
HDAC9 Contributes to Serous Ovarian Cancer Progression through Regulating Epithelial–Mesenchymal Transition

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Plasmids and Transfections
2.3. Ovarian Cancer Tissues Microarray and Immunohistochemistry
2.4. RNA Extraction and Quantitative Real-Time PCR
2.5. Western Blot
2.6. Wound-Healing Assay
2.7. Clonogenic Assay
2.8. Transwell Assay
2.9. Flow Cytometry
2.10. HDAC9 Enzymatic Activity Measurement
2.11. Immunofluorescence Staining
2.12. Statistical Analysis
3. Results
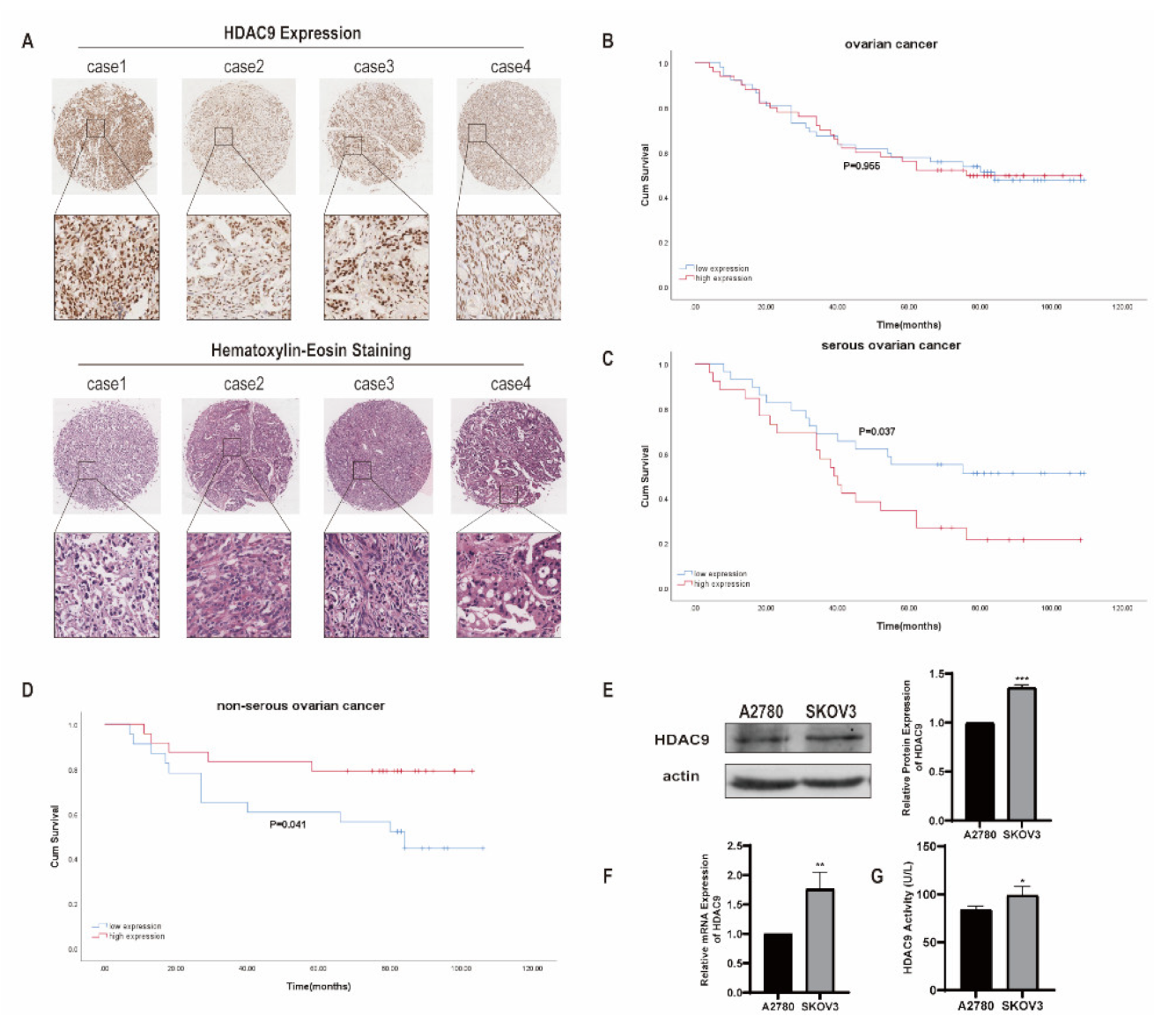
3.1. HDAC9 Correlates with the Prognosis of Ovarian Cancer
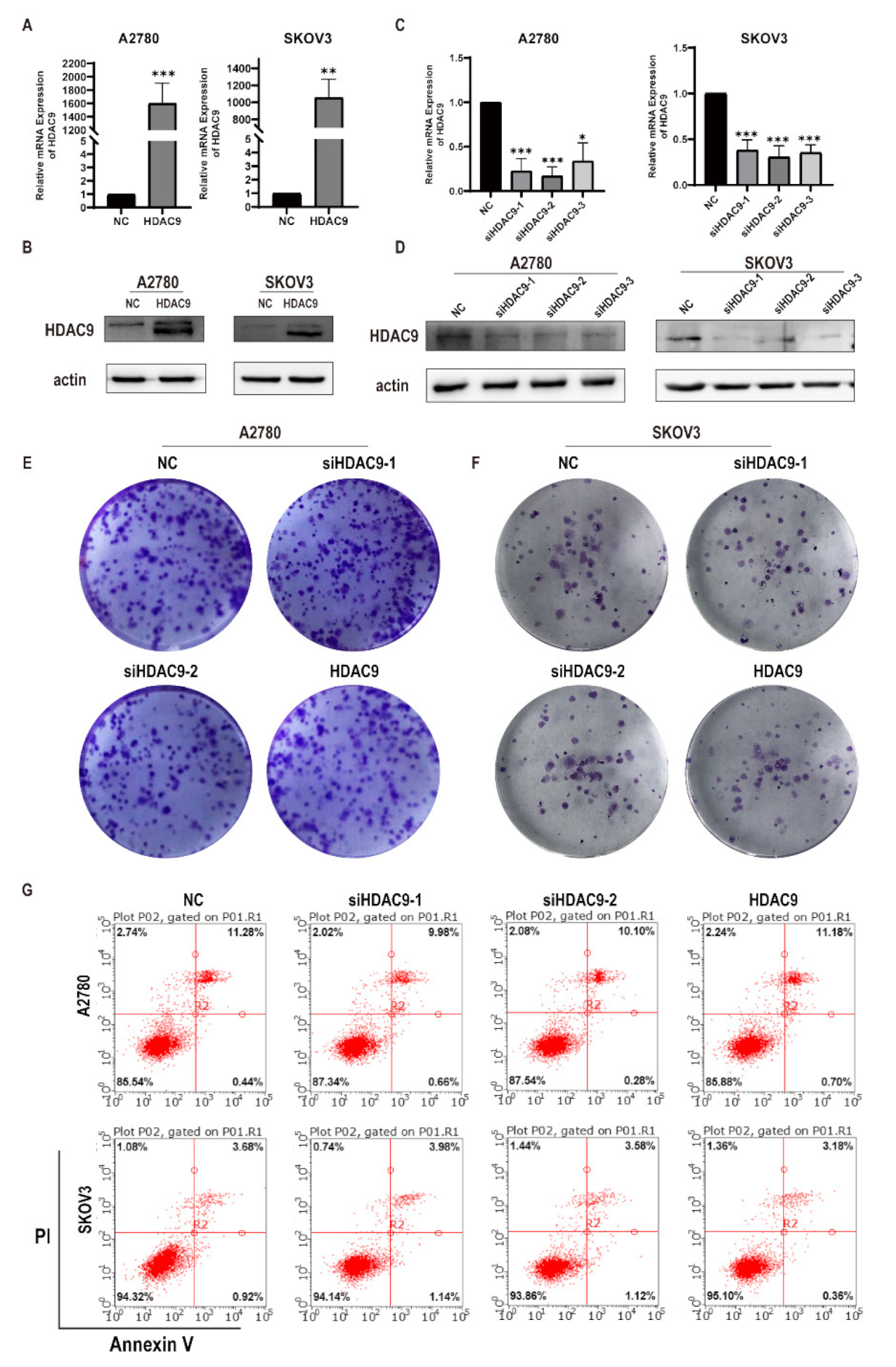
3.2. HDAC9 Does Not Significantly Affect the Proliferation of Ovarian Cancer Cells
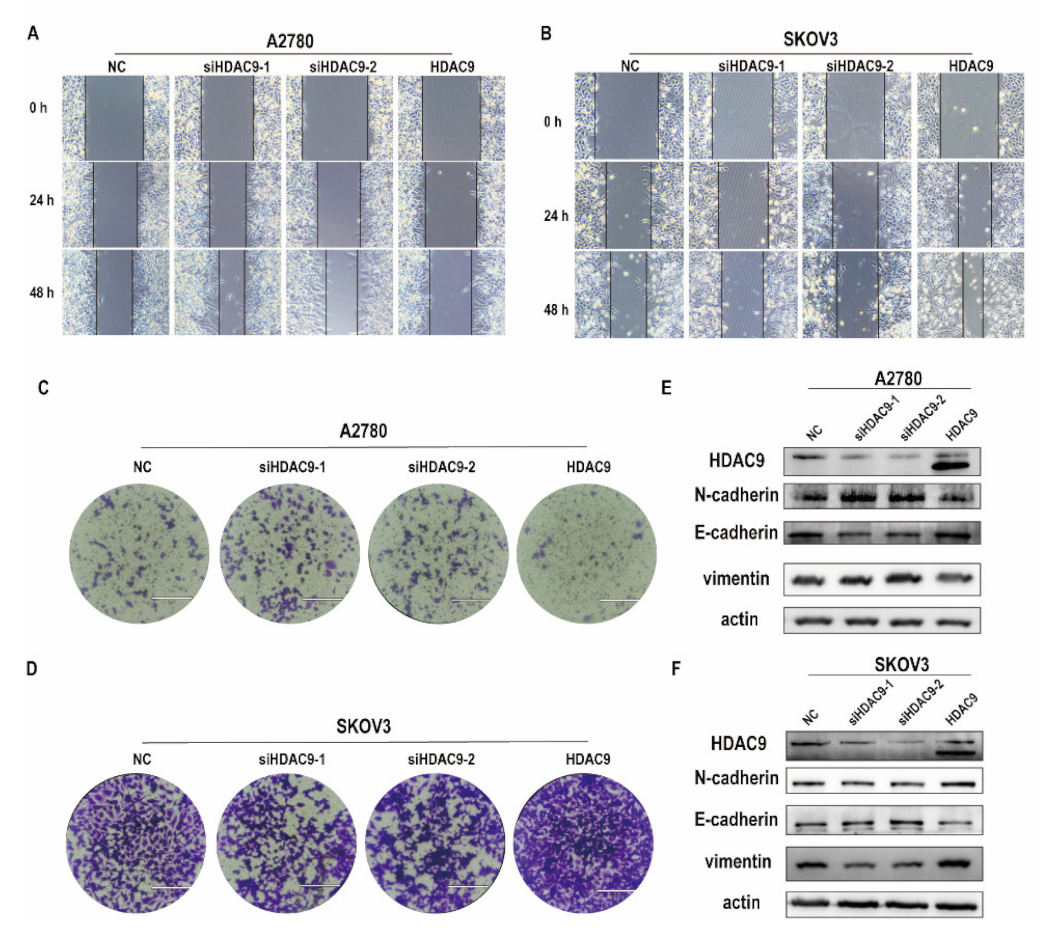
3.3. HDAC9 Is Involved in Cell Motility, Invasion, and EMT of Ovarian Cancer Cells
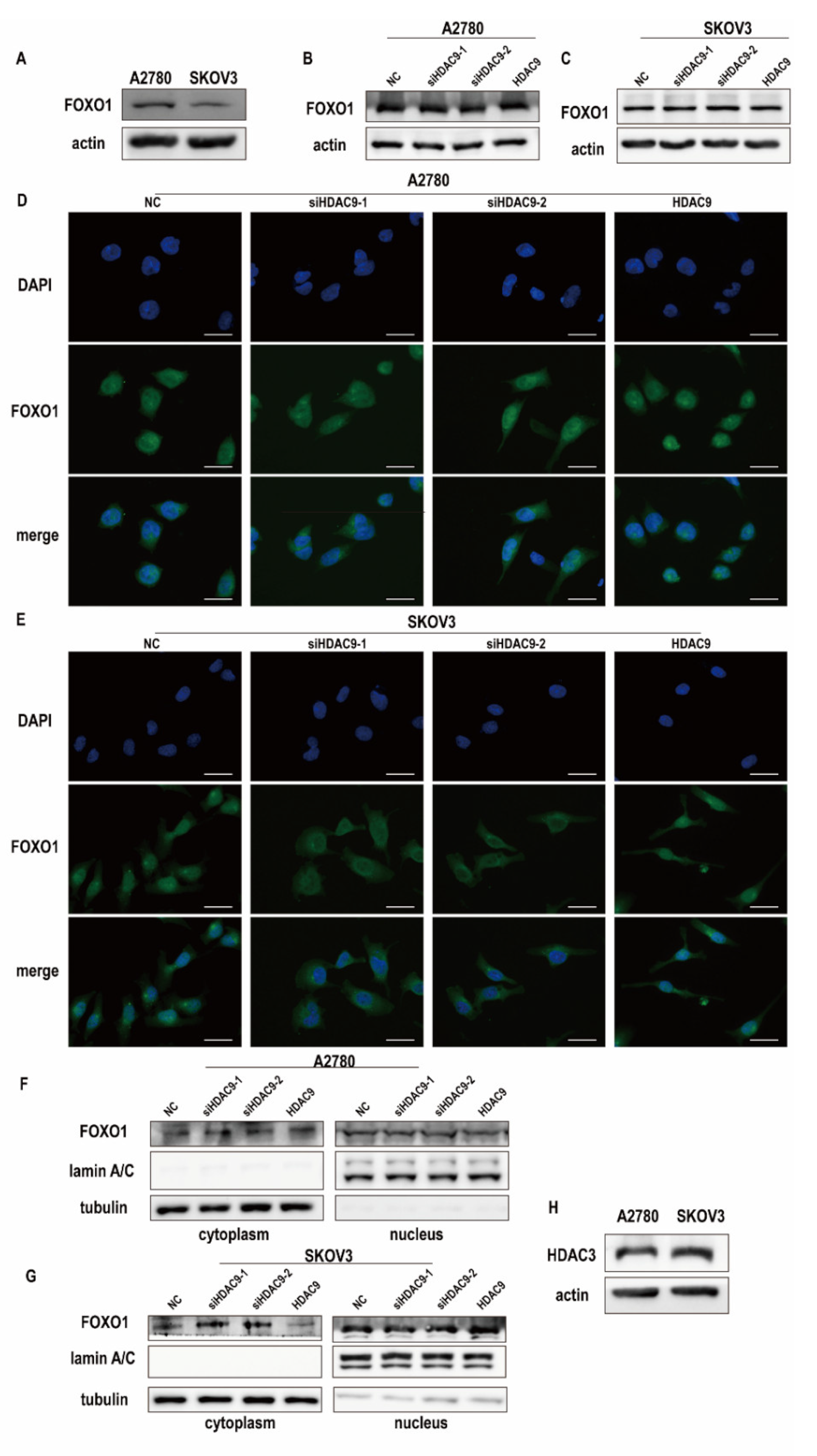
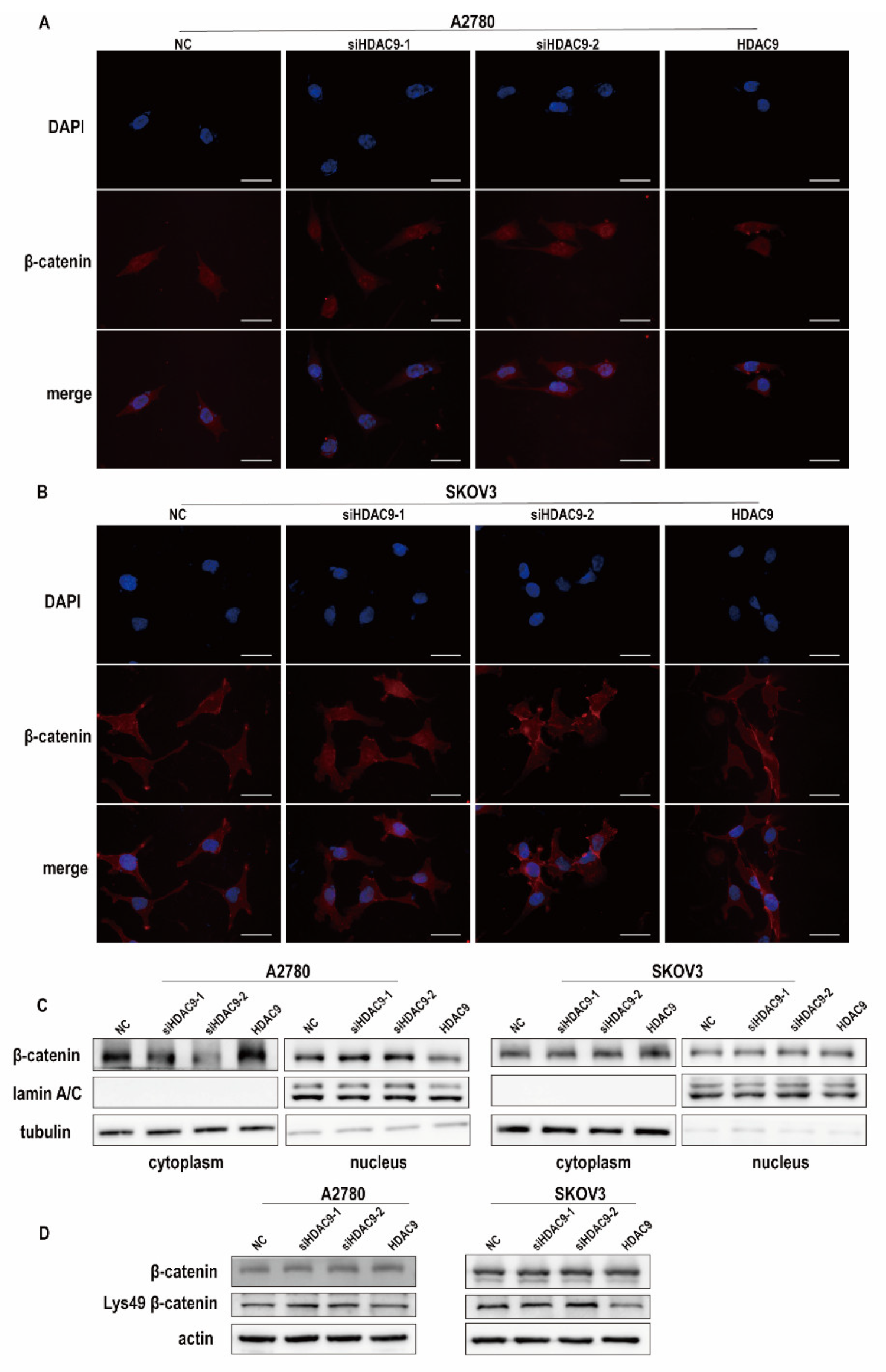
3.4. HDAC9 Regulates the Subcellular Localization of FOXO1 in SKOV3 Cells
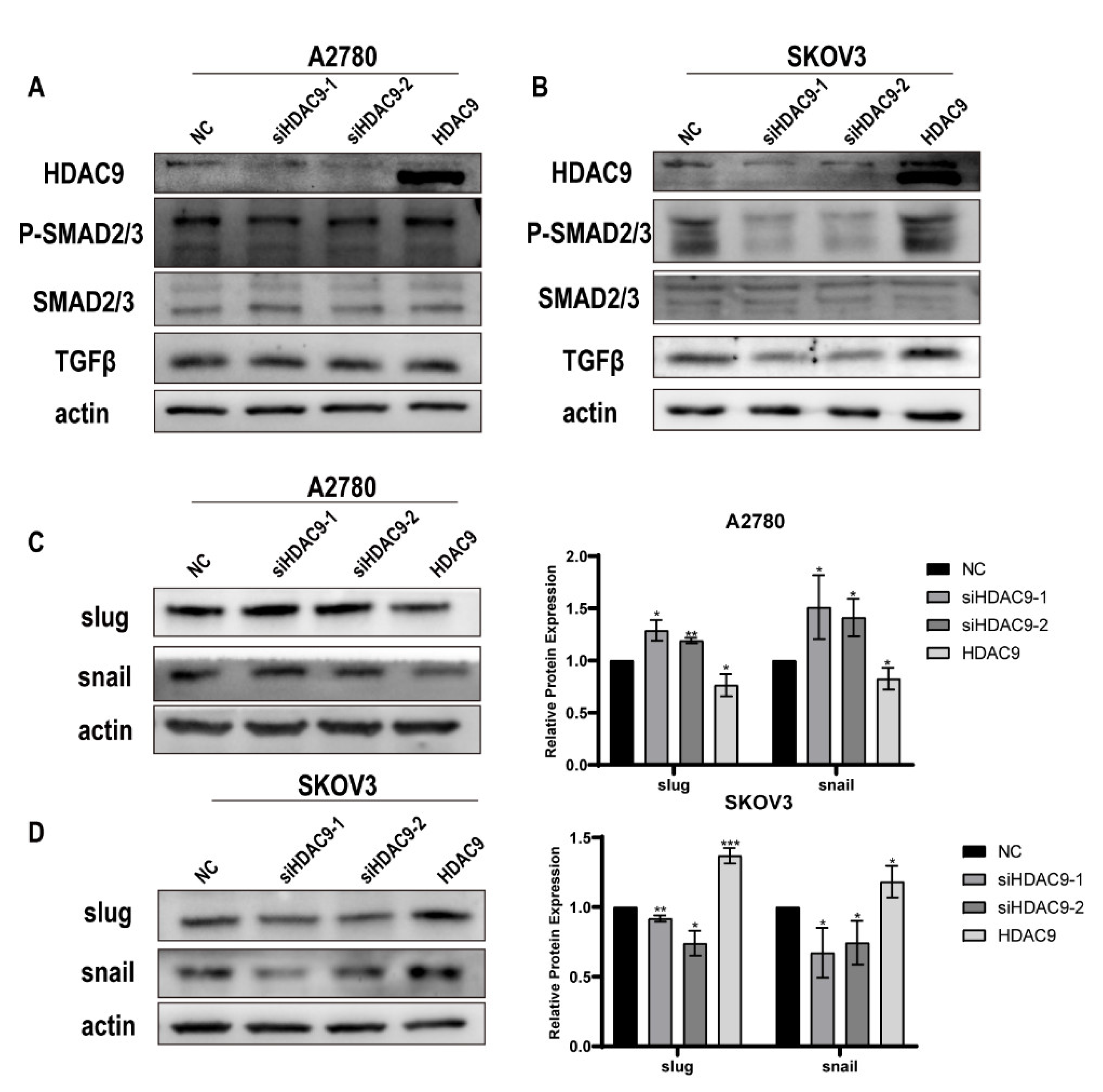
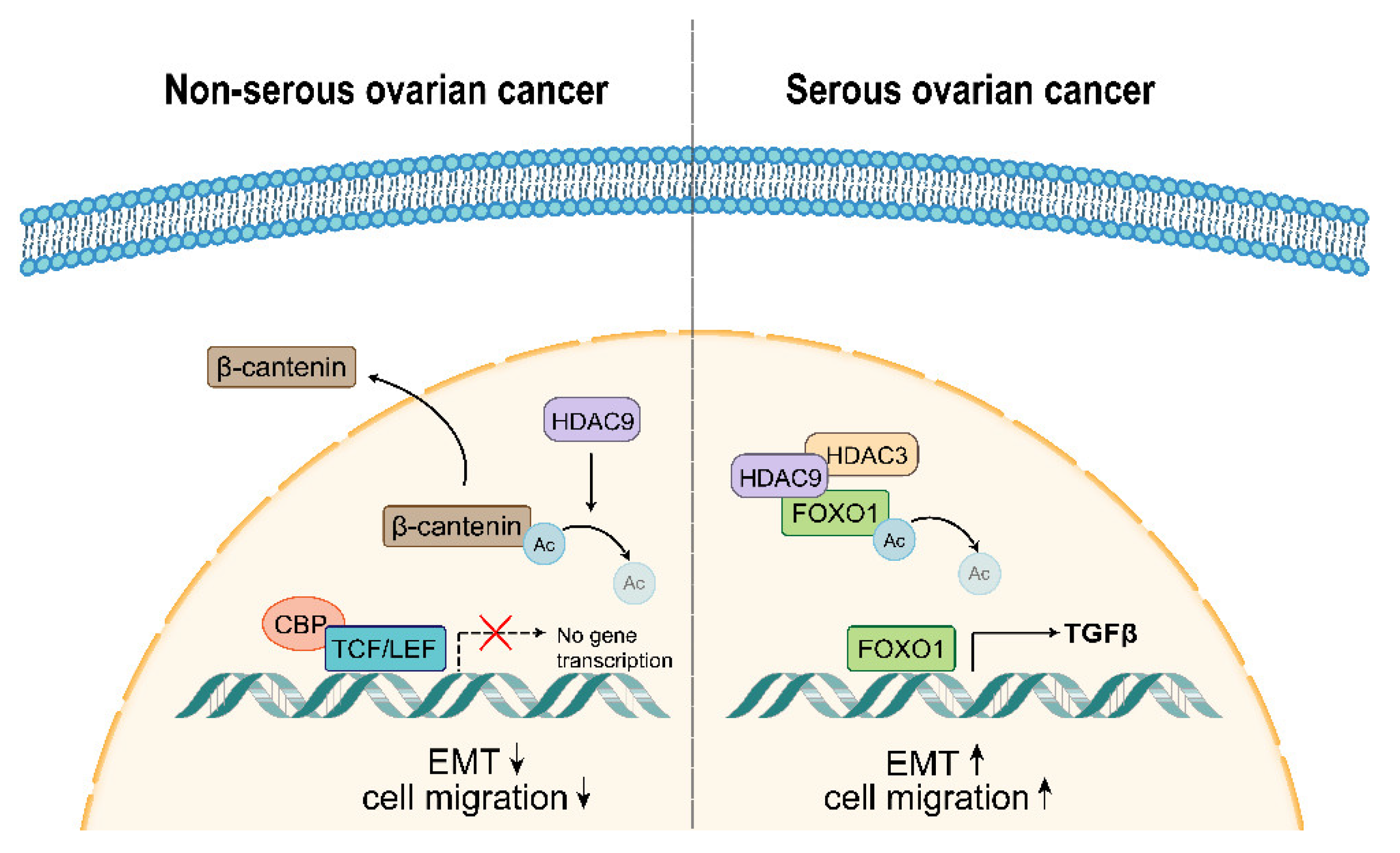
3.5. HDAC9 May Promote EMT in SKOV3 Cells by Upregulating TGF-β Signaling
3.6. HDAC9 May Inhibit EMT in A2780 Cells by Suppressing β-catenin Signaling
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Yan, K.; Cao, Q.; Reilly, C.M.; Young, N.L.; Garcia, B.A.; Mishra, N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wu, D.; Xia, F.; Xian, H.; Zhu, X.; Cui, H.; Huang, Z. Downregulation of HDAC9 inhibits cell proliferation and tumor formation by inducing cell cycle arrest in retinoblastoma. Biochem. Biophys. Res. Commun. 2016, 473, 600–606. [Google Scholar] [CrossRef]
- Rastogi, B.; Raut, S.K.; Panda, N.K.; Rattan, V.; Radotra, B.D.; Khullar, M. Overexpression of HDAC9 promotes oral squamous cell carcinoma growth, regulates cell cycle progression, and inhibits apoptosis. Mol. Cell Biochem. 2016, 415, 183–196. [Google Scholar] [CrossRef]
- Hutt, D.M.; Roth, D.M.; Vignaud, H.; Cullin, C.; Bouchecareilh, M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS ONE 2014, 9, e106224. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Li, W.; Shi, Q.; Wang, M.; Li, H.; Yang, X.; Zhang, J. MicroRNA936 inhibits the malignant phenotype of retinoblastoma by directly targeting HDAC9 and deactivating the PI3K/AKT pathway. Oncol Rep. 2020, 43, 635–645. [Google Scholar] [CrossRef]
- Huang, Y.; Jian, W.; Zhao, J.; Wang, G. Overexpression of HDAC9 is associated with poor prognosis and tumor progression of breast cancer in Chinese females. Onco Targets 2018, 11, 2177–2184. [Google Scholar] [CrossRef] [Green Version]
- Rui, Y.; Wu, Y.; Mei, W.; Sun, Z.; Cui, H. HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation. Oncotarget 2015, 6, 7644. [Google Scholar]
- Ma, Z.; Liu, D.; Di, S.; Zhang, Z.; Li, W.; Zhang, J.; Xu, L.; Guo, K.; Zhu, Y.; Li, X.; et al. Histone deacetylase 9 downregulation decreases tumor growth and promotes apoptosis in non-small cell lung cancer after melatonin treatment. J. Pineal Res. 2019, 67, e12587. [Google Scholar] [CrossRef]
- Okudela, K.; Mitsui, H.; Suzuki, T.; Woo, T.; Tateishi, Y.; Umeda, S.; Saito, Y.; Tajiri, M.; Masuda, M.; Ohashi, K. Expression of HDAC9 in lung cancer--potential role in lung carcinogenesis. Int J. Clin. Exp. Pathol 2014, 7, 213–220. [Google Scholar]
- Coomans de Brachene, A.; Demoulin, J.B. FOXO transcription factors in cancer development and therapy. Cell Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.Q.; Li, A.; Yang, Y.; Li, X.X.; Zhang, L.N.; Guo, H.C. The regulation of FOXO1 and its role in disease progression. Life Sci. 2018, 193, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arend, R.C.; Londono-Joshi, A.I.; Straughn, J.M., Jr.; Buchsbaum, D.J. The Wnt/beta-catenin pathway in ovarian cancer: A review. Gynecol. Oncol. 2013, 131, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Hua, F.; Hu, Z.W. The regulation of beta-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margariti, A.; Zampetaki, A.; Xiao, Q.; Zhou, B.; Karamariti, E.; Martin, D.; Yin, X.; Mayr, M.; Li, H.; Zhang, Z.; et al. Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ. Res. 2010, 106, 1202–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Ponugoti, B.; Xu, F.; Zhang, C.; Tian, C.; Pacios, S.; Graves, D.T. FOXO1 promotes wound healing through the up-regulation of TGF-beta1 and prevention of oxidative stress. J. Cell Biol. 2013, 203, 327–343. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Yan, R.; Zhang, X.; Wang, L.; Ke, X.; Qu, Y. Activating Wnt/beta-catenin signaling pathway for disease therapy: Challenges and opportunities. Pharm. Ther. 2019, 196, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Winer, I.S.; Bommer, G.T.; Gonik, N.; Fearon, E.R. Lysine residues Lys-19 and Lys-49 of beta-catenin regulate its levels and function in T cell factor transcriptional activation and neoplastic transformation. J. Biol. Chem. 2006, 281, 26181–26187. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Cho, E.H.; Lee, J.Y. Histone Deacetylase 9: Its Role in the Pathogenesis of Diabetes and Other Chronic Diseases. Diabetes Metab. J. 2020, 44, 234–244. [Google Scholar] [CrossRef]
- Giorgio, E.D.; Brancolini, C. Regulation of class IIa HDAC activities: It is not only matter of subcellular localization. Epigenomics 2016, 8, 251–269. [Google Scholar] [CrossRef] [Green Version]
- Parra, M. Class IIa HDACs—New insights into their functions in physiology and pathology. FEBS J. 2015, 282, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Petrie, K.; Guidez, F.; Howell, L.; Healy, L.; Waxman, S.; Greaves, M.; Zelent, A. The histone deacetylase 9 gene encodes multiple protein isoforms. J. Biol. Chem. 2003, 278, 16059–16072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.C.; Chiang, W.F.; Huang, H.H.; Chen, P.F.; Shen, Y.Y.; Chiang, H.C. Role of SIRT1 in regulation of epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis. Mol. Cancer 2014, 13, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wang, N.; Dong, M.; Guo, M.; Zhao, Y.; Zhuo, Z.; Zhang, C.; Chi, X.; Pan, Y.; Jiang, J.; et al. The Metabolic Regulator Histone Deacetylase 9 Contributes to Glucose Homeostasis Abnormality Induced by Hepatitis C Virus Infection. Diabetes 2015, 64, 4088–4098. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Zhai, Y.; Fearon, E.R.; Cho, K.R. Diverse mechanisms of beta-catenin deregulation in ovarian endometrioid adenocarcinomas. Cancer Res. 2001, 61, 8247–8255. [Google Scholar]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | HDAC9 Expression | p-Value | |
|---|---|---|---|
| High (n = 26) | Low (n = 29) | ||
| Age(y) | 0.148 | ||
| <55 | 11 | 18 | |
| ≥55 | 15 | 11 | |
| Tumor Size (cm) | 0.116 | ||
| <5 | 1 | 5 | |
| ≥5 | 25 | 24 | |
| Histological grade | 0.433 | ||
| I–II | 5 | 8 | |
| III | 20 | 19 | |
| T stage | 0.586 | ||
| T1 | 0 | 2 | |
| T2 | 5 | 5 | |
| T3 | 21 | 22 | |
| N stage | 0.023 * | ||
| N0 | 12 | 22 | |
| N1 | 14 | 7 | |
| M stage | 0.4 | ||
| M0 | 18 | 21 | |
| M1 | 8 | 8 | |
| Relapse | 0.179 | ||
| Absence | 2 | 6 | |
| Presence | 24 | 23 | |
| Follow-ups | 0.029 * | ||
| Dead | 20 | 14 | |
| Survival | 6 | 15 | |
| Characteristic | HDAC9 Expression | p-Value | |
|---|---|---|---|
| High (n = 24) | Low (n = 23) | ||
| Age(y) | 0.103 | ||
| <55 | 16 | 17 | |
| ≥55 | 8 | 6 | |
| Tumor Size (cm) | 0.966 | ||
| <5 | 7 | 4 | |
| ≥5 | 17 | 19 | |
| Histological grade | 0.688 | ||
| I–II | 3 | 2 | |
| III | 16 | 16 | |
| T stage | 0.773 | ||
| T1 | 2 | 0 | |
| T2 | 7 | 7 | |
| T3 | 15 | 16 | |
| N stage | 0.291 | ||
| N0 | 22 | 18 | |
| N1 | 2 | 5 | |
| M stage | 0.137 | ||
| M0 | 22 | 18 | |
| M1 | 2 | 5 | |
| Relapse | 0.445 | ||
| Absence | 6 | 3 | |
| Presence | 18 | 20 | |
| Follow-ups | 0.73 | ||
| Dead | 7 | 12 | |
| Survival | 17 | 11 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Wang, J.; Liu, B.; Fu, J.; Zhao, Y.; Yu, S.; Shen, L.; Yan, X.; Su, J. HDAC9 Contributes to Serous Ovarian Cancer Progression through Regulating Epithelial–Mesenchymal Transition. Biomedicines 2022, 10, 374. https://doi.org/10.3390/biomedicines10020374
Xu L, Wang J, Liu B, Fu J, Zhao Y, Yu S, Shen L, Yan X, Su J. HDAC9 Contributes to Serous Ovarian Cancer Progression through Regulating Epithelial–Mesenchymal Transition. Biomedicines. 2022; 10(2):374. https://doi.org/10.3390/biomedicines10020374
Chicago/Turabian StyleXu, Long, Jian Wang, Buhan Liu, Jiaying Fu, Yuanxin Zhao, Sihang Yu, Luyan Shen, Xiaoyu Yan, and Jing Su. 2022. "HDAC9 Contributes to Serous Ovarian Cancer Progression through Regulating Epithelial–Mesenchymal Transition" Biomedicines 10, no. 2: 374. https://doi.org/10.3390/biomedicines10020374
APA StyleXu, L., Wang, J., Liu, B., Fu, J., Zhao, Y., Yu, S., Shen, L., Yan, X., & Su, J. (2022). HDAC9 Contributes to Serous Ovarian Cancer Progression through Regulating Epithelial–Mesenchymal Transition. Biomedicines, 10(2), 374. https://doi.org/10.3390/biomedicines10020374