
Exercise-Boosted Mitochondrial Remodeling in Parkinson’s Disease
 ,
,  ,
,  and
and 
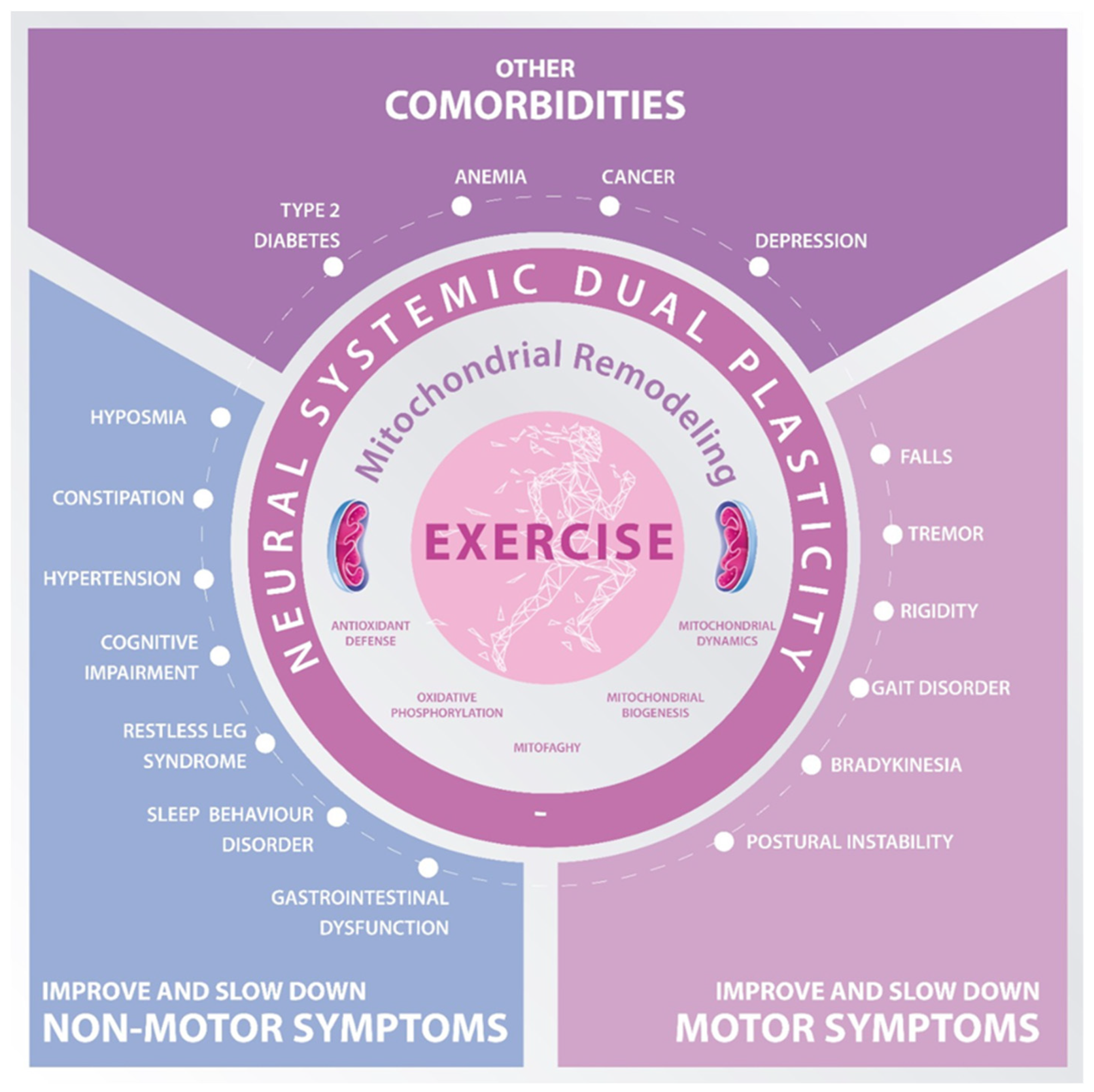{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neuronal and Muscular Alterations Found in Parkinson’s Disease
2.1. Muscular Function Impairments in Parkinson’s Disease
2.2. Neuronal Function Abnormalities: The Multi-Faceted Role of Mitochondria in Parkinson’s Disease
3. Ameliorating Mitochondrial Dysfunction in Parkinson’s Disease through Exercise
3.1. Types of Exercise Applied to Parkinson’s Disease Patients
3.2. Muscle–Brain Crosstalk in Parkinson’s Disease: The Role of Exercise Secretome
3.3. Exercised Mitochondria, a Cross-Optimization of Metabolic Pathways with Potential to Slow down PD Progression?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heemels, M.T. Neurodegenerative diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef] [Green Version]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. (Vienna) 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Brakedal, B.; Toker, L.; Haugarvoll, K.; Tzoulis, C. A nationwide study of the incidence, prevalence and mortality of Parkinson’s disease in the Norwegian population. NPJ Park. Dis. 2022, 8, 19. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic—A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global Trends in the Incidence, Prevalence, and Years Lived With Disability of Parkinson’s Disease in 204 Countries/Territories From 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef] [PubMed]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells Us about the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadastik-Eerme, L.; Rosenthal, M.; Paju, T.; Muldmaa, M.; Taba, P. Health-related quality of life in Parkinson’s disease: A cross-sectional study focusing on non-motor symptoms. Heal. Qual. Life Outcomes 2015, 13, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Berg, D.; Borghammer, P.; Fereshtehnejad, S.-M.; Heinzel, S.; Horsager, J.; Schaeffer, E.; Postuma, R.B. Prodromal Parkinson disease subtypes — key to understanding heterogeneity. Nat. Rev. Neurol. 2021, 17, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Orayj, K.; Akbari, A.; Lacey, A.; Smith, M.; Pickrell, O.; Lane, E.L. Factors affecting the choice of first-line therapy in Parkinson’s disease patients in Wales: A Population-Based study. Saudi Pharm. J. 2021, 29, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology (Basel) 2019, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Pereira, F.B.; Raimundo, N.; Oliveira, P.J. Mitochondrial remodeling in human skin fibroblasts from sporadic male Parkinson’s disease patients uncovers metabolic and mitochondrial bioenergetic defects. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165615. [Google Scholar] [CrossRef]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Teixeira, J.; Simões, R.F.; Cagide, F.; Benfeito, S.; Borges, F.; Raimundo, N.; Oliveira, P.J. A mitochondria-targeted caffeic acid derivative reverts cellular and mitochondrial defects in human skin fibroblasts from male sporadic Parkinson’s disease patients. Redox Biol. 2021, 45, 102037. [Google Scholar] [CrossRef]
- Milanese, C.; Payán-Gómez, C.; Galvani, M.; González, N.M.; Tresini, M.; Abdellah, S.N.; Van Roon-Mom, W.M.C.; Figini, S.; Marinus, J.; Van Hilten, J.J.; et al. Peripheral mitochondrial function correlates with clinical severity in idiopathic Parkinson’s disease. Mov. Disord. 2019, 34, 1192–1202. [Google Scholar] [CrossRef]
- World Health Organization. Global Action Plan on Physical Activity 2018–2030: More Active People for a Healthier World; World Health Organization: Geneva, Switzerland, 2018.
- Gualdi-Russo, E.; Zaccagni, L. Physical Activity for Health and Wellness. Int. J. Environ. Res. Public Health 2021, 18, 7823. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Barbirato, D.; Araujo, N.; Martins, J.V.; Cavalcanti, J.L.; Santos, T.M.; Coutinho, E.S.; Laks, J.; Deslandes, A.C. Comparison of strength training, aerobic training, and additional physical therapy as supplementary treatments for Parkinson’s disease: Pilot study. Clin. Interv. Aging 2015, 10, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalsing, K.S.; Abbas, M.M.; Tan, L.C.S. Role of Physical Activity in Parkinson’s Disease. Ann. Indian Acad. Neurol. 2018, 21, 242–249. [Google Scholar] [CrossRef]
- Caspersen, C.J.; Powell, K.E.; Christenson, G.M. Physical activity, exercise, and physical fitness: Definitions and distinctions for health-related research. Public Health Rep. 1985, 100, 126–131. [Google Scholar] [PubMed]
- Aguer, C.; Loro, E.; Di Raimondo, D. Editorial: The Role of the Muscle Secretome in Health and Disease. Front. Physiol. 2020, 11, 1101. [Google Scholar] [CrossRef]
- Liguori, G. ACSM’s Guidelines for Exercise Testing and Prescription, 11th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2021. [Google Scholar]
- Drake, J.C.; Laker, R.C.; Wilson, R.; Zhang, M.; Yan, Z. Exercise-induced mitophagy in skeletal muscle occurs in the absence of stabilization of Pink1 on mitochondria. Cell Cycle 2019, 18, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef]
- Vainshtein, A.; Tryon, L.D.; Pauly, M.; Hood, D.A. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Physiol. 2015, 308, C710–C719. [Google Scholar] [CrossRef] [Green Version]
- Morales-Martínez, A.; Martínez-Gómez, P.A.; Martinez-Fong, D.; Villegas-Rojas, M.M.; Pérez-Severiano, F.; Del Toro-Colín, M.A.; Delgado-Minjares, K.M.; Blanco-Alvarez, V.M.; Leon-Chavez, B.A.; Aparicio-Trejo, O.E.; et al. Oxidative Stress and Mitochondrial Complex I Dysfunction Correlate with Neurodegeneration in an α-Synucleinopathy Animal Model. Int. J. Mol. Sci. 2022, 23, 11394. [Google Scholar] [CrossRef]
- Keane, P.C.; Kurzawa, M.; Blain, P.G.; Morris, C.M. Mitochondrial Dysfunction in Parkinson’s Disease. Park. Dis. 2011, 2011, 1–18. [Google Scholar] [CrossRef]
- Yoo, S.-Z.; No, M.-H.; Heo, J.-W.; Park, D.-H.; Kang, J.-H.; Kim, J.-H.; Seo, D.-Y.; Han, J.; Jung, S.-J.; Kwak, H.-B. Effects of Acute Exercise on Mitochondrial Function, Dynamics, and Mitophagy in Rat Cardiac and Skeletal Muscles. Int. Neurourol. J. 2019, 23, S22–S31. [Google Scholar] [CrossRef] [PubMed]
- Han, G.-S.; Kim, S.-R. Effects of Endurance Exercise on Mitochondrial Function in Mice. J. Phys. Ther. Sci. 2013, 25, 1317–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef]
- Moustafa, A.A.; Chakravarthy, S.; Phillips, J.R.; Gupta, A.; Keri, S.; Polner, B.; Frank, M.J.; Jahanshahi, M. Motor symptoms in Parkinson’s disease: A unified framework. Neurosci. Biobehav. Rev. 2016, 68, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic Criteria for Parkinson Disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef]
- Marsili, L.; Rizzo, G.; Colosimo, C. Diagnostic Criteria for Parkinson’s Disease: From James Parkinson to the Concept of Prodromal Disease. Front. Neurol. 2018, 9, 156. [Google Scholar] [CrossRef]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Goetz, C.G.; Tilley, B.C.; Shaftman, S.R.; Stebbins, G.T.; Fahn, S.; Martinez-Martin, P.; Poewe, W.; Sampaio, C.; Stern, M.B.; Dodel, R.; et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov. Disord. 2008, 23, 2129–2170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Wee, H.L.; Chan, Y.-H.; Seah, S.H.; Au, W.L.; Lau, P.N.; Pica, E.C.; Li, S.C.; Luo, N.; Tan, L.C. Progression of Parkinson’s disease as evaluated by Hoehn and Yahr stage transition times. Mov. Disord. 2010, 25, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Cotogni, M.; Sacchi, L.; Sadikov, A.; Georgiev, D. Asymmetry at Disease Onset Is Not a Predictor of Parkinson’s Disease Progression. J. Park. Dis. 2021, 11, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression and mortality. Neurology 1967, 17, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Hurk, M.V.D.; Lau, S.; Marchetto, M.C.; Mertens, J.; Stern, S.; Corti, O.; Brice, A.; Winner, B.; Winkler, J.; Gage, F.H.; et al. Druggable transcriptomic pathways revealed in Parkinson’s patient-derived midbrain neurons. NPJ Park. Dis. 2022, 8, 1–18. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current Understanding of the Molecular Mechanisms in Parkinson’s Disease: Targets for Potential Treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122 Pt 8, 1437–1448. [Google Scholar] [CrossRef]
- Karasawa, N.; Hayashi, M.; Yamada, K.; Nagatsu, I.; Iwasa, M.; Takeuchi, T.; Uematsu, M.; Watanabe, K.; Onozuka, M. Tyrosine Hydroxylase (TH)- and Aromatic-L-Amino Acid Decarboxylase (AADC)-Immunoreactive Neurons of the Common Marmoset (Callithrix jacchus) Brain: An Immunohistochemical Analysis. Acta Histochem. Cytochem. 2007, 40, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Lohr, K.M.; Chen, M.; Hoffman, C.A.; McDaniel, M.J.; Stout, K.; Dunn, A.; Wang, M.; Bernstein, A.; Miller, G.W. Vesicular Monoamine Transporter 2 (VMAT2) Level Regulates MPTP Vulnerability and Clearance of Excess Dopamine in Mouse Striatal Terminals. Toxicol. Sci. 2016, 153, 79–88. [Google Scholar] [CrossRef]
- Berman, S.B.; Hastings, T.G. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: Implications for Parkinson’s disease. J. Neurochem. 1999, 73, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcia, C.; Barreiro, A.F.; Poza, M.; Herrero, M.-T. Parkinson’s disease and inflammatory changes. Neurotox. Res. 2003, 5, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Wang, W.; Hoppel, C.; Liu, J.; Zhu, X. Parkinson’s disease-associated pathogenic VPS35 mutation causes complex I deficits. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2017, 1863, 2791–2795. [Google Scholar] [CrossRef]
- Mullin, S.; Schapira, A. α-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 2020, 136, 104717. [Google Scholar] [CrossRef]
- Curtis, W.M.; Seeds, W.A.; Mattson, M.P.; Bradshaw, P.C. NADPH and Mitochondrial Quality Control as Targets for a Circadian-Based Fasting and Exercise Therapy for the Treatment of Parkinson’s Disease. Cells 2022, 11, 2416. [Google Scholar] [CrossRef] [PubMed]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin–proteasome system. Open Biol. 2017, 7, 170007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollville, E.; Joers, V.; Nakamura, A.; Swahari, V.; Tansey, M.G.; Moy, S.S.; Deshmukh, M. Characterization of a Cul9–Parkin double knockout mouse model for Parkinson’s disease. Sci. Rep. 2020, 10, 16886. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.B.; Rutter, J. Mitochondrial quality control by the ubiquitin–proteasome system. Biochem. Soc. Trans. 2011, 39, 1509–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, L.; Groh, C.; Herrmann, J.M. The proteasome: Friend and foe of mitochondrial biogenesis. FEBS Lett. 2021, 595, 1223–1238. [Google Scholar] [CrossRef] [PubMed]
- El-Saiy, K.A.; Sayed, R.H.; El-Sahar, A.E.; Kandil, E.A. Modulation of histone deacetylase, the ubiquitin proteasome system, and autophagy underlies the neuroprotective effects of venlafaxine in a rotenone-induced Parkinson’s disease model in rats. Chem. Biol. Interact. 2022, 354, 109841. [Google Scholar] [CrossRef]
- Jang, H.J.; Chung, K.C. The ubiquitin–proteasome system and autophagy mutually interact in neurotoxin-induced dopaminergic cell death models of Parkinson’s disease. FEBS Lett. 2022, 596, 2898–2913. [Google Scholar] [CrossRef]
- Sevenich, M.; Honold, D.; Willuweit, A.; Kutzsche, J.; Mohrluder, J.; Willbold, D. Development of an alpha-synuclein fibril and oligomer specific tracer for diagnosis of Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy. Neurochem. Int. 2022, 161, 105422. [Google Scholar] [CrossRef]
- Asghar, M.; Odeh, A.; Fattahi, A.J.; Henriksson, A.E.; Miglar, A.; Khosousi, S.; Svenningsson, P. Mitochondrial biogenesis, telomere length and cellular senescence in Parkinson’s disease and Lewy body dementia. Sci. Rep. 2022, 12, 17578. [Google Scholar] [CrossRef] [PubMed]
- Bridi, J.; Hirth, F. Mechanisms of α-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef]
- Oczkowska, A.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Mutations in PRKN and SNCA Genes Important for the Progress of Parkinson’s Disease. Curr. Genom. 2013, 14, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Speelman, A.D.; van de Warrenburg, B.P.; van Nimwegen, M.; Petzinger, G.M.; Munneke, M.; Bloem, B.R. How might physical activity benefit patients with Parkinson disease? Nat. Rev. Neurol. 2011, 7, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, L.; Yao, J.; Wang, N.; Liu, D.; Wang, Y.; Liu, D.; Wu, W.; Jiang, T.; Wang, Z. Early implementation of intended exercise improves quality of life in Parkinson’s disease patients. Neurol. Sci. 2022, 43, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Schenkman, M.; Cutson, T.M.; Kuchibhatla, M.; Chandler, J.; Pieper, C.F.; Ray, L.; Laub, K.C. Exercise to improve spinal flexibility and function for people with Parkinson’s disease: A randomized, controlled trial. J. Am. Geriatr. Soc. 1998, 46, 1207–1216. [Google Scholar] [CrossRef]
- Gaßner, H.; Steib, S.; Klamroth, S.; Pasluosta, C.F.; Adler, W.; Eskofier, B.M.; Pfeifer, K.; Winkler, J.; Klucken, J. Perturbation Treadmill Training Improves Clinical Characteristics of Gait and Balance in Parkinson’s Disease. J. Park. Dis. 2019, 9, 413–426. [Google Scholar] [CrossRef]
- Mehrholz, J.; Friis, R.; Kugler, J.; Twork, S.; Storch, A.; Pohl, M. Treadmill training for patients with Parkinson Disease. An abridged version of a Cochrane Review. Eur. J. Phys. Rehabil. Med. 2016, 52, 704–713. [Google Scholar]
- Mehrholz, J.; Kugler, J.; Storch, A.; Pohl, M.; Hirsch, K.; Elsner, B. Treadmill training for patients with Parkinson’s disease. Cochrane Database Syst. Rev. 2015, 2015, CD007830. [Google Scholar] [CrossRef]
- Herman, T.; Giladi, N.; Hausdorff, J.M. Treadmill training for the treatment of gait disturbances in people with Parkinson’s disease: A mini-review. J. Neural Transm. 2009, 116, 307–318. [Google Scholar] [CrossRef]
- Berra, E.; De Icco, R.; Avenali, M.; Dagna, C.; Cristina, S.; Pacchetti, C.; Fresia, M.; Sandrini, G.; Tassorelli, C. Body Weight Support Combined With Treadmill in the Rehabilitation of Parkinsonian Gait: A Review of Literature and New Data From a Controlled Study. Front. Neurol. 2018, 9, 1066. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, M.; Sathyaprabha, T.N.; Gupta, A.; Pal, P.K. Effect of Partial Weight-Supported Treadmill Gait Training on Balance in Patients With Parkinson Disease. PM&R 2014, 6, 22–33. [Google Scholar] [CrossRef]
- Toole, T.; Maitland, C.G.; Warren, E.; Hubmann, M.F.; Panton, L. The effects of loading and unloading treadmill walking on balance, gait, fall risk, and daily function in Parkinsonism. NeuroRehabilitation 2005, 20, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Ustinova, K.; Chernikova, L.; Bilimenko, A.; Telenkov, A.; Epstein, N. Effect of robotic locomotor training in an individual with Parkinson’s disease: A case report. Disabil. Rehabil. Assist. Technol. 2011, 6, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-G.; Yun, S.J.; Shin, H.I.; Kim, E.; Lee, H.H.; Oh, B.-M.; Gil Seo, H. Effects of robot-assisted gait training in patients with Parkinson’s disease: Study protocol for a randomized controlled trial. Trials 2019, 20, 15. [Google Scholar] [CrossRef]
- Van Diest, M.; Lamoth, C.J.C.; Stegenga, J.; Verkerke, G.J.; Postema, K. Exergaming for balance training of elderly: State of the art and future developments. J. Neuroeng. Rehabil. 2013, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirelman, A.; Maidan, I.; Deutsch, J.E. Virtual reality and motor imagery: Promising tools for assessment and therapy in Parkinson’s disease. Mov. Disord. 2013, 28, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Li, C.; Liu, J.; Wang, L.; Ma, J.; Li, G.; Gan, L.; Shang, X.; Wu, Z. Virtual Reality Rehabilitation Versus Conventional Physical Therapy for Improving Balance and Gait in Parkinson’s Disease Patients: A Randomized Controlled Trial. J. Pharmacol. Exp. Ther. 2019, 25, 4186–4192. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, A.; Lungu, O.; Duchesne, C.; Robillard, M.; Bore, A.; Bobeuf, F.; Plamondon, R.; Lafontaine, A.-L.; Gheysen, F.; Bherer, L.; et al. A 12-Week Cycling Training Regimen Improves Gait and Executive Functions Concomitantly in People with Parkinson’s Disease. Front. Hum. Neurosci. 2016, 10, 690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeldt, A.B.; Dey, T.; Alberts, J.L. Aerobic Exercise Preserves Olfaction Function in Individuals with Parkinson’s Disease. Park. Dis. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijders, A.H.; Toni, I.; Ružička, E.; Bloem, B.R. Bicycling breaks the ice for freezers of gait. Mov. Disord. 2011, 26, 367–371. [Google Scholar] [CrossRef]
- Fiorelli, C.M.; Ciolac, E.G.; Simieli, L.; Silva, F.A.; Fernandes, B.; Christofoletti, G.; Barbieri, F.A. Differential Acute Effect of High-Intensity Interval or Continuous Moderate Exercise on Cognition in Individuals With Parkinson’s Disease. J. Phys. Act. Health 2019, 16, 157–164. [Google Scholar] [CrossRef]
- Uygur, M.; Bellumori, M.; Knight, C.A. Effects of a low-resistance, interval bicycling intervention in Parkinson’s Disease. Physiother. Theory Pract. 2017, 33, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, C.; Lungu, O.; Nadeau, A.; Robillard, M.; Boré, A.; Bobeuf, F.; Lafontaine, A.; Gheysen, F.; Bherer, L.; Doyon, J. Enhancing both motor and cognitive functioning in Parkinson’s disease: Aerobic exercise as a rehabilitative intervention. Brain Cogn. 2015, 99, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, C.; Gheysen, F.; Bore, A.; Albouy, G.; Nadeau, A.; Robillard, M.; Bobeuf, F.; Lafontaine, A.; Lungu, O.; Bherer, L.; et al. Influence of aerobic exercise training on the neural correlates of motor learning in Parkinson’s disease individuals. NeuroImage Clin. 2016, 12, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steib, S.; Wanner, P.; Adler, W.; Winkler, J.; Klucken, J.; Pfeifer, K. A Single Bout of Aerobic Exercise Improves Motor Skill Consolidation in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 328. [Google Scholar] [CrossRef]
- Allen, N.E.; Sherrington, C.; Paul, S.S.; Canning, C.G. Balance and falls in Parkinson’s disease: A meta-analysis of the effect of exercise and motor training. Mov. Disord. 2011, 26, 1605–1615. [Google Scholar] [CrossRef]
- Shen, X.; Wong-Yu, I.S.; Mak, M.K. Effects of Exercise on Falls, Balance, and Gait Ability in Parkinson’s Disease: A Meta-analysis. Neurorehabil. Neural Repair 2016, 30, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Pompeu, J.E.; Arduini, L.A.; Botelho, A.R.; Fonseca, M.B.; Pompeu, S.M.; Torriani-Pasin, C.; Deutsch, J.E. Feasibility, safety and outcomes of playing Kinect Adventures! for people with Parkinson’s disease: A pilot study. Physiotherapy 2014, 100, 162–168. [Google Scholar] [CrossRef]
- Barry, G.; Galna, B.; Rochester, L. The role of exergaming in Parkinson’s disease rehabilitation: A systematic review of the evidence. J. Neuroeng. Rehabil. 2014, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Dibble, L.E.; Hale, T.F.; Marcus, R.; Gerber, J.P.; LaStayo, P.C. High intensity eccentric resistance training decreases bradykinesia and improves quality of life in persons with Parkinson’s disease: A preliminary study. Park. Relat. Disord. 2009, 15, 752–757. [Google Scholar] [CrossRef]
- Saltychev, M.; Bärlund, E.; Paltamaa, J.; Katajapuu, N.; Laimi, K. Progressive resistance training in Parkinson’s disease: A systematic review and meta-analysis. BMJ Open 2016, 6, e008756. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, W.; Gao, S. Effects of mind-body exercises on cognitive impairment in people with Parkinson’s disease: A mini-review. Front. Neurol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.P.; Earhart, G.M. Randomized Controlled Trial of Community-Based Dancing to Modify Disease Progression in Parkinson Disease. Neurorehabilit. Neural Repair 2012, 26, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkman, M.; Moore, C.G.; Kohrt, W.M.; Hall, D.A.; Delitto, A.; Comella, C.L.; Josbeno, D.A.; Christiansen, C.L.; Berman, B.D.; Kluger, B.M.; et al. Effect of High-Intensity Treadmill Exercise on Motor Symptoms in Patients With De Novo Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2018, 75, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariati, I.; Masuelli, L.; Bei, R.; Tancredi, V.; Frank, C.; D’Arcangelo, G. Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment. Int. J. Mol. Sci. 2021, 22, 6600. [Google Scholar] [CrossRef]
- Bloomer, R.J.; Schilling, B.K.; Karlage, R.E.; Ledoux, M.S.; Pfeiffer, R.F.; Callegari, J. Effect of Resistance Training on Blood Oxidative Stress in Parkinson Disease. Med. Sci. Sports Exerc. 2008, 40, 1385–1389. [Google Scholar] [CrossRef]
- van der Kolk, N.M.; de Vries, N.M.; Kessels, R.P.C.; Joosten, H.; Zwinderman, A.H.; Post, B.; Bloem, B.R. Effectiveness of home-based and remotely supervised aerobic exercise in Parkinson’s disease: A double-blind, randomised controlled trial. Lancet Neurol. 2019, 18, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Batouli, S.A.H.; Saba, V. At least eighty percent of brain grey matter is modifiable by physical activity: A review study. Behav. Brain Res. 2017, 332, 204–217. [Google Scholar] [CrossRef]
- Pereira, A.C.; Huddleston, D.E.; Brickman, A.M.; Sosunov, A.A.; Hen, R.; McKhann, G.M.; Sloan, R.; Gage, F.H.; Brown, T.R.; Small, S.A. An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. USA 2007, 104, 5638–5643. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. A Unifying Role for Prions in Neurodegenerative Diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Del Tredici, K.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution ofα-synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 33–50. [Google Scholar] [CrossRef]
- Villar-Conde, S.; Astillero-Lopez, V.; Gonzalez-Rodriguez, M.; Villanueva-Anguita, P.; Saiz-Sanchez, D.; Martinez-Marcos, A.; Flores-Cuadrado, A.; Ubeda-Bañon, I. The Human Hippocampus in Parkinson’s Disease: An Integrative Stereological and Proteomic Study. J. Park. Dis. 2021, 11, 1345–1365. [Google Scholar] [CrossRef]
- Yanagisawa, N. Functions and dysfunctions of the basal ganglia in humans. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2018, 94, 275–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzel, S.; Berg, D.; Gasser, T.; Chen, H.; Yao, C.; Postuma, R.B.; the MDS Task Force on the Definition of Parkinson’s Disease. Update of the MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2019, 34, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Tandon, S.D.; Colon, L.; Vega, P.; Murphy, J.; Alonso, A. Birth outcomes associated with receipt of group prenatal care among low-income Hispanic women. J. Midwifery Women’s Health 2012, 57, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Martí, M.J.; Hernández, I.; Valldeoriola, F.; Reñé, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. 2014, 29, 1010–1018. [Google Scholar] [CrossRef]
- Lim, S.-Y.; Fox, S.H.; Lang, A.E. Overview of the Extranigral Aspects of Parkinson Disease. Arch. Neurol. 2009, 66, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Leuchtmann, A.B.; Adak, V.; Dilbaz, S.; Handschin, C. The Role of the Skeletal Muscle Secretome in Mediating Endurance and Resistance Training Adaptations. Front. Physiol. 2021, 12, 1296. [Google Scholar] [CrossRef]
- Swain, R.A.; Berggren, K.L.; Kerr, A.L.; Patel, A.; Peplinski, C.; Sikorski, A.M. On Aerobic Exercise and Behavioral and Neural Plasticity. Brain Sci. 2012, 2, 709–744. [Google Scholar] [CrossRef] [Green Version]
- Hawley, J.A.; Hargreaves, M.; Joyner, M.J.; Zierath, J.R. Integrative Biology of Exercise. Cell 2014, 159, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Bassel-Duby, R.; Olson, E.N. Signaling Pathways in Skeletal Muscle Remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, R.; Cariati, I.; Tarantino, U.; D’Arcangelo, G.; Tancredi, V. Physical Exercise and Health: A Focus on Its Protective Role in Neurodegenerative Diseases. J. Funct. Morphol. Kinesiol. 2022, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Pedersen, M.; Krabbe, K.S.; Bruunsgaard, H.; Matthews, V.B.; Febbraio, M.A. Role of exercise-induced brain-derived neurotrophic factor production in the regulation of energy homeostasis in mammals. Exp. Physiol. 2009, 94, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Pinho, R.A.; Aguiar, A.S., Jr.; Radak, Z. Effects of Resistance Exercise on Cerebral Redox Regulation and Cognition: An Interplay Between Muscle and Brain. Antioxidants 2019, 8, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.-C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.; Greig, N.H.; Mattison, J.A.; et al. Running-Induced Systemic Cathepsin B Secretion Is Associated with Memory Function. Cell Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupr, B.; Handschin, C. Complex Coordination of Cell Plasticity by a PGC-1α-controlled Transcriptional Network in Skeletal Muscle. Front. Physiol. 2015, 6, 325. [Google Scholar] [CrossRef] [Green Version]
- Schnyder, S.; Handschin, C. Skeletal muscle as an endocrine organ: PGC-1alpha, myokines and exercise. Bone 2015, 80, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Delezie, J.; Handschin, C. Endocrine Crosstalk Between Skeletal Muscle and the Brain. Front. Neurol. 2018, 9, 698. [Google Scholar] [CrossRef]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Grilo, L.F.; Diniz, M.S.; Tocantins, C.; Areia, A.L.; Pereira, S.P. The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming? Obesities 2022, 2, 236–255. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, G.; Ghezzi, C.; Sepe, S.; Milanese, C.; Payan-Gomez, C.; Bombardieri, C.R.; Armentero, M.-T.; Zangaglia, R.; Pacchetti, C.; Mastroberardino, P.G.; et al. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1385–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teves, J.M.Y.; Bhargava, V.; Kirwan, K.R.; Corenblum, M.J.; Justiniano, R.; Wondrak, G.T.; Anandhan, A.; Flores, A.J.; Schipper, D.A.; Khalpey, Z.; et al. Parkinson’s Disease Skin Fibroblasts Display Signature Alterations in Growth, Redox Homeostasis, Mitochondrial Function, and Autophagy. Front. Neurosci. 2017, 11, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.; Vitorino, R.; Padrão, A.I.; Espadas, G.; Mancuso, F.M.; Moreira-Gonçalves, D.; Castro-Sousa, G.; Henriques-Coelho, T.; Oliveira, P.A.; Barros, A.S.; et al. Lifelong Exercise Training Modulates Cardiac Mitochondrial Phosphoproteome in Rats. J. Proteome Res. 2014, 13, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Chen, C.C.W.; Hood, D.A. Mitochondria, Muscle Health, and Exercise with Advancing Age. Physiology 2015, 30, 208–223. [Google Scholar] [CrossRef] [Green Version]
- Islam, H.; Hood, D.A.; Gurd, B.J. Looking beyond PGC-1α: Emerging regulators of exercise-induced skeletal muscle mitochondrial biogenesis and their activation by dietary compounds. Appl. Physiol. Nutr. Metab. 2020, 45, 11–23. [Google Scholar] [CrossRef]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef]
- Belardinelli, R.; Georgiou, D.; Cianci, G.; Purcaro, A. 10-year exercise training in chronic heart failure: A randomized controlled trial. J. Am. Coll. Cardiol. 2012, 60, 1521–1528. [Google Scholar] [CrossRef]
- Kachur, S.; Lavie, C.J.; Morera, R.; Ozemek, C.; Milani, R.V. Exercise training and cardiac rehabilitation in cardiovascular disease. Expert Rev. Cardiovasc. Ther. 2019, 17, 585–596. [Google Scholar] [CrossRef]
- Roh, J.; Rhee, J.; Chaudhari, V.; Rosenzweig, A. The Role of Exercise in Cardiac Aging: From Physiology to Molecular Mechanisms. Circ. Res. 2016, 118, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartoni, R.; Leger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Deriaz, O.; Zorzano, A.; et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567 Pt 1, 349–358. [Google Scholar] [CrossRef]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxidative Med. Cell. Longev. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Di Vaia, E.; Iaccarino, G. Physical Exercise: A Novel Tool to Protect Mitochondrial Health. Front. Physiol. 2021, 12, 660068. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K.; Fujita, R.; Seko, D.; Suematsu, T.; Miura, S.; Ono, Y. Distinct Roles of Zmynd17 and PGC1α in Mitochondrial Quality Control and Biogenesis in Skeletal Muscle. Front. Cell Dev. Biol. 2019, 7, 330. [Google Scholar] [CrossRef] [PubMed]
- Fujita, R.; Yoshioka, K.; Seko, D.; Suematsu, T.; Mitsuhashi, S.; Senoo, N.; Miura, S.; Nishino, I.; Ono, Y. Zmynd17 controls muscle mitochondrial quality and whole-body metabolism. FASEB J. 2018, 32, 5012–5025. [Google Scholar] [CrossRef] [Green Version]
- Casuso, R.A.; Huertas, J.R. The emerging role of skeletal muscle mitochondrial dynamics in exercise and ageing. Ageing Res. Rev. 2020, 58, 101025. [Google Scholar] [CrossRef]
- Huertas, J.R.; Ruiz-Ojeda, F.J.; Plaza-Díaz, J.; Nordsborg, N.B.; Martín-Albo, J.; Rueda-Robles, A.; Casuso, R.A. Human muscular mitochondrial fusion in athletes during exercise. FASEB J. 2019, 33, 12087–12098. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, M.; Gunnarsson, T.P.; Hostrup, M.; Iaia, F.M.; Schena, F.; Pilegaard, H.; Bangsbo, J. Metabolic stress-dependent regulation of the mitochondrial biogenic molecular response to high-intensity exercise in human skeletal muscle. J. Physiol. 2018, 596, 2823–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granata, C.; Oliveira, R.S.F.; Little, J.P.; Renner, K.; Bishop, D.J. Sprint-interval but not continuous exercise increases PGC-1α protein content and p53 phosphorylation in nuclear fractions of human skeletal muscle. Sci. Rep. 2017, 7, srep44227. [Google Scholar] [CrossRef] [Green Version]
- Place, N.; Ivarsson, N.; Venckunas, T.; Neyroud, D.; Brazaitis, M.; Cheng, A.J.; Ochala, J.; Kamandulis, S.; Girard, S.; Volungevičius, G.; et al. Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca 2+ leak after one session of high-intensity interval exercise. Proc. Natl. Acad. Sci. USA 2015, 112, 15492–15497. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: The plasticity model. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1837, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Roberts, F.L.; Markby, G.R. New Insights into Molecular Mechanisms Mediating Adaptation to Exercise; A Review Focusing on Mitochondrial Biogenesis, Mitochondrial Function, Mitophagy and Autophagy. Cells 2021, 10, 2639. [Google Scholar] [CrossRef]
- Greggio, C.; Jha, P.; Kulkarni, S.S.; Lagarrigue, S.; Broskey, N.T.; Boutant, M.; Wang, X.; Alonso, S.C.; Ofori, E.; Auwerx, J.; et al. Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab. 2017, 25, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Deus, C.M.; Teixeira, J.; Raimundo, N.; Tucci, P.; Borges, F.; Saso, L.; Oliveira, P.J. Modulation of cellular redox environment as a novel therapeutic strategy for Parkinson’s disease. Eur. J. Clin. Investig. 2022, 52. [Google Scholar] [CrossRef]
- Powers, S.K.; Radak, Z.; Ji, L.L. Exercise-induced oxidative stress: Past, present and future. J. Physiol. 2016, 594, 5081–5092. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magaña, J.C.; Deus, C.M.; Giné-Garriga, M.; Montané, J.; Pereira, S.P. Exercise-Boosted Mitochondrial Remodeling in Parkinson’s Disease. Biomedicines 2022, 10, 3228. https://doi.org/10.3390/biomedicines10123228
Magaña JC, Deus CM, Giné-Garriga M, Montané J, Pereira SP. Exercise-Boosted Mitochondrial Remodeling in Parkinson’s Disease. Biomedicines. 2022; 10(12):3228. https://doi.org/10.3390/biomedicines10123228
Chicago/Turabian StyleMagaña, Juan Carlos, Cláudia M. Deus, Maria Giné-Garriga, Joel Montané, and Susana P. Pereira. 2022. "Exercise-Boosted Mitochondrial Remodeling in Parkinson’s Disease" Biomedicines 10, no. 12: 3228. https://doi.org/10.3390/biomedicines10123228
APA StyleMagaña, J. C., Deus, C. M., Giné-Garriga, M., Montané, J., & Pereira, S. P. (2022). Exercise-Boosted Mitochondrial Remodeling in Parkinson’s Disease. Biomedicines, 10(12), 3228. https://doi.org/10.3390/biomedicines10123228