
Fats, Friends or Foes: Investigating the Role of Short- and Medium-Chain Fatty Acids in Alzheimer’s Disease


Abstract
:
1. Introduction
2. Alzheimer´s Disease Pathology Hallmarks
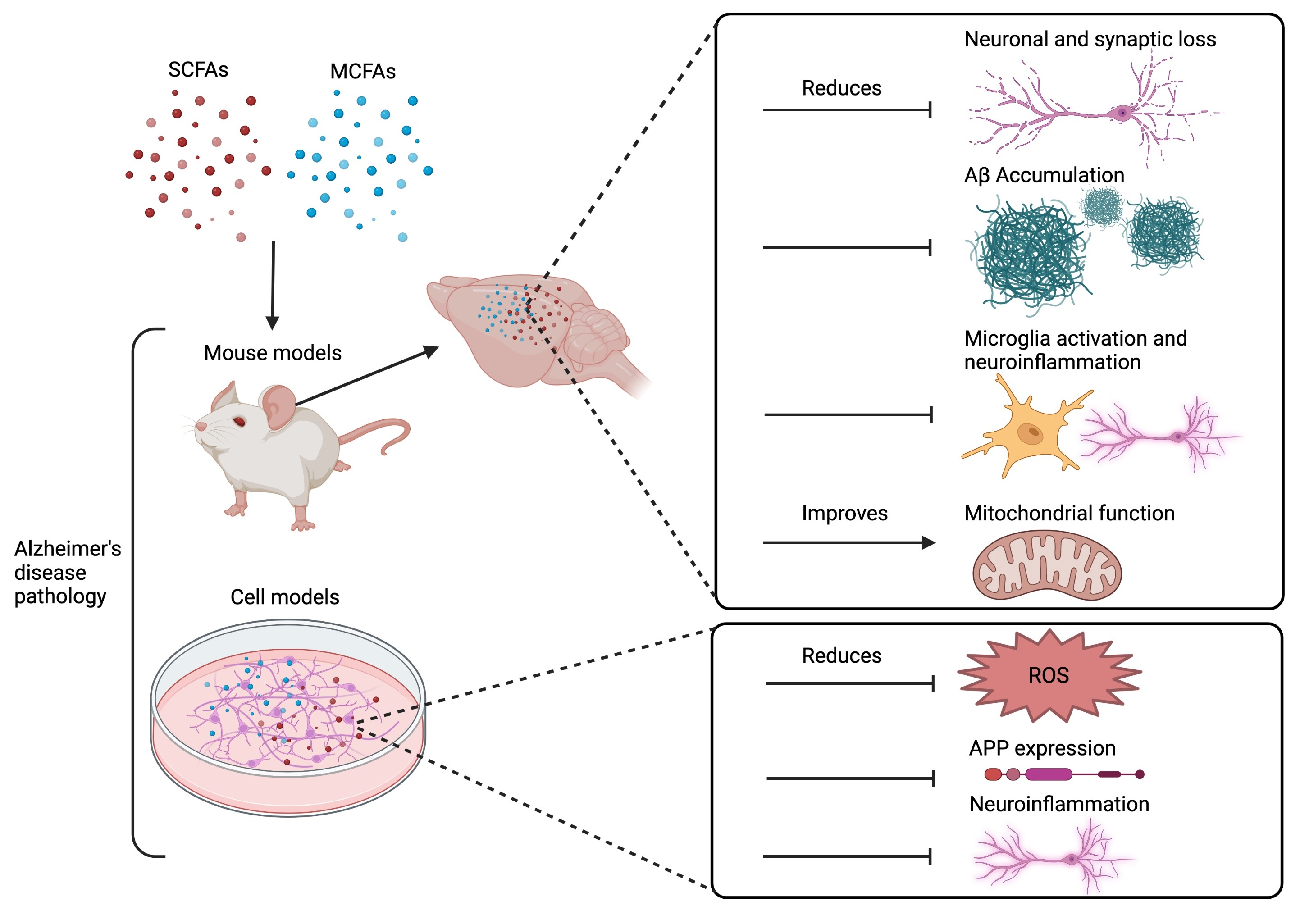
2.1. Animal Models of AD Pathology
2.2. Cellular Models of AD Pathology
3. SCFAs and Their Metabolites
Roles of SCFAs in AD
4. MCFAs and Their Metabolites
MCT-Based Metabolic Interventions for Energy Rescue in AD
5. Effect of SCFAs, MCFAs, and Their Metabolites on AD Mouse Models
5.1. Aβ Accumulation
5.2. Neuronal and Synaptic Loss/Function
5.3. Neuroinflammation and Glia Activation
5.4. Mitochondrial Function
5.5. Cognitive Function

{kind=link}
{kind=link}
| Model | Mutations | Neuropathology | Type of FA | Treatment Regime | Observations | Ref. |
|---|---|---|---|---|---|---|
| 5xFAD | APP: Swedish (K670N/M671L) + Florida (I716C) + London (V717I) PSEN1: M146L + L286V | Aβ deposition and plaques at 2 months of age; astrogliosis; neuronal and synaptic loss | MCFA | MCT triheptanoin at 3.5 months old for 8 months | Restored brain ATP; preserved mitochondrial function; protection against synaptic loss in hippocampus and entorhinal cortex; no changes in amyloid depositions | [52,108] |
| SCFA | Prebiotic mannan oligosaccharide at 6 months old for 8 weeks to increase SCFA production | Improved cognitive function and spatial memory; balanced brain redox status and suppressed neuroinflammatory responses; reduced the reduced Aβ in cortex, hippocampus, and amygdala | [52,104] | |||
| SCFA | Sodium butyrate (salt of SCFA) IP injection at 8 weeks old for 2 weeks | Reduced neuroinflammation; improved synaptic plasticity; reduced Aβ | [52,105] | |||
| 3xTgAD | APP: Swedish (K670M/N671L) PSEN1: M146V, MAPT: P301L | Aβ deposition at 3 months; plaques at 9 months; NFTs at 12 months; astrogliosis | KB | Ester of β -hydroxybutyrate and 1,3 butane diol at 8 months old for 8 months | Increased BHB levels in hippocampus and cortex; more reduced mitochondrial redox potential; lower level of oxidised lipids/proteins in hippocampus | [53,110] |
| APPswe/PS1dE9 | APP: Swedish (K670M/N671L), PSEN1: exon 9 deletion | Aβ deposition and gliosis at 6–9 months; neuronal and synaptic loss | MCFA | Ketogenic diet supplemented with MCT triheptanoin at 3 months old for 3 months | Reduced astroglia response in vicinity of Aβ plaques; reduced expression of the pro-inflammatory cytokines in astrocytes; transcriptional up-regulation of the ROS detoxifying mechanisms Sirt1 and Pparg; no changes in amyloid deposition | [50,109] |
| SCFA | Sodium acetate (salt of SCFA) oral gavage once daily for 4 weeks | Decreased cognitive impairment; anti-neuroinflammatory effects | [50,65] | |||
| APP/PS1 | APP: Swedish (K670M/N671L), PSEN1: L166P | Aβ deposition and astrogliosis at 1.5 months; synaptic loss | KB | BHB and pyruvate at age 12–13 weeks for 5 weeks | Increased brain NADPH; Reduced neuronal hyperexcitability | [29,51] |
| SCFA | Sodium butyrate (salt of SCFA) IP injection at 15 months old for 6 weeks | Improved associative memory; increased hippocampal histone acetylation and expression of genes linked to associative learning; no changes in amyloid deposition | [51,112] | |||
| SCFA | SCFA drinking water containing sodium propionate, sodium butyrate, and sodium acetate given to germ-free mice from 4 weeks and specific pathogen-free mice at 8 weeks until 12 weeks old. | Increased Aβ deposition and plaque formation; increased microglia activation | [51,107] | |||
| PS/APP | APP: Swedish (K670N/M671L), PSEN1: M146L | Aβ deposition at 3 months; Aβ plaques at 6 months; gliosis | MCFA | Ketogenic diet containing MCT oil at 5 months old for 3 months | Increased locomotor activity; improved motor function; cognition not improved; no changes in amyloid deposition | [49] |
| SIRT3+/−APP PS1 | APP: Swedish (K670M/N671L) PSEN1: exon 9 deletion, SIRT3: Heterozygous Knockout | Aβ deposition; degeneration of GABAergic neurons; seizure-related death before 5 months | KB | Ketogenic diet containing BHB at 4 months old for 2 weeks | Increased SIRT 3 expression; reduced loss of GABAergic neurons; prevented seizure related death | [54] |
| J2 | APP: Swedish (K670M/N671L) + Indiana (V717F) | Aβ deposition and plaques at 5–7 months; gliosis; neuronal and synaptic loss | KB | BHB daily injection at 4 months old for 2 months | Reduced intracellular Aβ levels; rescued mitochondrial complex I activity; reduced oxidative stress; improved synaptic plasticity and cognition | [24,46] |
| APP V717I | APP: London (V717I) | Increased levels of soluble Aβ at 3 months; plaques at 10 months | KD | Ketogenic diet at 3 months old for 43 days | Reduced Aβ levels; no change in behaviour or cognition | [45,113] |
| Tg2576 | APP: Swedish (K670M/N671L) | Aβ plaques at 11–13 months; synaptic loss at 4 months | SCFA | Phenylbutyrate (phenolic SCFA) injection at 6, 12 and 16 months old for 5 weeks | Increased clearance of intraneuronal Aβ; restored dendritic spine density in hippocampus; reduced ER stress | [44,106] |
| APP NL-G-F KI | APP homozygous knock-in: Swedish (K670M/N671L) + Iberian (I716F) + Arctic (E693G) | Aβ plaques at 2 months; gliosis | SCFA | probiotic VSL#3 supplement at 6–8 months old for 8 weeks | Increase in serum SCFAs acetate, butyrate, lactate, Propionate, and isobutyrate; increase in brain lactate and acetate; increased hippocampal c-Fos staining linked to increased neuronal activity | [55,68] |
| Tg4510 | MAPT: P301L | NFTs by 4 months; neuronal loss, and brain atrophy at 6 months | MCFA | Ketogenic diet containing MCT oil at 5 months old for 3 months | Increased locomotor activity; improved motor function; cognition not improved; no changes in amyloid deposition | [47,114] |
6. Effect of SCFAs and MCFAs on Cellular Models of Neurodegeneration and AD
6.1. Human Neuroblastoma
6.2. Mouse Neuroblastoma
6.3. hiPSC Astrocytes and Neurons
| Model | Type of FA | Treatment Regime | Observations | References |
|---|---|---|---|---|
| Human SH-SY5Y | MCFA | Decanoic acid | Reduced oxidative stress; decrease in H2O2 induced cell death independent of BHB levels | [115] |
| MCFA | Octanoic acid and decanoic acid | Higher rate of β-oxidation for C8 compared to C10; greater dependence of C10 on CPT1 | [116] | |
| Human SH-SY5Y + Aβ | SCFA | Sodium propionate (salt of SCFA) | Reduced inflammation; protected against cell damage from Aβ | [118] |
| Neuro2a cells | SCFA | Sodium butyrate (salt of SCFA) | Reduced oxidative stress; reduced expression of APP | [119] |
| hiPSC astrocytes | MCFA | MCFAs octanoic acid and decanoic acid | Greater extracellular concentrations and faster secretion rates of BHB and AcAc with C8 than C10 | [76] |
| MCFA | MCFAs octanoic acid and decanoic acid | Reduction in mitochondrial electrical potential; reduction in levels of NADPH; C10 increased glycolysis; C8 increased rate of astrocyte ketogenesis | [97] | |
| hiPSC neurons | MCFA | MCFAs octanoic acid and decanoic acid | No significant change in metabolic function | [97] |
| hiPSC astrocytes (from late-onset AD patients) | KB | BHB | No significant change in metabolic function | [122] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barnard, N.D.; Bush, A.I.; Ceccarelli, A.; Cooper, J.; de Jager, C.A.; Erickson, K.I.; Fraser, G.; Kesler, S.; Levin, S.M.; Lucey, B.; et al. Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiol. Aging 2014, 35 (Suppl. 2), S74–S78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Estes, R.E.; Lin, B.; Khera, A.; Davis, M.Y. Lipid Metabolism Influence on Neurodegenerative Disease Progression: Is the Vehicle as Important as the Cargo? Front. Mol. Neurosci. 2021, 14, 788695. [Google Scholar] [CrossRef] [PubMed]
- Deary, I.J.; Whiteman, M.C.; Pattie, A.; Starr, J.M.; Hayward, C.; Wright, A.F.; Carothers, A.; Whalley, L.J. Cognitive change and the APOE epsilon 4 allele. Nature 2002, 418, 932. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Meng, L.; Shen, L. Multiple roles of short-chain fatty acids in Alzheimer disease. Nutrition 2022, 93, 111499. [Google Scholar] [CrossRef]
- Marizzoni, M.; Cattaneo, A.; Mirabelli, P.; Festari, C.; Lopizzo, N.; Nicolosi, V.; Mombelli, E.; Mazzelli, M.; Luongo, D.; Naviglio, D.; et al. Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 78, 683–697. [Google Scholar] [CrossRef]
- Itzcovich, T.; Chrem-Méndez, P.; Vázquez, S.; Barbieri-Kennedy, M.; Niikado, M.; Martinetto, H.; Allegri, R.; Sevlever, G.; Surace, E.I. A novel mutation in PSEN1 (p.T119I) in an Argentine family with early- and late-onset Alzheimer’s disease. Neurobiol. Aging 2020, 85, e155–e159. [Google Scholar] [CrossRef]
- Lista, S.; O’Bryant, S.E.; Blennow, K.; Dubois, B.; Hugon, J.; Zetterberg, H.; Hampel, H. Biomarkers in Sporadic and Familial Alzheimer’s Disease. J. Alzheimers Dis. 2015, 47, 291–317. [Google Scholar] [CrossRef]
- D’Argenio, V.; Sarnataro, D. New Insights into the Molecular Bases of Familial Alzheimer’s Disease. J. Pers. Med. 2020, 10, 26. [Google Scholar] [CrossRef]
- Chen, G.-f.; Xu, T.-h.; Yan, Y.; Zhou, Y.-r.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Liu, C.-C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.-W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032.e1023. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Nuriel, T.; Peng, K.Y.; Ashok, A.; Dillman, A.A.; Figueroa, H.Y.; Apuzzo, J.; Ambat, J.; Levy, E.; Cookson, M.R.; Mathews, P.M.; et al. The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front. Neurosci. 2017, 11, 702. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Nikolakopoulou, A.M.; Huuskonen, M.T.; Sagare, A.P.; Lawson, E.J.; Lazic, D.; Rege, S.V.; Grond, A.; Zuniga, E.; Barnes, S.R.; et al. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat. Aging 2021, 1, 506–520. [Google Scholar] [CrossRef]
- Li, X.; Bao, X.; Wang, R. Experimental models of Alzheimer’s disease for deciphering the pathogenesis and therapeutic screening (Review). Int. J. Mol. Med. 2016, 37, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Frontzkowski, L.; Ewers, M.; Brendel, M.; Biel, D.; Ossenkoppele, R.; Hager, P.; Steward, A.; Dewenter, A.; Römer, S.; Rubinski, A.; et al. Earlier Alzheimer’s disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading. Nat. Commun. 2022, 13, 4899. [Google Scholar] [CrossRef]
- Rodriguez-Callejas, J.D.; Fuchs, E.; Perez-Cruz, C. Evidence of Tau Hyperphosphorylation and Dystrophic Microglia in the Common Marmoset. Front. Aging Neurosci. 2016, 8, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasanna, P.; Rathee, S.; Rahul, V.; Mandal, D.; Chandra Goud, M.S.; Yadav, P.; Hawthorne, S.; Sharma, A.; Gupta, P.K.; Ojha, S.; et al. Microfluidic Platforms to Unravel Mysteries of Alzheimer’s Disease: How Far Have We Come? Life 2021, 11, 1022. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Qi, G.; Brinton, R.D.; Yin, F. Mitochondria-Targeted Therapeutics for Alzheimer’s Disease: The Good, the Bad, the Potential. Antioxid. Redox Signal. 2021, 34, 611–630. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Cunnane, S.C.; Courchesne-Loyer, A.; Vandenberghe, C.; St-Pierre, V.; Fortier, M.; Hennebelle, M.; Croteau, E.; Bocti, C.; Fulop, T.; Castellano, C.-A. Can Ketones Help Rescue Brain Fuel Supply in Later Life? Implications for Cognitive Health during Aging and the Treatment of Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Croteau, E.; Castellano, C.A.; Fortier, M.; Bocti, C.; Fulop, T.; Paquet, N.; Cunnane, S.C. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 2018, 107, 18–26. [Google Scholar] [CrossRef]
- Zilberter, M.; Ivanov, A.; Ziyatdinova, S.; Mukhtarov, M.; Malkov, A.; Alpár, A.; Tortoriello, G.; Botting, C.H.; Fülöp, L.; Osypov, A.A.; et al. Dietary energy substrates reverse early neuronal hyperactivity in a mouse model of Alzheimer’s disease. J. Neurochem. 2013, 125, 157–171. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Nissen, J.D.; Waagepetersen, H.S. Improved cerebral energetics and ketone body metabolism in db/db mice. J. Cereb. Blood Flow Metab. 2017, 37, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Koczwara, J.B.; Gallelli, C.A.; Vergara, D.; Micioni Di Bonaventura, M.V.; Gaetani, S.; Giudetti, A.M. Fats for thoughts: An update on brain fatty acid metabolism. Int. J. Biochem. Cell Biol. 2017, 84, 40–45. [Google Scholar] [CrossRef] [PubMed]
- King, A. The search for better animal models of Alzheimer’s disease. Nature 2018, 559, S13–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Sanchez-Varo, R.; Mejias-Ortega, M.; Fernandez-Valenzuela, J.J.; Nuñez-Diaz, C.; Caceres-Palomo, L.; Vegas-Gomez, L.; Sanchez-Mejias, E.; Trujillo-Estrada, L.; Garcia-Leon, J.A.; Moreno-Gonzalez, I.; et al. Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis. Int. J. Mol. Sci. 2022, 23, 5404. [Google Scholar] [CrossRef]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Mckean, N.E.; Handley, R.R.; Snell, R.G. A Review of the Current Mammalian Models of Alzheimer’s Disease and Challenges That Need to Be Overcome. Int. J. Mol. Sci. 2021, 22, 13168. [Google Scholar] [CrossRef]
- Vitek, M.P.; Araujo, J.A.; Fossel, M.; Greenberg, B.D.; Howell, G.R.; Rizzo, S.J.S.; Seyfried, N.T.; Tenner, A.J.; Territo, P.R.; Windisch, M.; et al. Translational animal models for Alzheimer’s disease: An Alzheimer’s Association Business Consortium Think Tank. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12114. [Google Scholar] [CrossRef]
- Aigner, B.; Renner, S.; Kessler, B.; Klymiuk, N.; Kurome, M.; Wünsch, A.; Wolf, E. Transgenic pigs as models for translational biomedical research. J. Mol. Med. (Berl) 2010, 88, 653–664. [Google Scholar] [CrossRef]
- Han, F.Y.; Conboy-Schmidt, L.; Rybachuk, G.; Volk, H.A.; Zanghi, B.; Pan, Y.; Borges, K. Dietary medium chain triglycerides for management of epilepsy: New data from human, dog, and rodent studies. Epilepsia 2021, 62, 1790–1806. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Nagykery, N.; Wu, C.K. Amyloid-beta deposits in the cerebral cortex of the aged common marmoset (Callithrix jacchus): Incidence and chemical composition. Acta Neuropathol. 2002, 103, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, A beta elevation, and amyloid plaques in transgenic mice. Sci. (Wash.) 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. (Lond) 2005, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic Diet Improves Motor Performance but Not Cognition in Two Mouse Models of Alzheimer’s Pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [Green Version]
- Gamache, J.; Benzow, K.; Forster, C.; Kemper, L.; Hlynialuk, C.; Furrow, E.; Ashe, K.H.; Koob, M.D. Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat. Commun. 2019, 10, 2479. [Google Scholar] [CrossRef] [Green Version]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 2003, 13, 159–170. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Wang, J.; Ghena, N.; Zhao, Q.; Perone, I.; King, T.M.; Veech, R.L.; Gorospe, M.; Wan, R.; Mattson, M.P. SIRT3 Haploinsufficiency Aggravates Loss of GABAergic Interneurons and Neuronal Network Hyperexcitability in an Alzheimer’s Disease Model. J. Neurosci. 2020, 40, 694. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Shi, Y.; Kirwan, P.; Smith, J.; MacLean, G.; Orkin, S.H.; Livesey, F.J. A Human Stem Cell Model of Early Alzheimer’s Disease Pathology in Down Syndrome. Sci. Transl. Med. 2012, 4, 124ra129. [Google Scholar] [CrossRef] [Green Version]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Velázquez, O.C.; Seto, R.W.; Rombeau, J.L. The scientific rationale and clinical application of short-chain fatty acids and medium-chain triacylglycerols. Proc. Nutr. Soc. 1996, 55, 49–78. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Gibson, D.L.; Klegeris, A. Unhealthy gut, unhealthy brain: The role of the intestinal microbiota in neurodegenerative diseases. Neurochem. Int. 2018, 120, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Paley, E.L.; Merkulova-Rainon, T.; Faynboym, A.; Shestopalov, V.I.; Aksenoff, I. Geographical Distribution and Diversity of Gut Microbial NADH:Ubiquinone Oxidoreductase Sequence Associated with Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 61, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zheng, S.-J.; Cai, W.-J.; Yu, L.; Yuan, B.-F.; Feng, Y.-Q. Stable isotope labeling combined with liquid chromatography-tandem mass spectrometry for comprehensive analysis of short-chain fatty acids. Anal. Chim. Acta 2019, 1070, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Gong, T.; Chen, W.; Mao, S.; Kong, Y.; Yu, J.; Sun, J. Anti-neuroinflammatory Effect of Short-Chain Fatty Acid Acetate against Alzheimer’s Disease via Upregulating GPR41 and Inhibiting ERK/JNK/NF-κB. J. Agric. Food Chem. 2020, 68, 7152–7161. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Gates, E.J.; Ranger, A.L.; Klegeris, A. Short-chain fatty acids (SCFAs) alone or in combination regulate select immune functions of microglia-like cells. Mol. Cell. Neurosci. 2020, 105, 103493. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Kaur, H.; Golovko, S.; Golovko, M.Y.; Singh, S.; Darland, D.C.; Combs, C.K. Effects of Probiotic Supplementation on Short Chain Fatty Acids in the App NL-G-F Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 76, 1083–1102. [Google Scholar] [CrossRef]
- Dienel, G.A.; Liu, K.; Cruz, N.F. Local uptake of (14)C-labeled acetate and butyrate in rat brain in vivo during spreading cortical depression. J. Neurosci. Res. 2001, 66, 812–820. [Google Scholar] [CrossRef]
- Huang, L.; Gao, L.; Chen, C. Role of Medium-Chain Fatty Acids in Healthy Metabolism: A Clinical Perspective. Trends Endocrinol. Metab. 2021, 32, 351–366. [Google Scholar] [CrossRef]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Schönfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.A.; Johnson, R.A.; Corkey, B.; Kamp, F. Fatty acid transport: The diffusion mechanism in model and biological membranes. J. Mol. Neurosci. 2001, 16, 99–108, discussion 151–107. [Google Scholar] [CrossRef]
- Spector, R. Fatty acid transport through the blood-brain barrier. J. Neurochem. 1988, 50, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef]
- Sonnay, S.; Chakrabarti, A.; Thevenet, J.; Wiederkehr, A.; Christinat, N.; Masoodi, M. Differential Metabolism of Medium-Chain Fatty Acids in Differentiated Human-Induced Pluripotent Stem Cell-Derived Astrocytes. Front. Physiol. 2019, 10, 657. [Google Scholar] [CrossRef]
- Wlaź, P.; Socała, K.; Nieoczym, D.; Żarnowski, T.; Żarnowska, I.; Czuczwar, S.J.; Gasior, M. Acute anticonvulsant effects of capric acid in seizure tests in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 57, 110–116. [Google Scholar] [CrossRef]
- Tan, K.N.; Carrasco-Pozo, C.; McDonald, T.S.; Puchowicz, M.; Borges, K. Tridecanoin is anticonvulsant, antioxidant, and improves mitochondrial function. J. Cereb. Blood Flow Metab. 2017, 37, 2035–2048. [Google Scholar] [CrossRef]
- Andersen, J.V.; Westi, E.W.; Jakobsen, E.; Urruticoechea, N.; Borges, K.; Aldana, B.I. Astrocyte metabolism of the medium-chain fatty acids octanoic acid and decanoic acid promotes GABA synthesis in neurons via elevated glutamine supply. Mol. Brain 2021, 14, 132. [Google Scholar] [CrossRef]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef]
- Nehlig, A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Wheless, J.W. History of the ketogenic diet. Epilepsia 2008, 49, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.; Tyrlikova, I.; Mathews, G.C. Dietary treatment in adults with refractory epilepsy: A review. Neurology 2014, 83, 1978–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharm. 2006, 17, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, E.; Vacy, K.; Boon, W.C. Fatty acids and their therapeutic potential in neurological disorders. Neurochem. Int. 2016, 95, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 1971, 21, 1097. [Google Scholar] [CrossRef]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Borges, K.; Kaul, N.; Germaine, J.; Kwan, P.; O’Brien, T.J. Randomized trial of add-on triheptanoin vs medium chain triglycerides in adults with refractory epilepsy. Epilepsia Open 2019, 4, 153–163. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croteau, E.; Castellano, C.A.; Richard, M.A.; Fortier, M.; Nugent, S.; Lepage, M.; Duchesne, S.; Whittingstall, K.; Turcotte, É.E.; Bocti, C.; et al. Ketogenic Medium Chain Triglycerides Increase Brain Energy Metabolism in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avgerinos, K.I.; Egan, J.M.; Mattson, M.P.; Kapogiannis, D. Medium Chain Triglycerides induce mild ketosis and may improve cognition in Alzheimer’s disease. A systematic review and meta-analysis of human studies. Ageing Res. Rev. 2020, 58, 101001. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Aldana, B.I.; Nissen, J.D.; Tanila, H.; Waagepetersen, H.S. Alterations in Cerebral Cortical Glucose and Glutamine Metabolism Precedes Amyloid Plaques in the APPswe/PSEN1dE9 Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2017, 42, 1589–1598. [Google Scholar] [CrossRef]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. Faseb. J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [Green Version]
- Yonezawa, T.; Kurata, R.; Yoshida, K.; Murayama, A.M.; Cui, X.; Hasegawa, A. Free Fatty Acids-Sensing G Protein-Coupled Receptors in Drug Targeting and Therapeutics. Curr. Med. Chem. 2013, 20, 3855–3871. [Google Scholar] [CrossRef]
- Ichimura, A.; Hasegawa, S.; Kasubuchi, M.; Kimura, I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front. Pharmacol. 2014, 5, 236. [Google Scholar] [CrossRef]
- Kristinsson, H.; Bergsten, P.; Sargsyan, E. Free fatty acid receptor 1 (FFAR1/GPR40) signaling affects insulin secretion by enhancing mitochondrial respiration during palmitate exposure. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2015, 1853, 3248–3257. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.Z.; Zhuang, X.; He, L. GPR40 receptor activation leads to CREB phosphorylation and improves cognitive performance in an Alzheimer’s disease mouse model. Neurobiol. Learn. Mem. 2016, 131, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Xi, Y.; Wang, Q.; Liu, J.; Li, P.; Meng, X.; Liu, K.; Chen, W.; Liu, X.; Liu, Z. Mannan oligosaccharide attenuates cognitive and behavioral disorders in the 5xFAD Alzheimer’s disease mouse model via regulating the gut microbiota-brain axis. Brain Behav. Immun. 2021, 95, 330–343. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, K.; Li, X.; Xu, L.; Yang, Z. Sodium butyrate ameliorates the impairment of synaptic plasticity by inhibiting the neuroinflammation in 5XFAD mice. Chem.-Biol. Interact. 2021, 341, 109452. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Marco, S.; Pérez-Otaño, I.; García-Osta, A. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 2012, 22, 1040–1050. [Google Scholar] [CrossRef]
- Colombo, A.V.; Sadler, R.K.; Llovera, G.; Singh, V.; Roth, S.; Heindl, S.; Sebastian Monasor, L.; Verhoeven, A.; Peters, F.; Parhizkar, S.; et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 2021, 10, e59826. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, L.; Tandon, N.; Sun, H.; Tian, J.; Du, H.; Pascual, J.M.; Guo, L. Triheptanoin Mitigates Brain ATP Depletion and Mitochondrial Dysfunction in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2020, 78, 425–437. [Google Scholar] [CrossRef]
- Aso, E.; Semakova, J.; Joda, L.; Semak, V.; Halbaut, L.; Calpena, A.; Escolano, C.; Perales, J.C.; Ferrer, I. Triheptanoin supplementation to ketogenic diet curbs cognitive impairment in APP/PS1 mice used as a model of familial Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 290–297. [Google Scholar] [CrossRef]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindarajan, N.; Agis-Balboa, R.C.; Walter, J.; Sananbenesi, F.; Fischer, A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J. Alzheimers Dis. 2011, 26, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Moechars, D.; Dewachter, I.; Lorent, K.; Reversé, D.; Baekelandt, V.; Naidu, A.; Tesseur, I.; Spittaels, K.; Haute, C.V.; Checler, F.; et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999, 274, 6483–6492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef] [Green Version]
- Mett, J.; Müller, U. The medium-chain fatty acid decanoic acid reduces oxidative stress levels in neuroblastoma cells. Sci. Rep. 2021, 11, 6135. [Google Scholar] [CrossRef]
- Khabbush, A.; Orford, M.; Tsai, Y.C.; Rutherford, T.; O’Donnell, M.; Eaton, S.; Heales, S.J.R. Neuronal decanoic acid oxidation is markedly lower than that of octanoic acid: A mechanistic insight into the medium-chain triglyceride ketogenic diet. Epilepsia 2017, 58, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Toleikis, A.; Trumbeckaite, S.; Liobikas, J.; Pauziene, N.; Kursvietiene, L.; Kopustinskiene, D.M. Fatty Acid Oxidation and Mitochondrial Morphology Changes as Key Modulators of the Affinity for ADP in Rat Heart Mitochondria. Cells 2020, 9, 340. [Google Scholar] [CrossRef] [Green Version]
- Filippone, A.; Lanza, M.; Campolo, M.; Casili, G.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Protective effect of sodium propionate in Aβ1-42 -induced neurotoxicity and spinal cord trauma. Neuropharmacology 2020, 166, 107977. [Google Scholar] [CrossRef]
- Sun, J.; Yuan, B.; Wu, Y.; Gong, Y.; Guo, W.; Fu, S.; Luan, Y.; Wang, W. Sodium Butyrate Protects N2a Cells against Aβ Toxicity In Vitro. Mediat. Inflamm 2020, 2020, 7605160. [Google Scholar] [CrossRef] [Green Version]
- Felizardo, R.J.F.; de Almeida, D.C.; Pereira, R.L.; Watanabe, I.K.M.; Doimo, N.T.S.; Ribeiro, W.R.; Cenedeze, M.A.; Hiyane, M.I.; Amano, M.T.; Braga, T.T.; et al. Gut microbial metabolite butyrate protects against proteinuric kidney disease through epigenetic- and GPR109a-mediated mechanisms. FASEB J. 2019, 33, 11894–11908. [Google Scholar] [CrossRef]
- Valdebenito, R.; Ruminot, I.; Garrido-Gerter, P.; Fernández-Moncada, I.; Forero-Quintero, L.; Alegría, K.; Becker, H.M.; Deitmer, J.W.; Barros, L.F. Targeting of astrocytic glucose metabolism by beta-hydroxybutyrate. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2016, 36, 1813–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, W.-I.; Bormann, M.K.; Shen, M.; Kim, D.; Forester, B.; Park, Y.; So, J.; Seo, H.; Sonntag, K.-C.; Cohen, B.M. Brain cells derived from Alzheimer’s disease patients have multiple specific innate abnormalities in energy metabolism. Mol. Psychiatry 2021, 26, 5702–5714. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ameen, A.O.; Freude, K.; Aldana, B.I. Fats, Friends or Foes: Investigating the Role of Short- and Medium-Chain Fatty Acids in Alzheimer’s Disease. Biomedicines 2022, 10, 2778. https://doi.org/10.3390/biomedicines10112778
Ameen AO, Freude K, Aldana BI. Fats, Friends or Foes: Investigating the Role of Short- and Medium-Chain Fatty Acids in Alzheimer’s Disease. Biomedicines. 2022; 10(11):2778. https://doi.org/10.3390/biomedicines10112778
Chicago/Turabian StyleAmeen, Aishat O., Kristine Freude, and Blanca I. Aldana. 2022. "Fats, Friends or Foes: Investigating the Role of Short- and Medium-Chain Fatty Acids in Alzheimer’s Disease" Biomedicines 10, no. 11: 2778. https://doi.org/10.3390/biomedicines10112778
APA StyleAmeen, A. O., Freude, K., & Aldana, B. I. (2022). Fats, Friends or Foes: Investigating the Role of Short- and Medium-Chain Fatty Acids in Alzheimer’s Disease. Biomedicines, 10(11), 2778. https://doi.org/10.3390/biomedicines10112778