
Mitochondrial Role in Intrinsic Apoptosis Induced by a New Synthesized Chalcone in Hepatocellular Carcinoma Cells

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of (E)-1-(2,4,6-triethoxyphenyl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (ETTC)
2.2. Cell Culture
2.3. Isolation of Mitochondria
2.4. Cell Viability Assay
2.5. Cell Cycle Analysis
2.6. DAPI Staining
2.7. Caspase Assays
2.8. Real-Time PCR
2.9. Mitochondrial Mass Analysis
2.10. Mitochondrial Membrane Potential Analysis
2.11. Mitochondrial Complex I Activity Assay
2.12. Mitochondrial Citrate Synthase Activity Assay
2.13. Mitochondrial Superoxide Analysis
2.14. Statistical Analysis
3. Results
3.1. Design and Synthesis of Chalcone ETTC
3.2. ETTC Reduced HepG2 Cell Viability
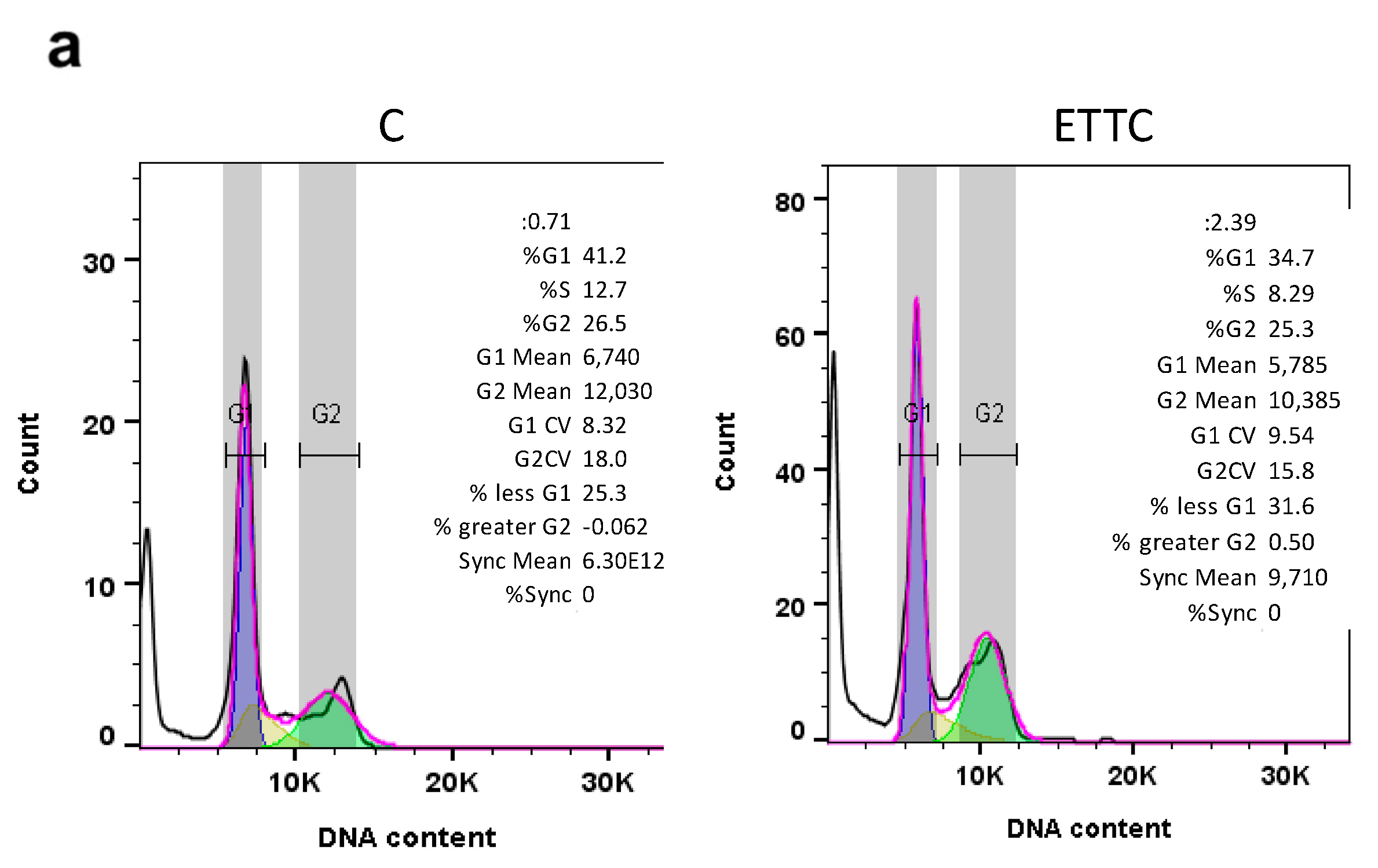
3.3. ETTC Caused subG1 Cell Cycle Arrest and Nuclear Fragmentation in HepG2 Cells
3.4. ETTC Induced Mitochondria-Mediated Apoptosis in HepG2 Cells
3.5. ETTC Caused Mitochondrial Dysfunctions in HepG2 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar]
- Yoon, S.K. Molecular mechanism of hepatocellular carcinoma. Hepatoma Res. 2018, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, R.; Bandoh, S.; Roberts, L.R. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research 2016, 5, 879. [Google Scholar] [CrossRef]
- Piñero, F.; Dirchwolf, M.; Pessôa, M.G. Biomarkers in Hepatocellular Carcinoma: Diagnosis, Prognosis and Treatment Response Assessment. Cells 2020, 9, 1370. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Harricharran, T.; Huaman, J.; Galuza, A.; Odumuwagun, O.; Tan, Y.; Ma, G.X.; Nguyen, M.T. Mechanisms of hepatocellular carcinoma progression. World J. Gastroenterol. 2019, 25, 2279–2293. [Google Scholar] [CrossRef]
- Koulouris, A.; Tsagkaris, C.; Spyrou, V.; Pappa, E.; Troullinou, A.; Nikolaou, M. Hepatocellular Carcinoma: An Overview of the Changing Landscape of Treatment Options. J. Hepatocell. Carcinoma 2021, 8, 387–401. [Google Scholar] [CrossRef]
- Lang, L. FDA Approves Sorafenib for Patients With Inoperable Liver Cancer. Gastroenterology 2008, 134, 379. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- Rudrapal, M.; Khan, J.; Dukhyil, A.A.B.; Alarousy, R.M.I.I.; Attah, E.I.; Sharma, T.; Khairnar, S.J.; Bendale, A.R. Chalcone Scaffolds, Bioprecursors of Flavonoids: Chemistry, Bioactivities, and Pharmacokinetics. Molecules 2021, 26, 7177. [Google Scholar] [CrossRef]
- Rozmer, Z.; Perjési, P. Naturally occurring chalcones and their biological activities. Phytochem. Rev. 2016, 15, 87–120. [Google Scholar]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A mini review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Amslinger, S. The Tunable Functionality of α,β-Unsaturated Carbonyl Compounds Enables Their Differential Application in Biological Systems. ChemMedChem 2010, 5, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Bandgar, B.P.; Gawande, S.S.; Bodade, R.G.; Totre, J.V.; Khobragade, C.N. Synthesis and biological evaluation of simple methoxylated chalcones as anticancer, anti-inflammatory and antioxidant agents. Bioorg. Med. Chem. 2010, 18, 1364–1370. [Google Scholar] [CrossRef]
- Gao, F.; Huang, G.; Xiao, J. Chalcone hybrids as potential anticancer agents: Current development, mechanism of action, and structure-activity relationship. Med. Res. Rev. 2020, 40, 2049–2084. [Google Scholar] [CrossRef]
- Constantinescu, T.; Lungu, C.N. Anticancer Activity of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2021, 22, 11306. [Google Scholar] [CrossRef]
- Rajendran, G.; Bhanu, D.; Aruchamy, B.; Ramani, P.; Pandurangan, N.; Bobba, K.N.; Oh, E.J.; Chung, H.Y.; Gangadaran, P.; Ahn, B.-C. Chalcone: A Promising Bioactive Scaffold in Medicinal Chemistry. Pharmaceuticals 2022, 15, 1250. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef]
- Kim, J.H.; Jung, C.H.; Jang, B.-H.; Go, H.Y.; Park, J.-H.; Choi, Y.-K.; Hong, S.I.; Shin, Y.C.; Ko, S.-G. Selective Cytotoxic Effects on Human Cancer Cell Lines of Phenolic-Rich Ethyl-Acetate Fraction from Rhus verniciflua Stokes. Am. J. Chin. Med. 2009, 37, 609–620. [Google Scholar] [CrossRef]
- Padmavathi, G.; Rathnakaram, S.R.; Monisha, J.; Bordoloi, D.; Roy, N.K.; Kunnumakkara, A.B. Potential of butein, a tetrahydroxychalcone to obliterate cancer. Phytomedicine 2015, 22, 1163–1171. [Google Scholar] [CrossRef]
- Kang, H.-M.; Kim, J.-H.; Lee, M.-Y.; Son, K.-H.; Yang, D.C.; Baek, N.-I.; Kwon, B.-M. Relationship Between Flavonoid Structure and Inhibition of Farnesyl Protein Transferase. Nat. Prod. Res. 2004, 18, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Di, S.; Fan, C.; Ma, Z.; Li, M.; Guo, K.; Han, D.; Li, X.; Mu, D.; Yan, X. PERK/eIF-2α/CHOP Pathway Dependent ROS Generation Mediates Butein-induced Non-small-cell Lung Cancer Apoptosis and G2/M Phase Arrest. Int. J. Biol. Sci. 2019, 15, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, M.; Yu, X.; Liu, T.; Li, T.; Zhou, L.; Liu, W.; Li, W.; Gao, F. Butein suppresses hepatocellular carcinoma growth via modulating Aurora B kinase activity. Int. J. Biol. Sci. 2018, 14, 1521–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, B.; Cho, S.-G.; Liu, M.; Aggarwal, B.B. Butein, a tetrahydroxychalcone, suppresses cancer-induced osteoclastogenesis through inhibition of receptor activator of nuclear factor-kappaB ligand signaling. Int. J. Cancer 2011, 129, 2062–2072. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, Z.; Akhtar, N.; Khan, A.; Khan, K.A.; Haqqi, T.M. Butrin, Isobutrin, and Butein from Medicinal Plant Butea monosperma Selectively Inhibit Nuclear Factor-κB in Activated Human Mast Cells: Suppression of Tumor Necrosis Factor-α, Interleukin (IL)-6, and IL-8. J. Pharmacol. Exp. Ther. 2010, 333, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [PubMed]
- Karthikeyan, C.; Moorthy, N.S.H.N.; Ramasamy, S.; Vanam, U.; Manivannan, E.; Karunagaran, D.; Trivedi, P. Advances in chalcones with anticancer activities. Recent Pat. Anticancer. Drug Discov. 2015, 10, 97–115. [Google Scholar] [CrossRef]
- Boumendjel, A.; Boccard, J.; Carrupt, P.-A.; Nicolle, E.; Blanc, M.; Geze, A.; Choisnard, L.; Wouessidjewe, D.; Matera, E.-L.; Dumontet, C. Antimitotic and Antiproliferative Activities of Chalcones: Forward Structure–Activity Relationship. J. Med. Chem. 2008, 51, 2307–2310. [Google Scholar] [CrossRef]
- Yadav, V.R.; Prasad, S.; Sung, B.; Aggarwal, B.B. The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int. Immunopharmacol. 2011, 11, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Santarsiero, A.; Convertini, P.; Vassallo, A.; Santoro, V.; Todisco, S.; Iacobazzi, D.; Fondufe-Mittendorf, Y.; Martelli, G.; de Oliveira, M.R.; Montanaro, R.; et al. Phenolic Compounds of Red Wine Aglianico del Vulture Modulate the Functional Activity of Macrophages via Inhibition of NF-κB and the Citrate Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Infantino, V.; Convertini, P.; Iacobazzi, F.; Pisano, I.; Scarcia, P.; Iacobazzi, V. Identification of a novel Sp1 splice variant as a strong transcriptional activator. Biochem. Biophys. Res. Commun. 2011, 412, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Convertini, P.; Tramutola, F.; Iacobazzi, V.; Lupattelli, P.; Chiummiento, L.; Infantino, V. Permethylated Anigopreissin A inhibits human hepatoma cell proliferation by mitochondria-induced apoptosis. Chem. Biol. Interact. 2015, 237, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Marsico, M.; Santarsiero, A.; Pappalardo, I.; Convertini, P.; Chiummiento, L.; Sardone, A.; Di Noia, M.A.; Infantino, V.; Todisco, S. Mitochondria-Mediated Apoptosis of HCC Cells Triggered by Knockdown of Glutamate Dehydrogenase 1: Perspective for Its Inhibition through Quercetin and Permethylated Anigopreissin A. Biomedicines 2021, 9, 1664. [Google Scholar] [CrossRef] [PubMed]
- Santarsiero, A.; Convertini, P.; Todisco, S.; Pierri, C.L.; De Grassi, A.; Williams, N.C.; Iacobazzi, D.; De Stefano, G.; O’Neill, L.A.J.; Infantino, V. ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-κB Acetylation. Cells 2021, 10, 2962. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L.; Cerchia, C.; Laghezza, A.; Ziccardi, P.; Sblano, S.; Tortorella, P.; Iacobazzi, V.; Infantino, V.; Convertini, P.; Dal Piaz, F.; et al. New diphenylmethane derivatives as peroxisome proliferator-activated receptor alpha/gamma dual agonists endowed with anti-proliferative effects and mitochondrial activity. Eur. J. Med. Chem. 2017, 127, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D. Detection of Mitochondrial Mass, Damage, and Reactive Oxygen Species by Flow Cytometry. Cold Spring Harb. Protoc. 2015, 2015, pdb.prot086298. [Google Scholar] [CrossRef]
- Belviso, S.; Cammarota, F.; Rossano, R.; Lelj, F. Effect of polyfluorination on self-assembling and electronic properties of thioalkyl-porphyrazines. J. Porphyr. Phthalocyanines 2016, 20, 223–233. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Giam, M.; Huang, D.C.S.; Bouillet, P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008, 27 (Suppl. S1), S128–S136. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [Green Version]
- Khodapasand, E.; Jafarzadeh, N.; Farrokhi, F.; Kamalidehghan, B.; Houshmand, M. Is Bax/Bcl-2 ratio considered as a prognostic marker with age and tumor location in colorectal cancer? Iran. Biomed. J. 2015, 19, 69–75. [Google Scholar] [PubMed]
- Agnello, M.; Morici, G.; Rinaldi, A.M. A method for measuring mitochondrial mass and activity. Cytotechnology 2008, 56, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Benz, R.; McLaughlin, S. The molecular mechanism of action of the proton ionophore FCCP (carbonylcyanide p-trifluoromethoxyphenylhydrazone). Biophys. J. 1983, 41, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Infantino, V.; Dituri, F.; Convertini, P.; Santarsiero, A.; Palmieri, F.; Todisco, S.; Mancarella, S.; Giannelli, G.; Iacobazzi, V. Epigenetic upregulation and functional role of the mitochondrial aspartate/glutamate carrier isoform 1 in hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Muñoz, F.; Lovy, A.; Cárdenas, C. The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 2017, 7, 118. [Google Scholar] [CrossRef]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma. Cancers 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasooriya, R.G.P.T.; Molagoda, I.M.N.; Park, C.; Jeong, J.-W.; Choi, Y.H.; Moon, D.-O.; Kim, M.-O.; Kim, G.-Y. Molecular chemotherapeutic potential of butein: A concise review. Food Chem. Toxicol. 2018, 112, 1–10. [Google Scholar] [CrossRef]
- Tuli, H.S.; Joshi, R.; Aggarwal, D.; Kaur, G.; Kaur, J.; Kumar, M.; Parashar, N.C.; Khan, M.A.; Sak, K. Molecular mechanisms underlying chemopreventive potential of butein: Current trends and future perspectives. Chem. Biol. Interact. 2021, 350, 109699. [Google Scholar] [CrossRef]
- Brown, S.; Griffiths, L.A. Metabolism and excretion of butein, 2′,3,4-trihydroxychalcone, 3-O-methylbutein, 4-O-methylbutein and 2′,4′,4-trihydroxychalcone in the rat. Xenobiotica 1983, 13, 669–682. [Google Scholar] [CrossRef]
- Toprak, M. Fluorescence study on the interaction of human serum albumin with Butein in liposomes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 154, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.-P.; Hsiao, P.-C.; Yang, S.-F.; Hsieh, S.-C.; Bau, D.-T.; Ling, C.-L.; Pai, C.-L.; Hsieh, Y.-H. Licochalcone A Suppresses Migration and Invasion of Human Hepatocellular Carcinoma Cells through Downregulation of MKK4/JNK via NF-κB Mediated Urokinase Plasminogen Activator Expression. PLoS ONE 2014, 9, e86537. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhao, S.; Long, J.; Su, J.; Wu, L.; Tao, J.; Zhou, J.; Zhang, J.; Chen, X.; Peng, C. A novel chalcone derivative has antitumor activity in melanoma by inducing DNA damage through the upregulation of ROS products. Cancer Cell Int. 2020, 20, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echeverria, C.; Santibañez, J.F.; Donoso-Tauda, O.; Escobar, C.A.; Ramirez-Tagle, R. Structural antitumoral activity relationships of synthetic chalcones. Int. J. Mol. Sci. 2009, 10, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-S.; Ahn, Y.; Lee, D.; Moon, S.W.; Kim, K.H.; Yamabe, N.; Hwang, G.S.; Jang, H.J.; Lee, H.; Kang, K.S.; et al. Synthesis of apoptotic chalcone analogues in HepG2 human hepatocellular carcinoma cells. Bioorg. Med. Chem. Lett. 2015, 25, 5705–5707. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Tagle, R.; Escobar, C.; Romero, V.; Montorfano, I.; Armisén, R.; Borgna, V.; Jeldes, E.; Pizarro, L.; Simon, F.; Echeverria, C. Chalcone-Induced Apoptosis through Caspase-Dependent Intrinsic Pathways in Human Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2016, 17, 260. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a008656. [Google Scholar] [CrossRef] [Green Version]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxia-induced stabilization of HIF-1α through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef] [PubMed]
- Frazier, A.E.; Thorburn, D.R. Biochemical Analyses of the Electron Transport Chain Complexes by Spectrophotometry. In Mitochondrial Disorders; Springer Science+Business Media, LLC: Humana Totowa, NJ, USA, 2012; pp. 49–62. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santarsiero, A.; Pappalardo, I.; Rosa, G.M.; Pisano, I.; Superchi, S.; Convertini, P.; Todisco, S.; Scafato, P.; Infantino, V. Mitochondrial Role in Intrinsic Apoptosis Induced by a New Synthesized Chalcone in Hepatocellular Carcinoma Cells. Biomedicines 2022, 10, 3120. https://doi.org/10.3390/biomedicines10123120
Santarsiero A, Pappalardo I, Rosa GM, Pisano I, Superchi S, Convertini P, Todisco S, Scafato P, Infantino V. Mitochondrial Role in Intrinsic Apoptosis Induced by a New Synthesized Chalcone in Hepatocellular Carcinoma Cells. Biomedicines. 2022; 10(12):3120. https://doi.org/10.3390/biomedicines10123120
Chicago/Turabian StyleSantarsiero, Anna, Ilaria Pappalardo, Gabriella Margherita Rosa, Isabella Pisano, Stefano Superchi, Paolo Convertini, Simona Todisco, Patrizia Scafato, and Vittoria Infantino. 2022. "Mitochondrial Role in Intrinsic Apoptosis Induced by a New Synthesized Chalcone in Hepatocellular Carcinoma Cells" Biomedicines 10, no. 12: 3120. https://doi.org/10.3390/biomedicines10123120
APA StyleSantarsiero, A., Pappalardo, I., Rosa, G. M., Pisano, I., Superchi, S., Convertini, P., Todisco, S., Scafato, P., & Infantino, V. (2022). Mitochondrial Role in Intrinsic Apoptosis Induced by a New Synthesized Chalcone in Hepatocellular Carcinoma Cells. Biomedicines, 10(12), 3120. https://doi.org/10.3390/biomedicines10123120