar] [CrossRef]
- Szabat, M.; Page, M.M.; Panzhinskiy, E.; Skovsø, S.; Mojibian, M.; Fernandez-Tajes, J.; Bruin, J.E.; Bround, M.J.; Lee, J.T.C.; Xu, E.E.; et al. Reduced Insulin Production Relieves Endoplasmic Reticulum Stress and Induces β Cell Proliferation. Cell Metab. 2016, 23, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arunagiri, A.; Alam, M.D.M.; Haataja, L.; Samy, P.J.; Soleimanpour, S.; Satin, L.S.; Itkin-Ansari, P.; Liu, M.; Arvan, P. 1461-P: Quantifying Proinsulin Misfolding in the Endoplasmic Reticulum. Diabetes 2022, 71, 1461. [Google Scholar] [CrossRef]
- Nakayama, M. Insulin as a Key Autoantigen in the Development of Type 1 Diabetes. Diabetes. Metab. Res. Rev. 2011, 27, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara-Imaizumi, M.; Cardozo, A.; Kikuta, T.; Eizirik, D.; Nagamatsu, S. The Cytokine Interleukin-1 Reduces the Docking and Fusion of Insulin Granules in Pancreatic -Cells, Preferentially Decreasing the First Phase of Exocytosis. J. Biol. Chem. 2004, 279, 41271–41274. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makam, A.A.; Biswas, A.; Kothegala, L.; Gandasi, N.R. Setting the Stage for Insulin Granule Dysfunction during Type-1-Diabetes: Is ER Stress the Culprit? Biomedicines 2022, 10, 2695. https://doi.org/10.3390/biomedicines10112695
Makam AA, Biswas A, Kothegala L, Gandasi NR. Setting the Stage for Insulin Granule Dysfunction during Type-1-Diabetes: Is ER Stress the Culprit? Biomedicines. 2022; 10(11):2695. https://doi.org/10.3390/biomedicines10112695
Chicago/Turabian StyleMakam, Aishwarya A., Anusmita Biswas, Lakshmi Kothegala, and Nikhil R. Gandasi. 2022. "Setting the Stage for Insulin Granule Dysfunction during Type-1-Diabetes: Is ER Stress the Culprit?" Biomedicines 10, no. 11: 2695. https://doi.org/10.3390/biomedicines10112695
APA StyleMakam, A. A., Biswas, A., Kothegala, L., & Gandasi, N. R. (2022). Setting the Stage for Insulin Granule Dysfunction during Type-1-Diabetes: Is ER Stress the Culprit? Biomedicines, 10(11), 2695. https://doi.org/10.3390/biomedicines10112695