
Unveiling Human Proteome Signatures of Heart Failure with Preserved Ejection Fraction
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Heart-Tissue Collection and Processing
2.2. Proteomics
2.2.1. Generation of the Spectral Reference Library
2.2.2. SWATH–MS Analysis and Targeted Data Extraction
2.2.3. Proteomic Data Analysis
3. Results and Discussion
3.1. Proteomic Signature of Myocardium Tissue from HFpEF Patients
3.2. Proteomic Signature of Myocardium Tissue from HFpEF Patients with Diabetes Mellitus
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Packer, M.; Butler, J.; Zannad, F.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Carson, P.; Anand, I.; Doehner, W.; Haass, M.; et al. Effect of Empagliflozin on Worsening Heart Failure Events in Patients With Heart Failure and Preserved Ejection Fraction: EMPEROR-Preserved Trial. Circulation 2021, 144, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Sanders-Van Wijk, S.; Tromp, J.; Beussink-Nelson, L.; Hage, C.; Svedlund, S.; Saraste, A.; Swat, S.A.; Sanchez, C.; Njoroge, J.; Tan, R.S.; et al. Proteomic Evaluation of the Comorbidity-Inflammation Paradigm in Heart Failure With Preserved Ejection Fraction: Results From the PROMIS-HFpEF Study. Circulation 2020, 142, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Komajda, M.; Carson, P.E.; Hetzel, S.; McKelvie, R.; McMurray, J.; Ptaszynska, A.; Zile, M.R.; DeMets, D.; Massie, B.M. Factors Associated with Outcome in Heart Failure with Preserved Ejection Fraction: Findings from the Irbesartan in Heart Failure with Preserved Ejection Fraction Study (I-PRESERVE). Circ. Heart Fail. 2011, 4, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, S.L.; Mogensen, U.M.; Jhund, P.S.; Petrie, M.C.; Preiss, D.; Win, S.; Køber, L.; McKelvie, R.S.; Zile, M.R.; Anand, I.S.; et al. Clinical and Echocardiographic Characteristics and Cardiovascular Outcomes According to Diabetes Status in Patients With Heart Failure and Preserved Ejection Fraction: A Report From the I-Preserve Trial (Irbesartan in Heart Failure with Preserved Ejection Fraction). Circulation 2017, 135, 724–735. [Google Scholar] [CrossRef] [Green Version]
- Ather, S.; Chan, W.; Bozkurt, B.; Aguilar, D.; Ramasubbu, K.; Zachariah, A.A.; Wehrens, X.H.T.; Deswal, A. Impact of Noncardiac Comorbidities on Morbidity and Mortality in a Predominantly Male Population with Heart Failure and Preserved versus Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2012, 59, 998–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.V.; Bronzwaer, J.G.F.; Van Der Velden, J.; Stienen, G.J.M.; Laarman, G.J.; Somsen, A.; et al. Low Myocardial Protein Kinase G Activity in Heart Failure with Preserved Ejection Fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Borbély, A.; Falcao-Pires, I.; Van Heerebeek, L.; Hamdani, N.; Édes, I.; Gavina, C.; Leite-Moreira, A.F.; Bronzwaer, J.G.F.; Papp, Z.; Van Der Velden, J.; et al. Hypophosphorylation of the Stiff N2B Titin Isoform Raises Cardiomyocyte Resting Tension in Failing Human Myocardium. Circ. Res. 2009, 104, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Falcão-Pires, I.; Hamdani, N.; Borbély, A.; Gavina, C.; Schalkwijk, C.G.; Van Der Velden, J.; Van Heerebeek, L.; Stienen, G.J.M.; Niessen, H.W.M.; Leite-Moreira, A.F.; et al. Diabetes Mellitus Worsens Diastolic Left Ventricular Dysfunction in Aortic Stenosis through Altered Myocardial Structure and Cardiomyocyte Stiffness. Circulation 2011, 124, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Kresoja, K.P.; Rommel, K.P.; Fengler, K.; Von Roeder, M.; Besler, C.; Lücke, C.; Gutberlet, M.; Desch, S.; Thiele, H.; Böhm, M.; et al. Renal Sympathetic Denervation in Patients With Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail. 2021, 14, 297–307. [Google Scholar] [CrossRef]
- Hanff, T.C.; Cohen, J.B.; Zhao, L.; Javaheri, A.; Zamani, P.; Prenner, S.B.; Rietzschel, E.; Jia, Y.; Walsh, A.; Maranville, J.; et al. Quantitative Proteomic Analysis of Diabetes Mellitus in Heart Failure With Preserved Ejection Fraction. JACC. Basic Transl. Sci. 2021, 6, 89–99. [Google Scholar] [CrossRef]
- AbouEzzeddine, O.F.; McKie, P.M.; Dunlay, S.M.; Stevens, S.R.; Felker, G.M.; Borlaug, B.A.; Chen, H.H.; Tracy, R.P.; Braunwald, E.; Redfield, M.M. Suppression of Tumorigenicity 2 in Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e004382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders-Van Wijk, S.; Van Empel, V.; Davarzani, N.; Maeder, M.T.; Handschin, R.; Pfisterer, M.E.; Brunner-La Rocca, H.P. Circulating Biomarkers of Distinct Pathophysiological Pathways in Heart Failure with Preserved vs. Reduced Left Ventricular Ejection Fraction. Eur. J. Heart Fail. 2015, 17, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropoulos, A.; Georgiopoulou, V.; Psaty, B.M.; Rodondi, N.; Smith, A.L.; Harrison, D.G.; Liu, Y.; Hoffmann, U.; Bauer, D.C.; Newman, A.B.; et al. Inflammatory Markers and Incident Heart Failure Risk in Older Adults: The Health ABC (Health, Aging, and Body Composition) Study. J. Am. Coll. Cardiol. 2010, 55, 2129–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hage, C.; Michaëlsson, E.; Linde, C.; Donal, E.; Daubert, J.C.; Gan, L.M.; Lund, L.H. Inflammatory Biomarkers Predict Heart Failure Severity and Prognosis in Patients with Heart Failure with Preserved Ejection Fraction: A Holistic Proteomic Approach. Circ. Cardiovasc. Genet. 2017, 10, e001633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure of the European Society of Cardiology (ESC)Developed with the Special Contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, D.; Silva, M.M.; Terrasso, A.P.; Arez, F.; Sousa, M.F.Q.; Mehrjardi, N.Z.; Šarić, T.; Gomes-Alves, P.; Raimundo, N.; Alves, P.M.; et al. Recapitulation of Human Neural Microenvironment Signatures in IPSC-Derived NPC 3D Differentiation. Stem Cell Rep. 2018, 11, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Sebastião, M.J.; Gomes-Alves, P.; Reis, I.; Sanchez, B.; Palacios, I.; Serra, M.; Alves, P.M. Bioreactor-Based 3D Human Myocardial Ischemia/Reperfusion in Vitro Model: A Novel Tool to Unveil Key Paracrine Factors upon Acute Myocardial Infarction. Transl. Res. 2020, 215, 57–74. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities Drive Myocardial Dysfunction and Remodeling through Coronary Microvascular Endothelial Inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Richards, A.M.; Pemberton, C.J. Progress in Proteomic Probing for Pathogenic Pathways in Heart Failure with Preserved Ejection Fraction. Eur. J. Heart Fail. 2021, 23, 1645–1647. [Google Scholar] [CrossRef] [PubMed]
- van Heerebeek, L.; Paulus, W.J. Understanding Heart Failure with Preserved Ejection Fraction: Where Are We Today? Neth. Heart J. 2016, 24, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; He, H.; Fu, Z.C.; Huang, S.; Chen, T.; Papasian, C.J.; Morse, L.R.; Xu, Y.; Battaglino, R.A.; Yang, X.F.; et al. Adipocyte-Derived PAMM Suppresses Macrophage Inflammation by Inhibiting MAPK Signalling. Biochem. J. 2015, 472, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Morse, L.R.; da Silva, R.A.B.; Wang, D.; Battaglino, R.A. A Short Report: PAMM, a Novel Antioxidant Protein, Induced by Oxidative Stress. Redox Biol. 2015, 6, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, L.R.; Nguyen, N.; Xu, Y.; Jha, P.; Battaglino, R.A. Circulating PAMM, a Novel Antioxidant and Anti-Inflammatory Protein, Is Elevated in Acute SCI. J. Transl. Med. 2020, 18, 135. [Google Scholar] [CrossRef] [Green Version]
- Kolwicz, S.C.; Purohit, S.; Tian, R. Cardiac Metabolism and Its Interactions with Contraction, Growth, and Survival of Cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Adams, V.; Wunderlich, S.; Mangner, N.; Hommel, J.; Esefeld, K.; Gielen, S.; Halle, M.; Ellingsen, Ø.; Van Craenenbroeck, E.M.; Wisløff, U.; et al. Ubiquitin-Proteasome-System and Enzymes of Energy Metabolism in Skeletal Muscle of Patients with HFpEF and HFrEF. ESC Heart Fail. 2021, 8, 2556–2568. [Google Scholar] [CrossRef]
- Ke, B.X.; Pepe, S.; Grubb, D.R.; Komen, J.C.; Laskowski, A.; Rodda, F.A.; Hardman, B.M.; Pitt, J.J.; Ryan, M.T.; Lazarou, M.; et al. Tissue-Specific Splicing of an Ndufs6 Gene-Trap Insertion Generates a Mitochondrial Complex I Deficiency-Specific Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2012, 109, 6165–6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, I.; Hershkovitz, E.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. Mitochondrial Complex I Deficiency Caused by a Deleterious NDUFA11 Mutation. Ann. Neurol. 2008, 63, 405–408. [Google Scholar] [CrossRef]
- Wischhof, L.; Gioran, A.; Sonntag-Bensch, D.; Piazzesi, A.; Stork, M.; Nicotera, P.; Bano, D. A Disease-Associated Aifm1 Variant Induces Severe Myopathy in Knockin Mice. Mol. Metab. 2018, 13, 10. [Google Scholar] [CrossRef]
- Zhou, W.; Ji, L.; Liu, X.; Tu, D.; Shi, N.; Yangqu, W.; Chen, S.; Gao, P.; Zhu, H.; Ruan, C. AIFM1, Negatively Regulated by MiR-145-5p, Aggravates Hypoxia-Induced Cardiomyocyte Injury. Biomed. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Bae, S.; Kumar, S.R.; Ke, Q.; Yalamarti, B.; Choi, J.H.; Kirshenbaum, L.A.; Kang, P.M. Role of AIF in Cardiac Apoptosis in Hypertrophic Cardiomyocytes from Dahl Salt-Sensitive Rats. Cardiovasc. Res. 2010, 85, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, J.H.; Patton, B.L.; Lentz, S.I.; Gilbert, D.J.; Snider, W.D.; Jenkins, N.A.; Copeland, N.G.; Sanes, J.R. The Laminin Alpha Chains: Expression, Developmental Transitions, and Chromosomal Locations of Alpha1-5, Identification of Heterotrimeric Laminins 8-11, and Cloning of a Novel Alpha3 Isoform. J. Cell Biol. 1997, 137, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hoshijima, M.; Lam, J.; Zhou, Z.; Jokiel, A.; Dalton, N.D.; Hultenby, K.; Ruiz-Lozano, P.; Ross, J.; Tryggvason, K.; et al. Cardiomyopathy Associated with Microcirculation Dysfunction in Laminin Alpha4 Chain-Deficient Mice. J. Biol. Chem. 2006, 281, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Tian, X.; Xie, W.; Jones, P.P.; Cai, S.; Wang, X.; Jiang, D.; Kong, H.; Zhang, L.; Chen, K.; et al. Functional Consequence of Protein Kinase A-Dependent Phosphorylation of the Cardiac Ryanodine Receptor: Sensitization of Store Overload-Induced Ca2+ Release. J. Biol. Chem. 2007, 282, 30256–30264. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Yang, R.; Awad, T.A.; Wang, F.; Li, W.; Williams, S.P.; Ogasawara, A.; Shimada, B.; Williams, P.M.; De Feo, G.; et al. Effects of Early Angiotensin-Converting Enzyme Inhibition on Cardiac Gene Expression after Acute Myocardial Infarction. Circulation 2001, 103, 736–742. [Google Scholar] [CrossRef] [Green Version]
- Adamo, L.; Yu, J.; Rocha-Resende, C.; Javaheri, A.; Head, R.D.; Mann, D.L. Proteomic Signatures of Heart Failure in Relation to Left Ventricular Ejection Fraction. J. Am. Coll. Cardiol. 2020, 76, 1982–1994. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Sharma, K. Mitochondrial Hormesis and Diabetic Complications. Diabetes 2015, 64, 663–672. [Google Scholar] [CrossRef] [Green Version]
- De Jong, K.A.; Lopaschuk, G.D. Complex Energy Metabolic Changes in Heart Failure With Preserved Ejection Fraction and Heart Failure With Reduced Ejection Fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, Y.; Kim, I.M.; Weintraub, N.L.; Tang, Y. The Impaired Bioenergetics of Diabetic Cardiac Microvascular Endothelial Cells. Front. Endocrinol. 2021, 12, 642857. [Google Scholar] [CrossRef] [PubMed]
- Rohm, M.; Savic, D.; Ball, V.; Curtis, M.K.; Bonham, S.; Fischer, R.; Legrave, N.; MacRae, J.I.; Tyler, D.J.; Ashcroft, F.M. Cardiac Dysfunction and Metabolic Inflexibility in a Mouse Model of Diabetes Without Dyslipidemia. Diabetes 2018, 67, 1057–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grynberg, A.; Demaison, L. Fatty Acid Oxidation in the Heart. J. Cardiovasc. Pharmacol. 1996, 28 (Suppl. 1), S11–S18. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- de las Fuentes, L.; Soto, P.F.; Cupps, B.P.; Pasque, M.K.; Herrero, P.; Gropler, R.J.; Waggoner, A.D.; Dávila-Román, V.G. Hypertensive Left Ventricular Hypertrophy Is Associated with Abnormal Myocardial Fatty Acid Metabolism and Myocardial Efficiency. J. Nucl. Cardiol. 2006, 13, 369–377. [Google Scholar] [CrossRef]
- Dávila-Román, V.G.; Vedala, G.; Herrero, P.; De Las Fuentes, L.; Rogers, J.G.; Kelly, D.P.; Gropler, R.J. Altered Myocardial Fatty Acid and Glucose Metabolism in Idiopathic Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Zhang, H.; Montano, S.; Hegestam, J.; Ekberg, N.R.; Holmgren, A.; Brismar, K.; Ungerstedt, J.S. Plasma Glutaredoxin Activity in Healthy Subjects and Patients with Abnormal Glucose Levels or Overt Type 2 Diabetes. Acta Diabetol. 2014, 51, 225–232. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Khan, M.M.; Ghosh, C.; Bank, S.; Maiti, S. The Role of Dermcidin Isoform-2 in the Occurrence and Severity of Diabetes. Sci. Rep. 2017, 7, 8252. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Guo, Z.; Wang, B.; Wu, Y.; Li, Z.; Yao, H.; Fang, R.; Yang, H.; Cao, H.; Cui, Y. Risk Prediction in Patients With Heart Failure With Preserved Ejection Fraction Using Gene Expression Data and Machine Learning. Front. Genet. 2021, 12, 652315. [Google Scholar] [CrossRef]



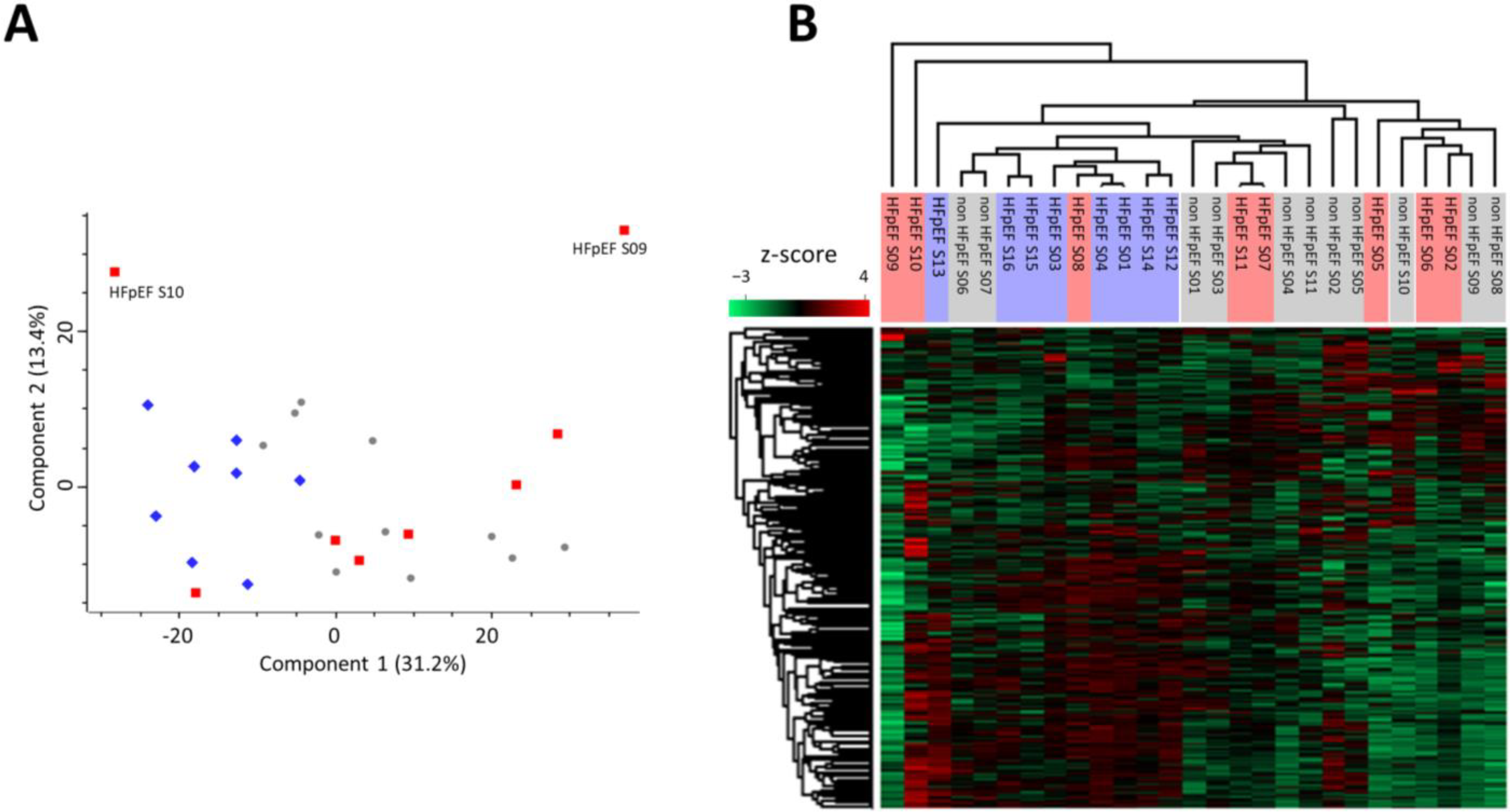{kind=link}
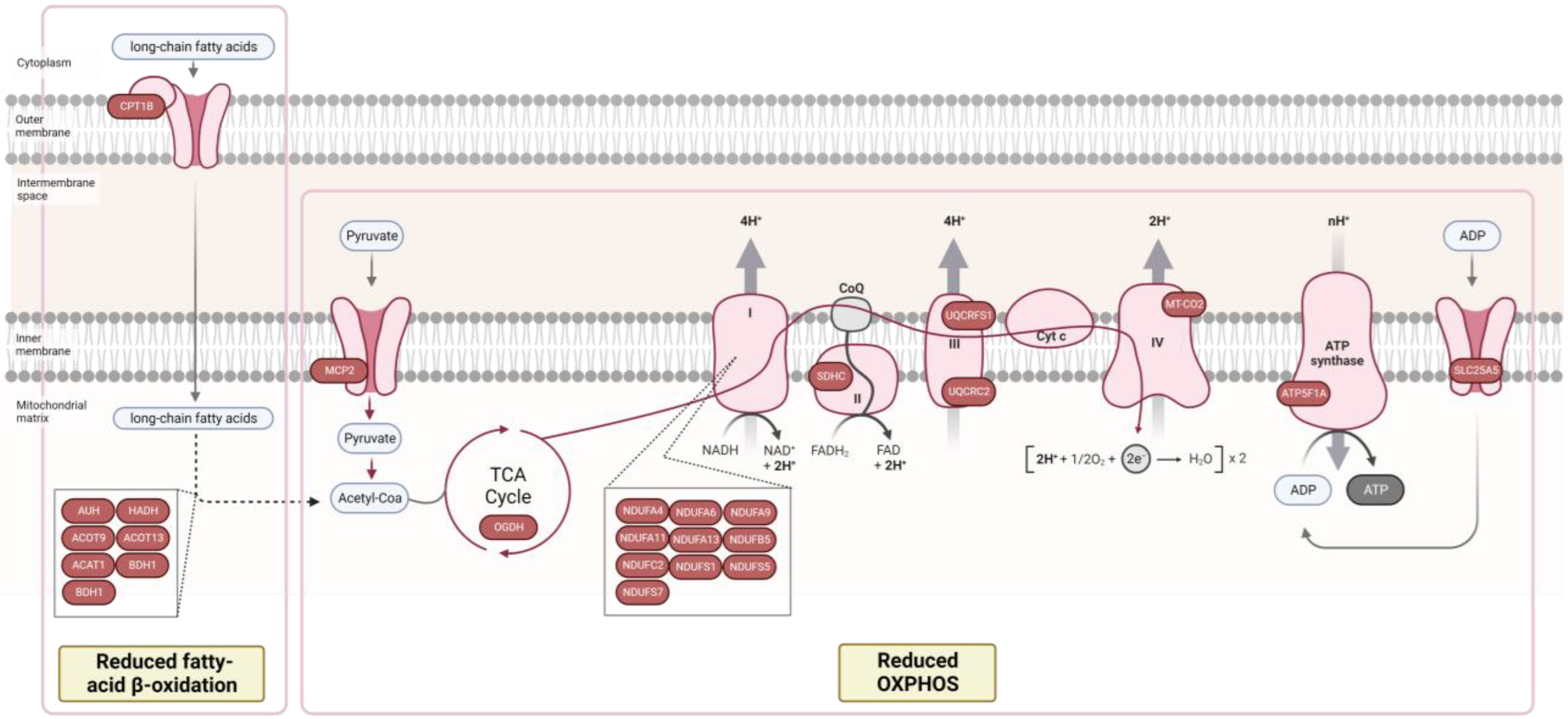{kind=link}
| Parameters | HFpEF (n = 16) | Non-HFpEF (n = 11) | p-Value |
|---|---|---|---|
| Age, y | 70 (67, 74) | 58 (40, 68) | 0.018 |
| Male (%) | 63 | 27 | 0.239 |
| Diabetes mellitus (%) | 67 | 27 | 0.047 |
| Hypertension (%) | 87 | 100 | >0.999 |
| Obesity (%) | 40 | 43 | >0.999 |
| Anemia (%) | 18 | 14 | >0.999 |
| Dyslipidemia (%) | 91 | 86 | >0.999 |
| ID | Name | FC | p-Value |
|---|---|---|---|
| EIF4A2 | Eukaryotic initiation factor 4A-II | 3.00 | 0.028 |
| ACTG1 | Actin, cytoplasmic 2 | 2.27 | 0.049 |
| TUBA4A | Tubulin alpha-4A chain | 2.10 | 0.035 |
| NDUFA11 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 11 | 2.06 | 0.049 |
| PRXL2A | Peroxiredoxin-like 2A | 2.00 | 0.038 |
| AIFM1 | Apoptosis-inducing factor 1, mitochondrial | 1.84 | 0.045 |
| IGKV3D-20 | Immunoglobulin kappa variable 3D-20 | 1.78 | 0.048 |
| PRKACA | cAMP-dependent protein kinase catalytic subunit alpha | 1.77 | 0.048 |
| LAMA4 | Laminin subunit alpha-4 | 1.67 | 0.045 |
| COMT | Catechol O-methyltransferase | 1.46 | 0.029 |
| EHD1 | EH domain-containing protein 1 | 1.36 | 0.035 |
| SAA1 | Serum amyloid A-1 protein | 0.42 | 0.046 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebastião, M.J.; Almeida, H.V.; Serra, M.; Hamdani, N.; Saraiva, F.; Lourenço, A.P.; Barros, A.S.; Vasques-Nóvoa, F.; Leite-Moreira, A.; Alves, P.M.; et al. Unveiling Human Proteome Signatures of Heart Failure with Preserved Ejection Fraction. Biomedicines 2022, 10, 2943. https://doi.org/10.3390/biomedicines10112943
Sebastião MJ, Almeida HV, Serra M, Hamdani N, Saraiva F, Lourenço AP, Barros AS, Vasques-Nóvoa F, Leite-Moreira A, Alves PM, et al. Unveiling Human Proteome Signatures of Heart Failure with Preserved Ejection Fraction. Biomedicines. 2022; 10(11):2943. https://doi.org/10.3390/biomedicines10112943
Chicago/Turabian StyleSebastião, Maria J., Henrique V. Almeida, Margarida Serra, Nazha Hamdani, Francisca Saraiva, André P. Lourenço, António S. Barros, Francisco Vasques-Nóvoa, Adelino Leite-Moreira, Paula M. Alves, and et al. 2022. "Unveiling Human Proteome Signatures of Heart Failure with Preserved Ejection Fraction" Biomedicines 10, no. 11: 2943. https://doi.org/10.3390/biomedicines10112943
APA StyleSebastião, M. J., Almeida, H. V., Serra, M., Hamdani, N., Saraiva, F., Lourenço, A. P., Barros, A. S., Vasques-Nóvoa, F., Leite-Moreira, A., Alves, P. M., Falcão-Pires, I., & Gomes-Alves, P. (2022). Unveiling Human Proteome Signatures of Heart Failure with Preserved Ejection Fraction. Biomedicines, 10(11), 2943. https://doi.org/10.3390/biomedicines10112943