
An Innovative PTD-IVT-mRNA Delivery Platform for CAR Immunotherapy of ErbB(+) Solid Tumor Neoplastic Cells
 ,
,  and
and
Abstract
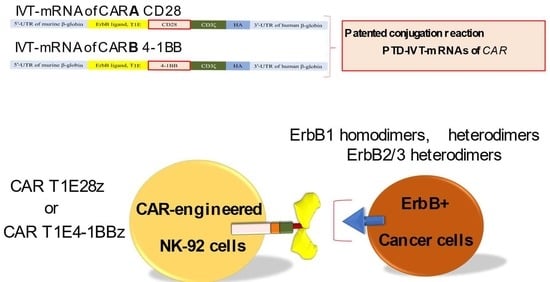
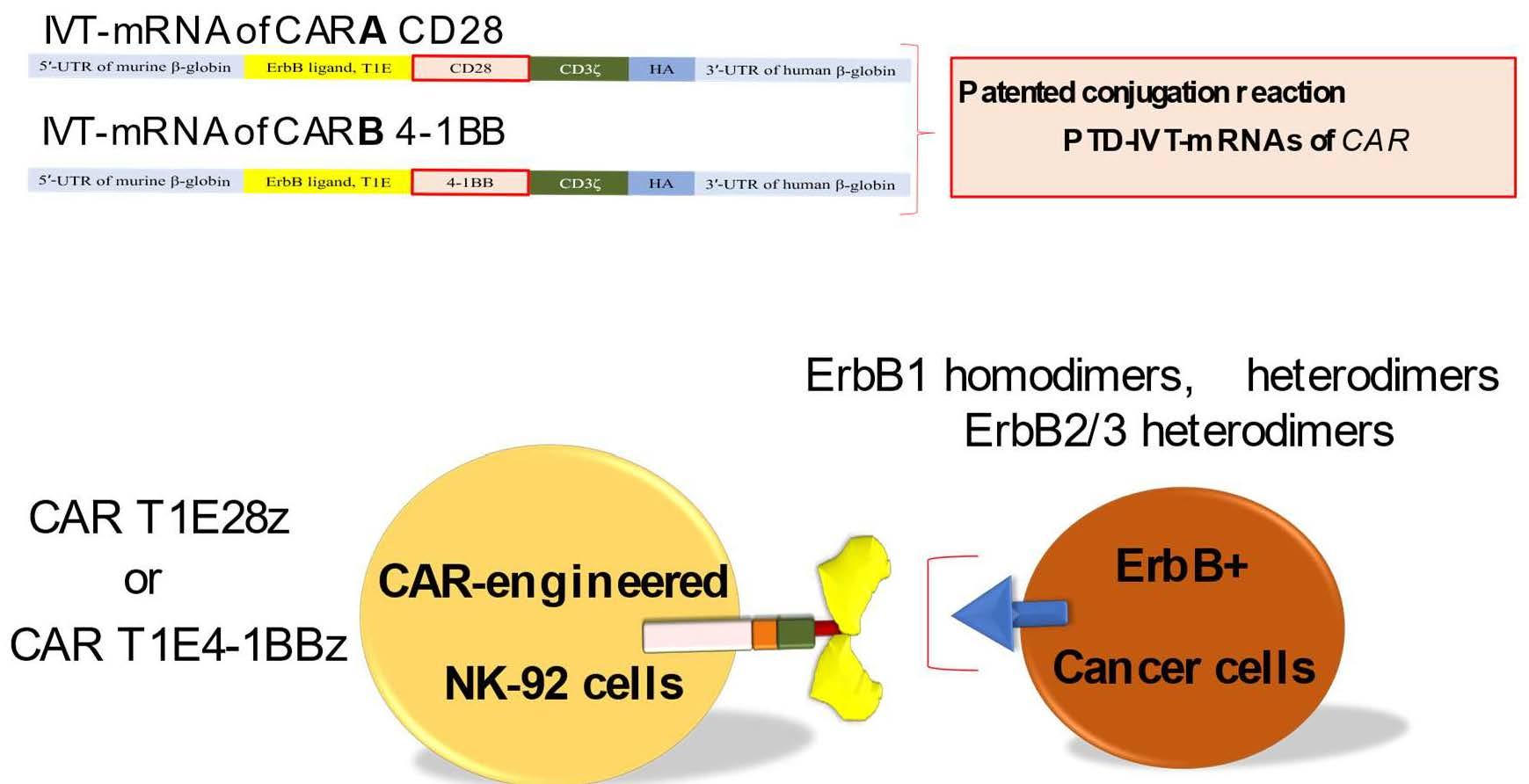
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Design, Construction and Cloning of CAR T1E28z and CAR T1E4-1BBz Sequences
2.3. Production of IVT mRNAs
2.4. Conjugation of the Selected PTD to the IVT-mRNAs of CARA and CARB
2.5. Transfection of NK-92 Cells (Effector Cells, Ε) with the PTD-IVT-mRNAs for CARA and CARB
2.6. RNA Isolation, Reverse Transcription (RT)-PCR and Quantitative (q)PCR
2.7. Preparation of Protein Cellular Lysates, Subcellular Fractionation and Western Blot Analysis
2.8. Assessment of Cell Growth and Viability of NK-92 Cells (Εffector Cells, Ε)
2.9. Co-Incubation Experiments of NK-92 Cells (Effector Cells, E) with HSC-3 or MCF-7 Cells (Target Cells, T)
2.10. Determination of Cell Death in the Co-Incubation Experiments
2.11. Statistical Analysis and Software
3. Results
3.1. Synthesis of the PTD-IVT-mRNAs of CARA and CARB
3.2. Stability Assays of the PTD-IVT-mRNAs of CARA and CARB
3.3. Assessment of the Intracellular Transduction and Expression of the IVT-mRNAs of CAR into the Respective CAR Receptors, Complemented by Their Subcellular Localization in K562 Cells, Employed as a Model Cell Line
3.4. Transduction of the PTD-IVT-mRNAs of CARA and CARB in NK-92 Cells Serving as the Effector Cells
3.5. Cytotoxic Potential of the PTD-IVT-mRNA-Engineered CAR-NK-92 Cells on Oral Squamous HSC-3 Cells
3.6. MCF-7 Breast Metastatic Adenocarcinoma Cells Expressed Significantly Lower ErbB2 Receptors but Were Targeted and Killed by the PTD-IVT-mRNA-Engineered CAR-NK-92 Cells to almost the Same Levels with HSC-3 OSCC Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Frey, N.V.; Grupp, S.A.; Maude, S.L. CAR T Cell Therapy in Acute Lymphoblastic Leukemia and Potential for Chronic Lymphocytic Leukemia. Curr. Treat. Options Oncol. 2016, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Huang, S.; Xiao, X.; Sun, Q.; Liang, X.; Chen, S.; Zhao, Z.; Huo, Z.; Tu, S.; Li, Y. Challenges and Clinical Strategies of CAR T-Cell Therapy for Acute Lymphoblastic Leukemia: Overview and Developments. Front. Immunol. 2020, 11, 569117. [Google Scholar] [CrossRef]
- Maakaron, J.E.; Hu, M.; El Jurdi, N. Chimeric antigen receptor T cell therapy for cancer: Clinical applications and practical considerations. BMJ 2022, 378, e068956. [Google Scholar] [CrossRef]
- Cao, B.; Liu, M.; Wang, L.; Liang, B.; Feng, Y.; Chen, X.; Shi, Y.; Zhang, J.; Ye, X.; Tian, Y.; et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 524, 96–102. [Google Scholar] [CrossRef]
- Munisvaradass, R.; Kumar, S.; Govindasamy, C.; Alnumair, K.S.; Mok, P.L. Human CD3+ T-Cells with the Anti-ERBB2 Chimeric Antigen Receptor Exhibit Efficient Targeting and Induce Apoptosis in ERBB2 Overexpressing Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1797. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Thaci, B.; Brown, C.E.; Binello, E.; Werbaneth, K.; Sampath, P.; Sengupta, S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014, 16, 1304–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.; Yang, Y.; McCloskey, J.E.; Zaman, M.; Vedvyas, Y.; Zhang, X.; Stefanova, D.; Gray, K.D.; Min, I.M.; Zarnegar, R.; et al. Chimeric Antigen Receptor T Cell Therapy Targeting ICAM-1 in Gastric Cancer. Mol. Ther. Oncolytics. 2020, 18, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. OncoImmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wang, X.; Li, W.; Yu, X.; Flores-Villanueva, P.; Xu-Monette, Z.Y.; Li, L.; Zhang, M.; Young, K.H.; Ma, X.; et al. Targeting PD-L1 in non-small cell lung cancer using CAR T cells. Oncogenesis 2020, 9, 72. [Google Scholar] [CrossRef]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.-F.; Bulsara, S.; et al. Glypican-3–Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. eBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fang, Y.; Chen, X.; Wang, Z.; Liang, X.; Zhang, T.; Liu, M.; Zhou, N.; Lv, J.; Tang, K.; et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020, 5, aax7969. [Google Scholar] [CrossRef]
- Rice, J.; Nagle, S.; Randall, J.; Hinson, H.E. Chimeric Antigen Receptor T Cell-Related Neurotoxicity: Mechanisms, Clinical Presentation, and Approach to Treatment. Curr. Treat Options Neurol. 2019, 21, 40. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Kenderian, S.S. Next-Generation Chimeric Antigen Receptor T-Cell Therapy: Going off the Shelf. BioDrugs 2017, 31, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.; Ho, N.; Buchholz, C.J. Precision medicine: In vivo CAR therapy as a showcase for receptor-targeted vector platforms. Mol. Ther. 2022, 30, 2401–2415. [Google Scholar] [CrossRef]
- Sun, Z.; Li, R.; Shen, Y.; Tan, S.; Ding, N.; Xu, R.; Wang, X.; Wei, J.; Liu, B.; Meng, F. In situ antigen modification-based target-redirected universal chimeric antigen receptor T (TRUE CAR-T) cell therapy in solid tumors. J. Hematol. Oncol. 2022, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Biederstädt, A.; Rezvani, K. Engineering the next generation of CAR-NK immunotherapies. Int. J. Hematol. 2021, 114, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Khawar, M.B.; Sun, H. CAR-NK Cells: From Natural Basis to Design for Kill. Front. Immunol. 2021, 12, 707542. [Google Scholar] [CrossRef]
- Sabbah, M.; Jondreville, L.; Lacan, C.; Norol, F.; Vieillard, V.; Roos-Weil, D.; Nguyen, S. CAR-NK Cells: A Chimeric Hope or a Promising Therapy? Cancers 2022, 14, 3839. [Google Scholar] [CrossRef]
- Suerth, J.D.; Schambach, A.; Baum, C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr. Opin. Immunol. 2012, 24, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Miliotou, A.N.; Papadopoulou, L.C. In Vitro-Transcribed(IVT)-mRNA CAR Therapy Development. Methods Mol. Biol. 2020, 2086, 87–117. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, O.V.; Koteliansky, V.E.; Zatsepin, T.S. mRNA-Based Therapeutics—Advances and Perspectives. Biochemistry 2016, 81, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Stepinski, J.; Jemielity, J.; Zuberek, J.; Stolarski, R.; Rhoads, R.E.; Darzynkiewicz, E. Synthesis of Anti-Reverse Cap Analogs (ARCAs) and their Applications in mRNA Translation and Stability. Methods Enzymol. 2007, 431, 203–227. [Google Scholar]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Kariko, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.; Sullivan, T.D. Half-lives of beta and gamma globin messenger RNAs and of protein synthetic capacity in cultured human reticulocytes. Blood 1985, 66, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Broderick, K.E.; Humeau, L.M. Electroporation-enhanced delivery of nucleic acid vaccines. Expert Rev. Vaccines 2015, 14, 195–204. [Google Scholar] [CrossRef]
- Okur, N.U.; Siafaka, P.I.; Gokce, E.H. Challenges in Oral Drug Delivery and Applications of Lipid Nanoparticles as Potent Oral Drug Carriers for Managing Cardiovascular Risk Factors. Curr. Pharm. Biotechnol. 2021, 22, 892–905. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef]
- Brasseur, R.; Divita, G. Happy birthday cell penetrating peptides: Already 20 years. Biochim. Biophys. Acta 2010, 1798, 2177–2181. [Google Scholar] [CrossRef] [PubMed]
- Miliotou, A.N.; Pappas, I.S.; Spyroulias, G.; Vlachaki, E.; Tsiftsoglou, A.S.; Vizirianakis, I.S.; Papadopoulou, L.C. Development of a novel PTD-mediated IVT-mRNA delivery platform for potential protein replacement therapy of metabolic/genetic disorders. Mol. Ther. Nucleic Acids 2021, 26, 694–710. [Google Scholar] [CrossRef] [PubMed]
- Miliotou, A.N.; Papagiannopoulou, D.; Vlachaki, E.; Samiotaki, M.; Laspa, D.; Theodoridou, S.; Tsiftsoglou, A.S. PTD-mediated delivery of alpha-globin chain into Kappa-562 erythroleukemia cells and alpha-thalassemic (HBH) patients’ RBCs ex vivo in the frame of Protein Replacement Therapy. J. Biol. Res. 2021, 28, 16. [Google Scholar]
- Kaiafas, G.C.; Papagiannopoulou, D.; Miliotou, A.N.; Tsingotjidou, A.S.; Chalkidou, P.C.; Tsika, A.C.; Spyroulias, G.A.; Tsiftsoglou, A.S.; Papadopoulou, L.C. In vivo biodistribution study of TAT-L-Sco2 fusion protein, developed as protein therapeutic for mitochondrial disorders attributed to SCO2 mutations. Mol. Genet Metab Rep. 2020, 25, 100683. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Ingendoh-Tsakmakidis, A.; Mpoutoureli, C.N.; Tzikalou, L.D.; Spyridou, E.D.; Gavriilidis, G.I.; Kaiafas, G.C.; Ntaska, A.T.; Vlachaki, E.; Panayotou, G.; et al. Production and Transduction of a Human Recombinant beta-Globin Chain into Proerythroid K-562 Cells To Replace Missing Endogenous beta-Globin. Mol. Pharm. 2018, 15, 5665–5677. [Google Scholar] [CrossRef] [PubMed]
- Foltopoulou, P.F.; Tsiftsoglou, A.S.; Bonovolias, I.D.; Ingendoh, A.T.; Papadopoulou, L.C. Intracellular delivery of full length recombinant human mitochondrial L-Sco2 protein into the mitochondria of permanent cell lines and SCO2 deficient patient’s primary cells. Biochim. Biophys. Acta 2010, 1802, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; DE Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK Cells: A New Non-viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef] [Green Version]
- Lozzio, B.B.; Lozzio, C.B. Properties of the K562 cell line derived from a patient with chronic myeloid leukemia. Int. J. Cancer 1977, 19, 136. [Google Scholar] [CrossRef]
- Georgiou-Siafis, S.K.; Samiotaki, M.K.; Demopoulos, V.J.; Panayotou, G.; Tsiftsoglou, A.S. Glutathione-Hemin/Hematin Adduct Formation to Disintegrate Cytotoxic Oxidant Hemin/Hematin in Human K562 Cells and Red Blood Cells’ Hemolysates: Impact of Glutathione on the Hemolytic Disorders and Homeostasis. Antioxidants 2022, 11, 1959. [Google Scholar] [CrossRef]
- Davies, D.M.; Foster, J.; Van Der Stegen, S.J.C.; Parente-Pereira, A.C.; Chiapero-Stanke, L.; Delinassios, G.J.; Burbridge, S.E.; Kao, V.; Liu, Z.; Bosshard-Carter, L.; et al. Flexible Targeting of ErbB Dimers That Drive Tumorigenesis by Using Genetically Engineered T Cells. Mol. Med. 2012, 18, 565–576. [Google Scholar] [CrossRef]
- Jemielity, J.; Fowler, T.; Zuberek, J.; Stepinski, J.; Lewdorowicz, M.; Niedzwiecka, A.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel “anti-reverse” cap analogs with superior translational properties. RNA 2003, 9, 1108–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Muinao, T.; Pal, M.; Boruah, H.P.D. Cytosolic and Transmembrane Protein Extraction Methods of Breast and Ovarian Cancer Cells: A Comparative Study. J. Biomol. Tech. 2018, 29, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Suski, J.M.; Lebiedzinska, M.; Wojtala, A.; Duszynski, J.; Giorgi, C.; Pinton, P.; Wieckowski, M.R. Isolation of plasma membrane–associated membranes from rat liver. Nat. Protoc. 2014, 9, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Lai, H.-C.; Chang, C.-J.; Yang, C.-H.; Hsu, Y.-J.; Chen, C.-C.; Lin, C.-S.; Tsai, Y.-H.; Huang, T.-T.; Ojcius, D.M.; Tsai, Y.-H.; et al. Activation of NK cell cytotoxicity by the natural compound 2,3-butanediol. J. Leukoc. Biol. 2012, 92, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Sarma, K.D.; Ray, D.; Antony, A. Improved sensitivity of trypan blue dye exclusion assay with Ni2+ or Co2+ salts. Cytotechnology 2000, 32, 93–95. [Google Scholar] [CrossRef]
- Komatsu, F.; Kajiwara, M. Relation of natural killer cell line NK-92-mediated cytolysis (NK-92-lysis) with the surface markers of major histocompatibility complex class I antigens, adhesion molecules, and Fas of target cells. Oncol. Res. 1998, 10, 483–489. [Google Scholar]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Minamino, Y.; Kakudo, K.; Nozaki, M. Resistance of oral squamous cell carcinoma cells to cetuximab is associated with EGFR insensitivity and enhanced stem cell-like potency. Oncol. Rep. 2014, 32, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C.-X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef] [PubMed]
- Whittington, M.D.; McQueen, R.B.; Campbell, J.D. Valuing Chimeric Antigen Receptor T-Cell Therapy: Current Evidence, Uncertainties, and Payment Implications. J. Clin. Oncol. 2020, 38, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Patel, U.; Ms, J.A.; Savani, B.N.; Oluwole, O.; Sengsayadeth, S.; Dholaria, B. CAR T cell therapy in solid tumors: A review of current clinical trials. eJHaem 2021, 3 (Suppl. S1), 24–31. [Google Scholar] [CrossRef] [PubMed]
- Larcombe-Young, D.; Papa, S.; Maher, J. PanErbB-targeted CAR T-cell immunotherapy of head and neck cancer. Expert Opin. Biol. Ther. 2020, 20, 965–970. [Google Scholar] [CrossRef]
- Futaki, S.; Nakase, I. Cell-Surface Interactions on Arginine-Rich Cell-Penetrating Peptides Allow for Multiplex Modes of Internalization. Accounts Chem. Res. 2017, 50, 2449–2456. [Google Scholar] [CrossRef]
- Bolhassani, A.; Jafarzade, B.S.; Mardani, G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides 2017, 87, 50–63. [Google Scholar] [CrossRef]
- Hu, J.W.; Liu, B.R.; Wu, C.-Y.; Lu, S.-W.; Lee, H.-J. Protein transport in human cells mediated by covalently and noncovalently conjugated arginine-rich intracellular delivery peptides. Peptides 2009, 30, 1669–1678. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Tsiftsoglou, A.S. The potential role of cell penetrating peptides in the intracellular delivery of proteins for therapy of erythroid related disorders. Pharmaceuticals 2013, 6, 32–53. [Google Scholar] [CrossRef] [Green Version]
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3’ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef] [Green Version]
- Fritz, S.E.; Haque, N.; Hogg, J.R. Highly efficient in vitro translation of authentic affinity-purified messenger ribonucleoprotein complexes. RNA 2018, 24, 982–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.R.; Kim, T.-D.; Choi, I. Understanding of molecular mechanisms in natural killer cell therapy. Exp. Mol. Med. 2015, 47, e141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Yang, P.; Ma, L. Prognostic and clinical implications of c-erbB-2 expression in patients with oral cancer: A meta-analysis. Medicine 2020, 99, e20575. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, Q.; Han, X.; Qin, S.; Zhao, W.; Li, A.; Wu, K. Development and clinical application of anti-HER2 monoclonal and bispecific antibodies for cancer treatment. Exp. Hematol. Oncol. 2017, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpela, S.P.; Hinz, T.K.; Oweida, A.; Kim, J.; Calhoun, J.; Ferris, R.; Nemenoff, R.A.; Karam, S.D.; Clambey, E.T.; Heasley, L.E. Role of epidermal growth factor receptor inhibitor-induced interferon pathway signaling in the head and neck squamous cell carcinoma therapeutic response. J. Transl. Med. 2021, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Nozaki, M.; Yasui, H.; Ohnishi, Y. Ligand-Independent EGFR Activation by Anchorage-Stimulated Src Promotes Cancer Cell Proliferation and Cetuximab Resistance via ErbB3 Phosphorylation. Cancers 2019, 11, 1552. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, Y.; Yasui, H.; Nozaki, M.; Nakajima, M. Molecularly-targeted therapy for the oral cancer stem cells. Jpn. Dent. Sci. Rev. 2018, 54, 88–103. [Google Scholar] [CrossRef]
- Zamora, A.E.; Grossenbacher, S.K.; Aguilar, E.G.; Murphy, W.J. Models to Study NK Cell Biology and Possible Clinical Application. Curr. Protoc. Immunol. 2015, 110, 14.37.1–14.37.14. [Google Scholar] [CrossRef] [Green Version]
- Masuda, H.; Alev, C.; Akimaru, H.; Ito, R.; Shizuno, T.; Kobori, M.; Horii, M.; Ishihara, T.; Isobe, K.; Isozaki, M.; et al. Methodological Development of a Clonogenic Assay to Determine Endothelial Progenitor Cell Potential. Circ. Res. 2011, 109, 20–37. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgiou-Siafis, S.K.; Miliotou, A.N.; Ntenti, C.; Pappas, I.S.; Papadopoulou, L.C. An Innovative PTD-IVT-mRNA Delivery Platform for CAR Immunotherapy of ErbB(+) Solid Tumor Neoplastic Cells. Biomedicines 2022, 10, 2885. https://doi.org/10.3390/biomedicines10112885
Georgiou-Siafis SK, Miliotou AN, Ntenti C, Pappas IS, Papadopoulou LC. An Innovative PTD-IVT-mRNA Delivery Platform for CAR Immunotherapy of ErbB(+) Solid Tumor Neoplastic Cells. Biomedicines. 2022; 10(11):2885. https://doi.org/10.3390/biomedicines10112885
Chicago/Turabian StyleGeorgiou-Siafis, Sofia K., Androulla N. Miliotou, Charikleia Ntenti, Ioannis S. Pappas, and Lefkothea C. Papadopoulou. 2022. "An Innovative PTD-IVT-mRNA Delivery Platform for CAR Immunotherapy of ErbB(+) Solid Tumor Neoplastic Cells" Biomedicines 10, no. 11: 2885. https://doi.org/10.3390/biomedicines10112885
APA StyleGeorgiou-Siafis, S. K., Miliotou, A. N., Ntenti, C., Pappas, I. S., & Papadopoulou, L. C. (2022). An Innovative PTD-IVT-mRNA Delivery Platform for CAR Immunotherapy of ErbB(+) Solid Tumor Neoplastic Cells. Biomedicines, 10(11), 2885. https://doi.org/10.3390/biomedicines10112885