
Autophagy in Extracellular Matrix and Wound Healing Modulation in the Cornea

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
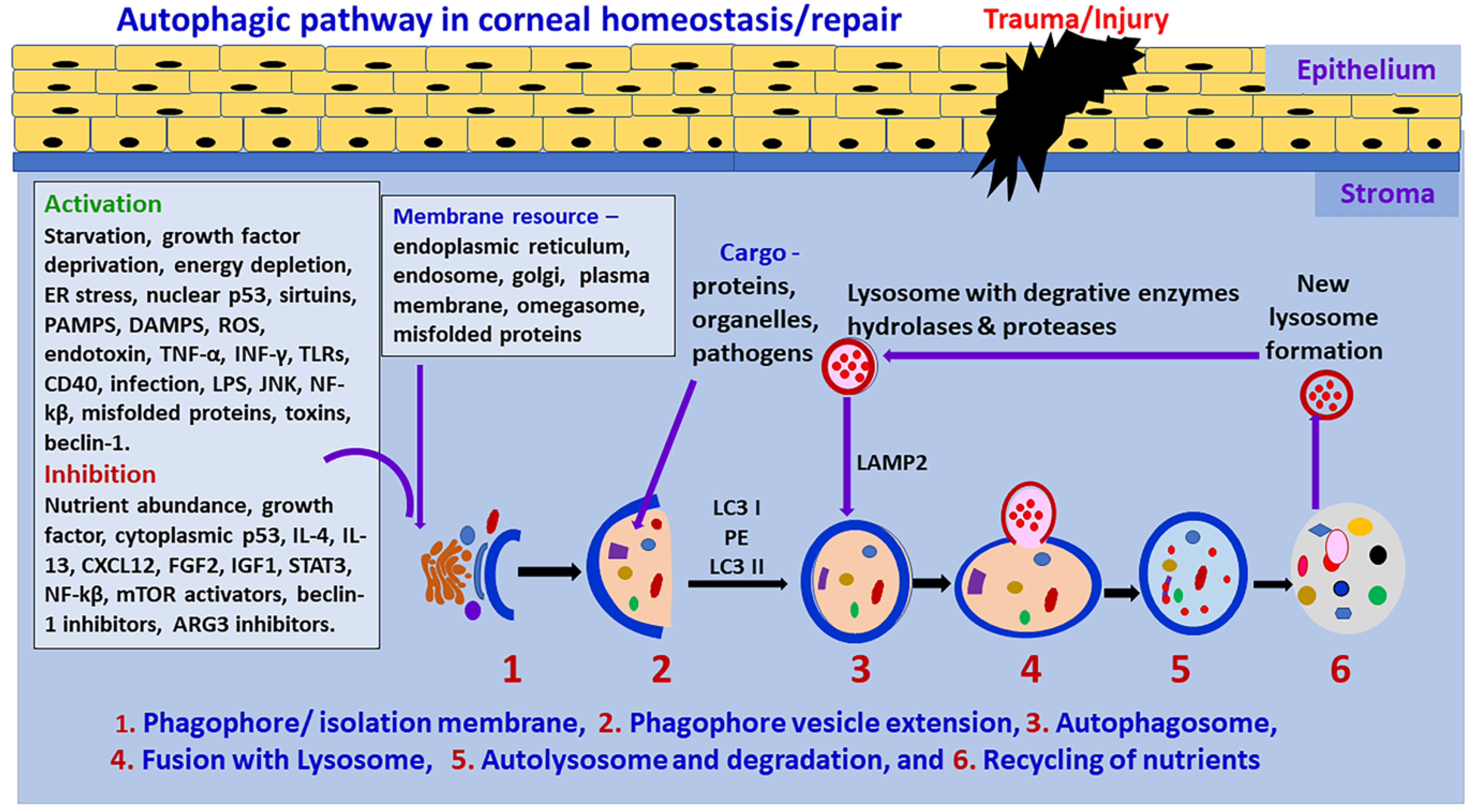
2. Autophagy
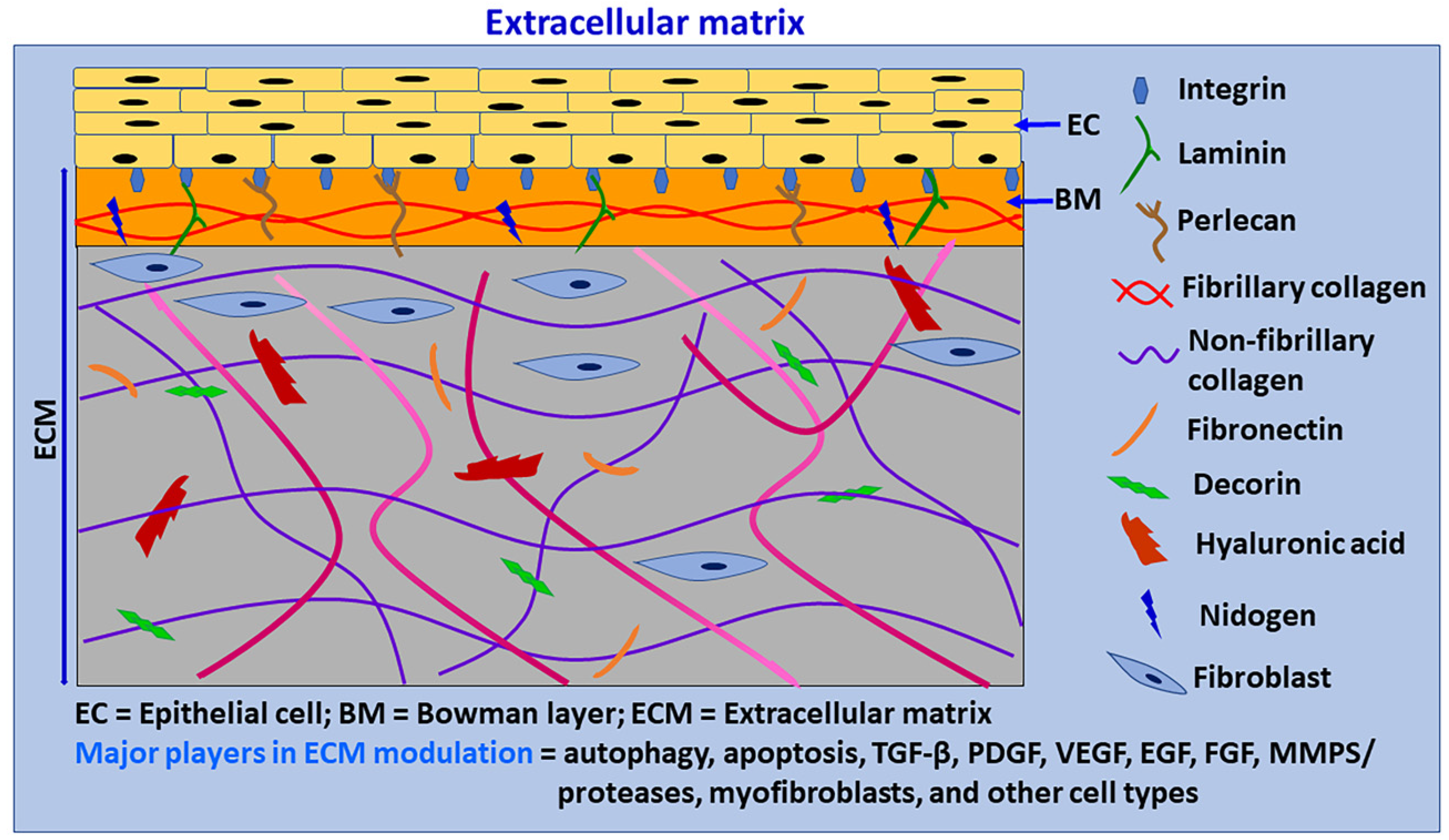
3. Extracellular Matrix
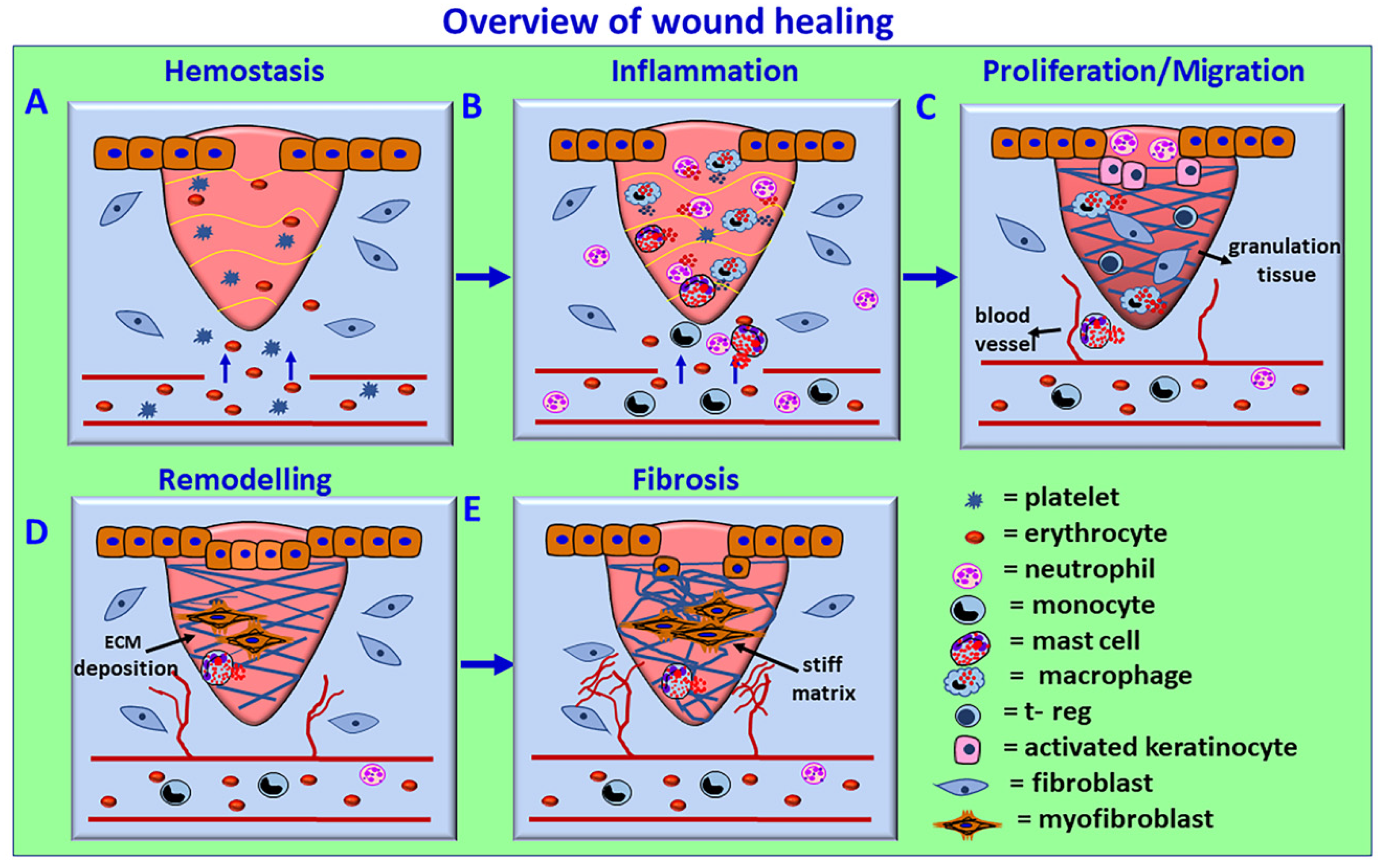
4. Wound Healing
5. Mast Cells in Wound Healing and Fibrosis
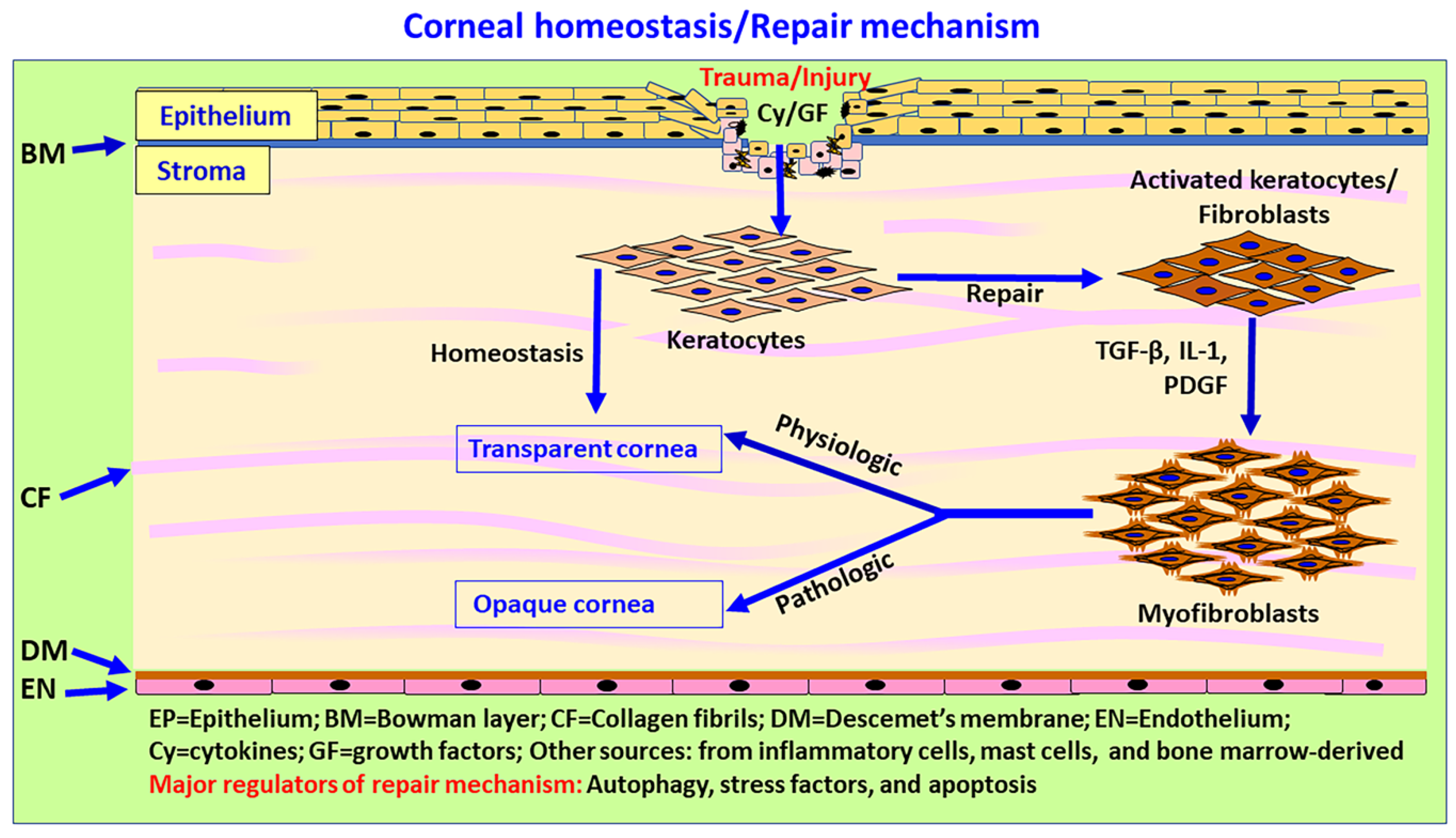
6. Corneal Wound Healing and Repair
7. Dysregulated or Delayed Autophagy and Its Late Effects
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chang, N.C. Autophagy and Stem Cells: Self-Eating for Self-Renewal. Front. Cell Dev. Biol. 2020, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Das, J.K.; Kumar, A.; Peng, H.Y.; Ren, Y.; Xiong, X.; Yang, J.M.; Song, J. Autophagy in T-cell differentiation, survival and memory. Immunol. Cell Biol. 2021, 99, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef]
- Belgrad, J.; De Pace, R.; Fields, R.D. Autophagy in Myelinating Glia. J. Neurosci. 2020, 40, 256–266. [Google Scholar] [CrossRef]
- Fernandez-Albarral, J.A.; de Julian-Lopez, E.; Soler-Dominguez, C.; de Hoz, R.; Lopez-Cuenca, I.; Salobrar-Garcia, E.; Ramirez, J.M.; Pinazo-Duran, M.D.; Salazar, J.J.; Ramirez, A.I. The Role of Autophagy in Eye Diseases. Life 2021, 11, 189. [Google Scholar] [CrossRef]
- Ma, S.; Yu, Z.; Feng, S.; Chen, H.; Chen, H.; Lu, X. Corneal autophagy and ocular surface inflammation: A new perspective in dry eye. Exp. Eye Res. 2019, 184, 126–134. [Google Scholar] [CrossRef]
- Martin, L.M.; Jeyabalan, N.; Tripathi, R.; Panigrahi, T.; Johnson, P.J.; Ghosh, A.; Mohan, R.R. Autophagy in corneal health and disease: A concise review. Ocul. Surf. 2019, 17, 186–197. [Google Scholar] [CrossRef]
- Frost, L.S.; Mitchell, C.H.; Boesze-Battaglia, K. Autophagy in the eye: Implications for ocular cell health. Exp. Eye Res. 2014, 124, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Chai, P.; Ni, H.; Zhang, H.; Fan, X. The Evolving Functions of Autophagy in Ocular Health: A Double-edged Sword. Int. J. Biol. Sci. 2016, 12, 1332–1340. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Esteban-Martinez, L.; Serrano-Puebla, A.; Gomez-Sintes, R.; Villarejo-Zori, B. Autophagy in the eye: Development, degeneration, and aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef] [PubMed]
- Kamil, S.; Mohan, R.R. Corneal stromal wound healing: Major regulators and therapeutic targets. Ocul. Surf. 2021, 19, 290–306. [Google Scholar] [CrossRef]
- Netto, M.V.; Mohan, R.R.; Ambrosio, R., Jr.; Hutcheon, A.E.; Zieske, J.D.; Wilson, S.E. Wound healing in the cornea: A review of refractive surgery complications and new prospects for therapy. Cornea 2005, 24, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Pompili, S.; Latella, G.; Gaudio, E.; Sferra, R.; Vetuschi, A. The Charming World of the Extracellular Matrix: A Dynamic and Protective Network of the Intestinal Wall. Front. Med. 2021, 8, 610189. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123 Pt 24, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Chaurasia, S.S.; Lim, R.R.; Lakshminarayanan, R.; Mohan, R.R. Nanomedicine approaches for corneal diseases. J. Funct. Biomater. 2015, 6, 277–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, R.R.; Martin, L.M.; Sinha, N.R. Novel insights into gene therapy in the cornea. Exp. Eye Res. 2021, 202, 108361. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Sharma, A.; Netto, M.V.; Sinha, S.; Wilson, S.E. Gene therapy in the cornea. Prog. Retin. Eye Res. 2005, 24, 537–559. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Rodier, J.T.; Sharma, A. Corneal gene therapy: Basic science and translational perspective. Ocul. Surf. 2013, 11, 150–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espana, E.M.; Birk, D.E. Composition, structure and function of the corneal stroma. Exp. Eye Res. 2020, 198, 108137. [Google Scholar] [CrossRef]
- Chen, S.; Mienaltowski, M.J.; Birk, D.E. Regulation of corneal stroma extracellular matrix assembly. Exp. Eye Res. 2015, 133, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Pouw, A.E.; Greiner, M.A.; Coussa, R.G.; Jiao, C.; Han, I.C.; Skeie, J.M.; Fingert, J.H.; Mullins, R.F.; Sohn, E.H. Cell-Matrix Interactions in the Eye: From Cornea to Choroid. Cells 2021, 10, 687. [Google Scholar] [CrossRef]
- Sylakowski, K.; Wells, A. ECM-regulation of autophagy: The yin and the yang of autophagy during wound healing. Matrix Biol. 2021, 100–101, 197–206. [Google Scholar] [CrossRef]
- Almadani, Y.H.; Vorstenbosch, J.; Davison, P.G.; Murphy, A.M. Wound Healing: A Comprehensive Review. Semin. Plast. Surg. 2021, 35, 141–144. [Google Scholar] [CrossRef]
- Flanagan, G.; Velez, T.; Gu, W.; Singman, E. The Relationship between Severe Visual Acuity Loss, Traumatic Brain Injuries, and Ocular Injuries in American Service Members from 2001 to 2015. Mil. Med. 2020, 185, e1576–e1583. [Google Scholar] [CrossRef]
- Balne, P.K.; Sinha, N.R.; Hofmann, A.C.; Martin, L.M.; Mohan, R.R. Characterization of hydrogen sulfide toxicity to human corneal stromal fibroblasts. Ann. N. Y. Acad. Sci. 2020, 1480, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Fink, M.K.; Martin, L.M.; Sinha, P.R.; Rodier, J.T.; Sinha, N.R.; Hesemann, N.P.; Chaurasia, S.S.; Mohan, R.R. A rabbit model for evaluating ocular damage from acrolein toxicity in vivo. Ann. N. Y. Acad. Sci. 2020, 1480, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Giuliano, E.A.; Sinha, N.R.; Mohan, R.R. Ocular toxicity of mustard gas: A concise review. Toxicol. Lett. 2021, 343, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Rasiah, P.K.; Geier, B.; Jha, K.A.; Gangaraju, R. Visual deficits after traumatic brain injury. Histol. Histopathol. 2021, 36, 711–724. [Google Scholar] [PubMed]
- Tripathi, R.; Balne, P.K.; Sinha, N.R.; Martin, L.M.; Kamil, S.; Landreneau, J.R.; Gupta, S.; Rodier, J.T.; Sinha, P.R.; Hesemann, N.P.; et al. A Novel Topical Ophthalmic Formulation to Mitigate Acute Mustard Gas Keratopathy in vivo: A Pilot Study. Transl. Vis. Sci. Technol. 2020, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- McNutt, P.M.; Mohan, R.R. The Need for Improved Therapeutic Approaches to Protect the Cornea against Chemotoxic Injuries. Transl. Vis. Sci. Technol. 2020, 9, 2. [Google Scholar] [CrossRef]
- Mohan, R.R.; Tovey, J.C.; Gupta, R.; Sharma, A.; Tandon, A. Decorin biology, expression, function and therapy in the cornea. Curr. Mol. Med. 2011, 11, 110–128. [Google Scholar] [CrossRef]
- Ljubimov, A.V.; Saghizadeh, M. Progress in corneal wound healing. Prog. Retin. Eye Res. 2015, 49, 17–45. [Google Scholar] [CrossRef] [Green Version]
- De Munck, D.G.; De Meyer, G.R.; Martinet, W. Autophagy as an emerging therapeutic target for age-related vascular pathologies. Expert Opin. Ther. Targets 2020, 24, 131–145. [Google Scholar] [CrossRef]
- Wu, J.; Lipinski, M.M. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells 2019, 8, 693. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Park, J.K.; Lavker, R.M. Autophagy and Macropinocytosis: Keeping an Eye on the Corneal/Limbal Epithelia. Investig. Ophthalmol. Vis. Sci. 2017, 58, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Santeford, A.; Wiley, L.A.; Park, S.; Bamba, S.; Nakamura, R.; Gdoura, A.; Ferguson, T.A.; Rao, P.K.; Guan, J.L.; Saitoh, T.; et al. Impaired autophagy in macrophages promotes inflammatory eye disease. Autophagy 2016, 12, 1876–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, T.A.; Laurie, G.W. Introduction to Autophagy in the Eye (or “What’s Eatin’ You?”). Exp. Eye Res. 2016, 144, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio, H.; Ueno, T.; Kojima, Y.; Komatsu, M.; Tanaka, S.; Yamamoto, A.; Ichimura, Y.; Ezaki, J.; Nishida, K.; Komazawa-Sakon, S.; et al. Crucial role for autophagy in degranulation of mast cells. J. Allergy Clin. Immunol. 2011, 127, 1267–1276.e1266. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Ushio, H. An unexpected role for autophagy in degranulation of mast cells. Autophagy 2011, 7, 657–659. [Google Scholar] [CrossRef] [Green Version]
- Rozman, S.; Yousefi, S.; Oberson, K.; Kaufmann, T.; Benarafa, C.; Simon, H.U. The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Differ. 2015, 22, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Botbol, Y.; Guerrero-Ros, I.; Macian, F. Key roles of autophagy in regulating T-cell function. Eur. J. Immunol. 2016, 46, 1326–1334. [Google Scholar] [CrossRef] [Green Version]
- Stepp, M.A.; Menko, A.S. Immune responses to injury and their links to eye disease. Transl. Res. 2021, 236, 52–71. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, D.; Chen, X.; Bian, F.; Gao, N.; Li, J.; Pflugfelder, S.C.; Li, D.Q. Autophagy Activation Protects Ocular Surface from Inflammation in a Dry Eye Model in vitro. Int. J. Mol. Sci. 2020, 21, 8966. [Google Scholar] [CrossRef]
- Wu, T.T.; Li, W.M.; Yao, Y.M. Interactions between Autophagy and Inhibitory Cytokines. Int. J. Biol. Sci. 2016, 12, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Kim, J.H.; Byun, S. Modulation of Autophagy for Controlling Immunity. Cells 2019, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portillo, J.A.; Okenka, G.; Reed, E.; Subauste, A.; Van Grol, J.; Gentil, K.; Komatsu, M.; Tanaka, K.; Landreth, G.; Levine, B.; et al. The CD40-autophagy pathway is needed for host protection despite IFN-Gamma-dependent immunity and CD40 induces autophagy via control of P21 levels. PLoS ONE 2010, 5, e14472. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, M.S. Anatomy of cornea and ocular surface. Indian J. Ophthalmol. 2018, 66, 190–194. [Google Scholar] [PubMed]
- Schaefer, L.; Dikic, I. Autophagy: Instructions from the extracellular matrix. Matrix Biol. 2021, 100–101, 1–8. [Google Scholar] [CrossRef]
- Mohan, R.R.; Tandon, A.; Sharma, A.; Cowden, J.W.; Tovey, J.C. Significant inhibition of corneal scarring in vivo with tissue-selective, targeted AAV5 decorin gene therapy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4833–4841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balne, P.K.; Gupta, S.; Zhang, J.; Bristow, D.; Faubion, M.; Heil, S.D.; Sinha, P.R.; Green, S.L.; Iozzo, R.V.; Mohan, R.R. The functional role of decorin in corneal neovascularization in vivo. Exp. Eye Res. 2021, 207, 108610. [Google Scholar] [CrossRef]
- Mohan, R.R.; Tripathi, R.; Sharma, A.; Sinha, P.R.; Giuliano, E.A.; Hesemann, N.P.; Chaurasia, S.S. Decorin antagonizes corneal fibroblast migration via caveolae-mediated endocytosis of epidermal growth factor receptor. Exp. Eye Res. 2019, 180, 200–207. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin causes autophagy in endothelial cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef] [Green Version]
- Mobaraki, M.; Abbasi, R.; Omidian Vandchali, S.; Ghaffari, M.; Moztarzadeh, F.; Mozafari, M. Corneal Repair and Regeneration: Current Concepts and Future Directions. Front. Bioeng. Biotechnol. 2019, 7, 135. [Google Scholar] [CrossRef] [Green Version]
- Shah, J.M.; Omar, E.; Pai, D.R.; Sood, S. Cellular events and biomarkers of wound healing. Indian J. Plast. Surg. 2012, 45, 220–228. [Google Scholar]
- Sabbatini, M.; Magnelli, V.; Reno, F. NETosis in Wound Healing: When Enough Is Enough. Cells 2021, 10, 494. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, Y.; Aziz, M.; Wang, P. Role of reverse transendothelial migration of neutrophils in inflammation. Biol. Chem. 2016, 397, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huang, Y.; Ji, Q.; Fu, S.; Gu, J.; Tai, N.; Wang, X. Interplay between Extracellular Matrix and Neutrophils in Diseases. J. Immunol. Res. 2021, 2021, 8243378. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.L.; Vannan, D.T.; Eksteen, B.; Avelar, I.J.; Rodriguez, T.; Gonzalez, M.I.; Mendoza, A.V. Innate and Adaptive Cell Populations Driving Inflammation in Dry Eye Disease. Mediat. Inflamm. 2018, 2018, 2532314. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Zheng, J.; Huang, X.; Zhang, Y.; Han, Y.; Hu, R.; Jin, X. Neutrophil extracellular traps promote corneal neovascularization-induced by alkali burn. Int. Immunopharmacol. 2020, 88, 106902. [Google Scholar] [CrossRef]
- Lorenzo-Martin, E.; Gallego-Munoz, P.; Mar, S.; Fernandez, I.; Cidad, P.; Martinez-Garcia, M.C. Dynamic changes of the extracellular matrix during corneal wound healing. Exp. Eye Res. 2019, 186, 107704. [Google Scholar] [CrossRef]
- Stuard, W.L.; Titone, R.; Robertson, D.M. The IGF/Insulin-IGFBP Axis in Corneal Development, Wound Healing, and Disease. Front. Endocrinol. 2020, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Smolin, G. Cellular response to inflammation at the limbus. Eye 1989, 3 Pt 2, 167–171. [Google Scholar] [CrossRef]
- Seyed-Safi, A.G.; Daniels, J.T. The limbus: Structure and function. Exp. Eye Res. 2020, 197, 108074. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Zhang, Z.; Kurashima, Y. Two Sides of the Coin: Mast Cells as a Key Regulator of Allergy and Acute/Chronic Inflammation. Cells 2021, 10, 1615. [Google Scholar] [CrossRef]
- Elieh Ali Komi, D.; Wohrl, S.; Bielory, L. Mast Cell Biology at Molecular Level: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2020, 58, 342–365. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.B.; Stahl, J.L.; Barney, N.P.; Graziano, F.M. Ocular mast cells. Characterization in normal and disease states. Clin. Rev. Allergy Immunol. 2001, 20, 243–268. [Google Scholar] [CrossRef]
- Levin, L.A.; Albert, D.M.; Johnson, D. Mast cells in human optic nerve. Investig. Ophthalmol. Vis. Sci. 1993, 34, 3147–3153. [Google Scholar]
- Cho, W.; Mittal, S.K.; Elbasiony, E.; Chauhan, S.K. Activation of ocular surface mast cells promotes corneal neovascularization. Ocul. Surf. 2020, 18, 857–864. [Google Scholar] [CrossRef]
- Irani, A.M. Ocular mast cells and mediators. Immunol. Allergy Clin. N. Am. 2008, 28, 25–42. [Google Scholar] [CrossRef]
- Liu, J.; Li, Z. Resident Innate Immune Cells in the Cornea. Front. Immunol. 2021, 12, 620284. [Google Scholar] [CrossRef]
- Micera, A.; Jirsova, K.; Esposito, G.; Balzamino, B.O.; Di Zazzo, A.; Bonini, S. Mast Cells Populate the Corneoscleral Limbus: New Insights for Our Understanding of Limbal Microenvironment. Investig. Ophthalmol. Vis. Sci. 2020, 61, 43. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Selvakumar, G.P.; Thangavel, R.; Ahmed, M.E.; Zaheer, S.; Raikwar, S.P.; Iyer, S.S.; Bhagavan, S.M.; Beladakere-Ramaswamy, S.; Zaheer, A. Mast Cell Activation in Brain Injury, Stress, and Post-traumatic Stress Disorder and Alzheimer’s Disease Pathogenesis. Front. Neurosci. 2017, 11, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhang, X.; Qian, Y. Mast cells and neuroinflammation. Med. Sci. Monit. Basic Res. 2014, 20, 200–206. [Google Scholar] [PubMed] [Green Version]
- Tsai, M.; Grimbaldeston, M.; Galli, S.J. Mast cells and immunoregulation/immunomodulation. Adv. Exp. Med. Biol. 2011, 716, 186–211. [Google Scholar] [PubMed]
- Church, M.K.; McGill, J.I. Human ocular mast cells. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 419–422. [Google Scholar] [CrossRef]
- Patel, D.; Sarala, N.; Datti, N.P. Topical Olopatadine Hydrochloride versus Ketotifen Fumarate for Allergic Conjunctivitis. J. Ophthalmic. Vis. Res. 2018, 13, 119–123. [Google Scholar]
- Mello-Bosnic, C.; Gimenes, A.D.; Oliani, S.M.; Gil, C.D. Treatment with galectin-1 eye drops regulates mast cell degranulation and attenuates the severity of conjunctivitis. Eur. J. Pharmacol. 2018, 833, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Elieh Ali Komi, D.; Rambasek, T.; Bielory, L. Clinical implications of mast cell involvement in allergic conjunctivitis. Allergy 2018, 73, 528–539. [Google Scholar] [CrossRef]
- Bielory, L.; Kempuraj, D.; Theoharides, T. Topical immunopharmacology of ocular allergies. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 435–445. [Google Scholar] [CrossRef]
- Kempuraj, D.; Huang, M.; Kandere, K.; Boucher, W.; Letourneau, R.; Jeudy, S.; Fitzgerald, K.; Spear, K.; Athanasiou, A.; Theoharides, T.C. Azelastine is more potent than olopatadine in inhibiting interleukin-6 and tryptase release from human umbilical cord blood-derived cultured mast cells. Ann. Allergy Asthma Immunol. 2002, 88, 501–506. [Google Scholar] [CrossRef]
- Mounsey, A.L.; Gray, R.E. Topical Antihistamines and Mast Cell Stabilizers for Treating Allergic Conjunctivitis. Am. Fam. Physician 2016, 93, 915–916. [Google Scholar]
- Cho, W.; Mittal, S.K.; Elbasiony, E.; Chauhan, S.K. Spatial Distribution of Mast Cells Regulates Asymmetrical Angiogenesis at the Ocular Surface. Am. J. Pathol. 2021, 191, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.K.; Mittal, S.K.; Foulsham, W.; Li, M.; Sangwan, V.S.; Chauhan, S.K. Mast Cells Initiate the Recruitment of Neutrophils Following Ocular Surface Injury. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Wulff, B.C.; Wilgus, T.A. Mast cell activity in the healing wound: More than meets the eye? Exp. Dermatol. 2013, 22, 507–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Khomtchouk, K.; Santa Maria, P.L. A Review of the Contribution of Mast Cells in Wound Healing: Involved Molecular and Cellular Mechanisms. Clin. Rev. Allergy Immunol. 2020, 58, 298–312. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Ronconi, G.; Kritas, S.K.; Mastrangelo, F.; Tettamanti, L.; Frydas, I.; Theoharides, T.C. Mast cells participate in allograft rejection: Can IL-37 play an inhibitory role? Inflamm. Res. 2018, 67, 747–755. [Google Scholar] [CrossRef]
- Strattan, E.; Hildebrandt, G.C. Mast Cell Involvement in Fibrosis in Chronic Graft-Versus-Host Disease. Int. J. Mol. Sci. 2021, 22, 2385. [Google Scholar] [CrossRef]
- Crivellato, E.; Nico, B.; Ribatti, D. Mast cells and tumour angiogenesis: New insight from experimental carcinogenesis. Cancer Lett. 2008, 269, 1–6. [Google Scholar] [CrossRef]
- Ng, M.F. The role of mast cells in wound healing. Int. Wound J. 2010, 7, 55–61. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef]
- Barrientez, B.; Nicholas, S.E.; Whelchel, A.; Sharif, R.; Hjortdal, J.; Karamichos, D. Corneal injury: Clinical and molecular aspects. Exp. Eye Res. 2019, 186, 107709. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E. Corneal wound healing. Exp. Eye Res. 2020, 197, 108089. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E. Corneal myofibroblasts and fibrosis. Exp. Eye Res. 2020, 201, 108272. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.R.; Du, J.H. Autophagy: A potential target for the treatment of intraocular neovascularization. Int. J. Ophthalmol. 2018, 11, 695–698. [Google Scholar] [PubMed]
- Mohan, R.R.; Tovey, J.C.; Sharma, A.; Schultz, G.S.; Cowden, J.W.; Tandon, A. Targeted decorin gene therapy delivered with adeno-associated virus effectively retards corneal neovascularization in vivo. PLoS ONE 2011, 6, e26432. [Google Scholar] [CrossRef] [Green Version]
- Vlasov, A.; Ryan, D.S.; Ludlow, S.; Coggin, A.; Weichel, E.D.; Stutzman, R.D.; Bower, K.S.; Colyer, M.H. Corneal and Corneoscleral Injury in Combat Ocular Trauma from Operations Iraqi Freedom and Enduring Freedom. Mil. Med. 2017, 182 (Suppl. 1), 114–119. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Ahmed, M.E.; Selvakumar, G.P.; Thangavel, R.; Dhaliwal, A.S.; Dubova, I.; Mentor, S.; Premkumar, K.; Saeed, D.; Zahoor, H.; et al. Brain Injury-Mediated Neuroinflammatory Response and Alzheimer’s Disease. Neuroscientist 2019, 26, 134–155. [Google Scholar] [CrossRef]
- DePalma, R.G.; Hoffman, S.W. Combat blast related traumatic brain injury (TBI): Decade of recognition; promise of progress. Behav. Brain Res. 2018, 340, 102–105. [Google Scholar] [CrossRef]
- Edwards, G., 3rd; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Kokiko-Cochran, O.N.; Godbout, J.P. The Inflammatory Continuum of Traumatic Brain Injury and Alzheimer’s Disease. Front. Immunol. 2018, 9, 672. [Google Scholar] [CrossRef]
- Hay, J.R.; Johnson, V.E.; Young, A.M.; Smith, D.H.; Stewart, W. Blood-Brain Barrier Disruption Is an Early Event That May Persist for Many Years after Traumatic Brain Injury in Humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar] [PubMed] [Green Version]
- Cockerham, G.C.; Lemke, S.; Rice, T.A.; Wang, G.; Glynn-Milley, C.; Zumhagen, L.; Cockerham, K.P. Closed-globe injuries of the ocular surface associated with combat blast exposure. Ophthalmology 2014, 121, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Cockerham, G.C.; Goodrich, G.L.; Weichel, E.D.; Orcutt, J.C.; Rizzo, J.F.; Bower, K.S.; Schuchard, R.A. Eye and visual function in traumatic brain injury. J. Rehabil. Res. Dev. 2009, 46, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Felix, E.R.; Levitt, R.C.; Eddy, C.; Vanner, E.A.; Feuer, W.J.; Sarantopoulos, C.D.; Galor, A. Traumatic brain injury, dry eye and comorbid pain diagnoses in US veterans. Br. J. Ophthalmol. 2018, 102, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. Visual problems associated with traumatic brain injury. Clin. Exp. Optom. 2018, 101, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kamil, S.; Sinha, P.R.; Rodier, J.T.; Chaurasia, S.S.; Mohan, R.R. Glutathione is a potential therapeutic target for acrolein toxicity in the cornea. Toxicol. Lett. 2021, 340, 33–42. [Google Scholar] [CrossRef]
- Rai, D.K.; Sharma, B. Carbofuran-induced oxidative stress in mammalian brain. Mol. Biotechnol. 2007, 37, 66–71. [Google Scholar] [CrossRef]
- Charkoftaki, G.; Jester, J.V.; Thompson, D.C.; Vasiliou, V. Nitrogen mustard-induced corneal injury involves the sphingomyelin-ceramide pathway. Ocul. Surf. 2018, 16, 154–162. [Google Scholar] [CrossRef]
- Gupta, R.C. Carbofuran toxicity. J. Toxicol. Environ. Health 1994, 43, 383–418. [Google Scholar] [CrossRef]
- Goswami, D.G.; Tewari-Singh, N.; Agarwal, R. Corneal toxicity induced by vesicating agents and effective treatment options. Ann. N. Y. Acad. Sci. 2016, 1374, 193–201. [Google Scholar] [CrossRef]
- Achanta, S.; Jordt, S.E. Toxic effects of chlorine gas and potential treatments: A literature review. Toxicol. Mech. Methods 2021, 31, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Govier, P.; Coulson, J.M. Civilian exposure to chlorine gas: A systematic review. Toxicol. Lett. 2018, 293, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.K.; Hom, G.G.; Fernandez, C.; Hom, L.G. Ocular effects of exposure to industrial chemicals: Clinical management and proteomic approaches to damage assessment. Cutan. Ocul. Toxicol. 2007, 26, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, M. Chemical warfare agents. Classes and targets. Toxicol. Lett. 2018, 293, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Kim, E.K. Autophagy in granular corneal dystrophy type 2. Exp. Eye Res. 2016, 144, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, S.; Panigrahi, T.; Shetty, R.; Subramani, M.; Ghosh, A.; Jeyabalan, N. Chloroquine Protects Human Corneal Epithelial Cells from Desiccation Stress Induced Inflammation without Altering the Autophagy Flux. Biomed. Res. Int. 2018, 2018, 7627329. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Chacon, G.; Vela, F.J.; Campos, J.L.; Abellan, E.; Yakhine-Diop, S.M.S.; Ballestin, A. Autophagy modulation in animal models of corneal diseases: A systematic review. Mol. Cell Biochem. 2020, 474, 41–55. [Google Scholar] [CrossRef]
- Shetty, R.; Sharma, A.; Pahuja, N.; Chevour, P.; Padmajan, N.; Dhamodaran, K.; Jayadev, C.; Nuijts, R.M.M.A.; Ghosh, A.; Nallathambi, J. Oxidative stress induces dysregulated autophagy in corneal epithelium of keratoconus patients. PLoS ONE 2017, 12, e0184628. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Wu, X.; Feng, D.; Luo, J.; Shi, Y.; Guo, W.; Liu, H.; Wang, Q.; Wang, L.; Ge, S.; et al. Acrolein Induces Systemic Coagulopathy via Autophagy-dependent Secretion of von Willebrand Factor in Mice after Traumatic Brain Injury. Neurosci. Bull. 2021, 37, 1160–1175. [Google Scholar] [CrossRef]
- Zhang, Y.B.; Li, S.X.; Chen, X.P.; Yang, L.; Zhang, Y.G.; Liu, R.; Tao, L.Y. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci. Bull. 2008, 24, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Au, A.K.; Aneja, R.K.; Bayir, H.; Bell, M.J.; Janesko-Feldman, K.; Kochanek, P.M.; Clark, R.S.B. Autophagy Biomarkers Beclin 1 and p62 are Increased in Cerebrospinal Fluid after Traumatic Brain Injury. Neurocrit. Care 2017, 26, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Zhao, Z.; Aungst, S.; Sabirzhanov, B.; Faden, A.I.; Lipinski, M.M. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 2014, 10, 2208–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, C.; Jones, J.W.; Hegdekar, N.; Thayer, J.A.; Kumar, A.; Faden, A.I.; Kane, M.A.; Lipinski, M.M. PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma. Autophagy 2020, 16, 466–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kempuraj, D.; Mohan, R.R. Autophagy in Extracellular Matrix and Wound Healing Modulation in the Cornea. Biomedicines 2022, 10, 339. https://doi.org/10.3390/biomedicines10020339
Kempuraj D, Mohan RR. Autophagy in Extracellular Matrix and Wound Healing Modulation in the Cornea. Biomedicines. 2022; 10(2):339. https://doi.org/10.3390/biomedicines10020339
Chicago/Turabian StyleKempuraj, Duraisamy, and Rajiv R. Mohan. 2022. "Autophagy in Extracellular Matrix and Wound Healing Modulation in the Cornea" Biomedicines 10, no. 2: 339. https://doi.org/10.3390/biomedicines10020339
APA StyleKempuraj, D., & Mohan, R. R. (2022). Autophagy in Extracellular Matrix and Wound Healing Modulation in the Cornea. Biomedicines, 10(2), 339. https://doi.org/10.3390/biomedicines10020339