
Poxviruses Bearing DNA Polymerase Mutations Show Complex Patterns of Cross-Resistance

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell and Virus Culture
2.2. Materials
2.3. Selection and Purification of Drug-Resistance Viruses
2.4. Cytopathic Effect (CPE) Reduction Assay
2.5. Genotyping of the VACV DNA Polymerase
3. Results
3.1. Characterization of Drug-Resistant VACV and CPXV Emerging under Different HPMP and HPMPO-DAPy Derivatives
3.1.1. VACV Resistance to Cyclic-HPMPC, HPMPA, and Cyclic-HPMPA
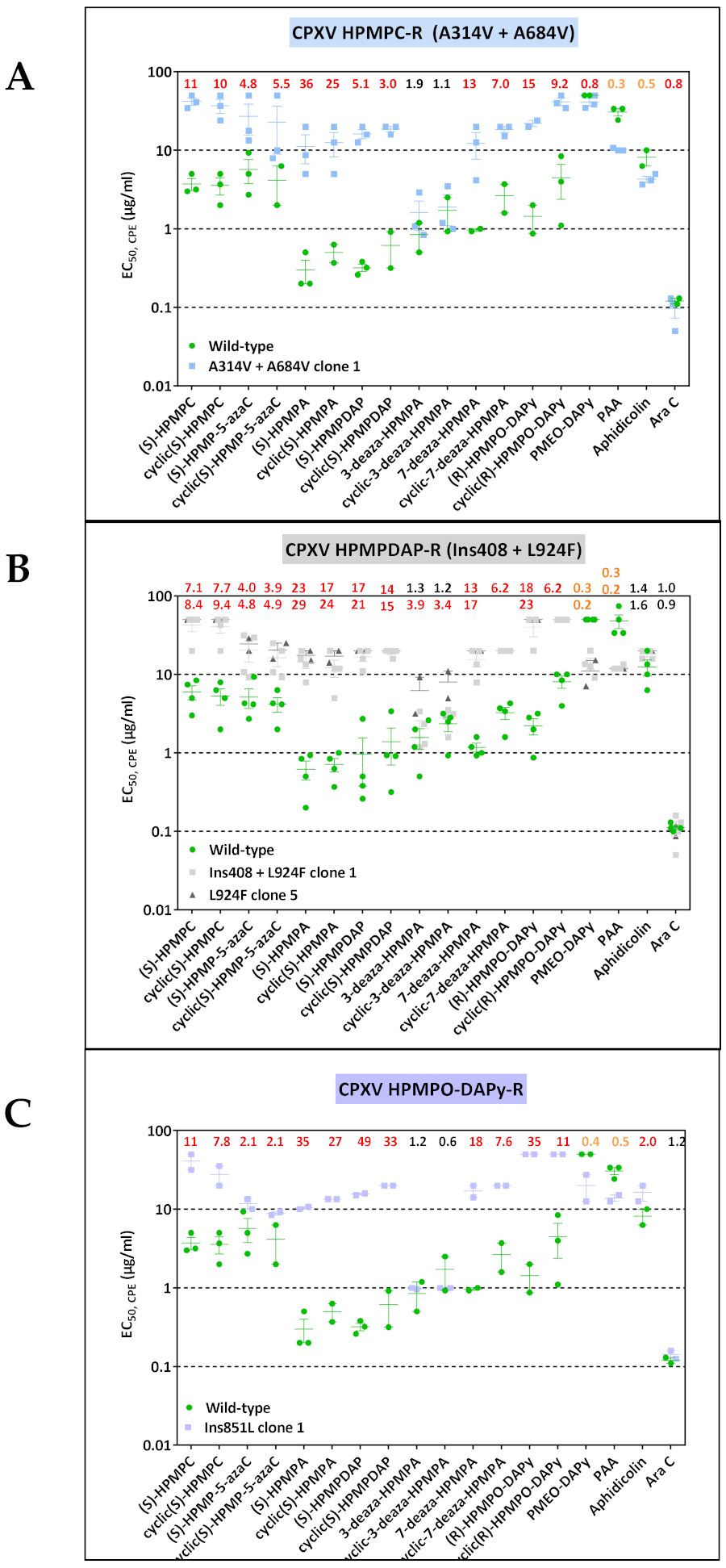
3.1.2. CPXV Resistance to HPMPC, HPMPDAP, and HPMPO-DAPy
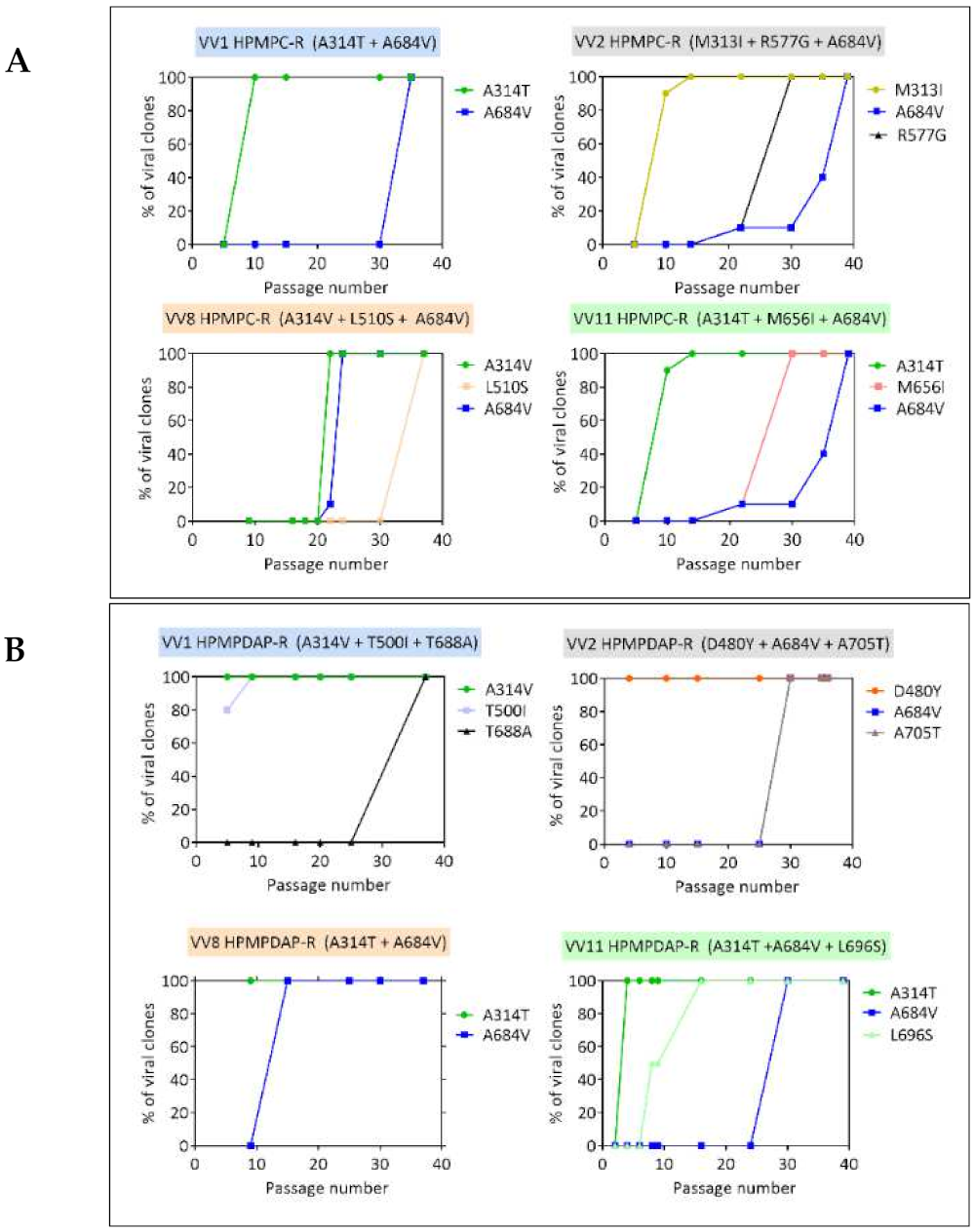
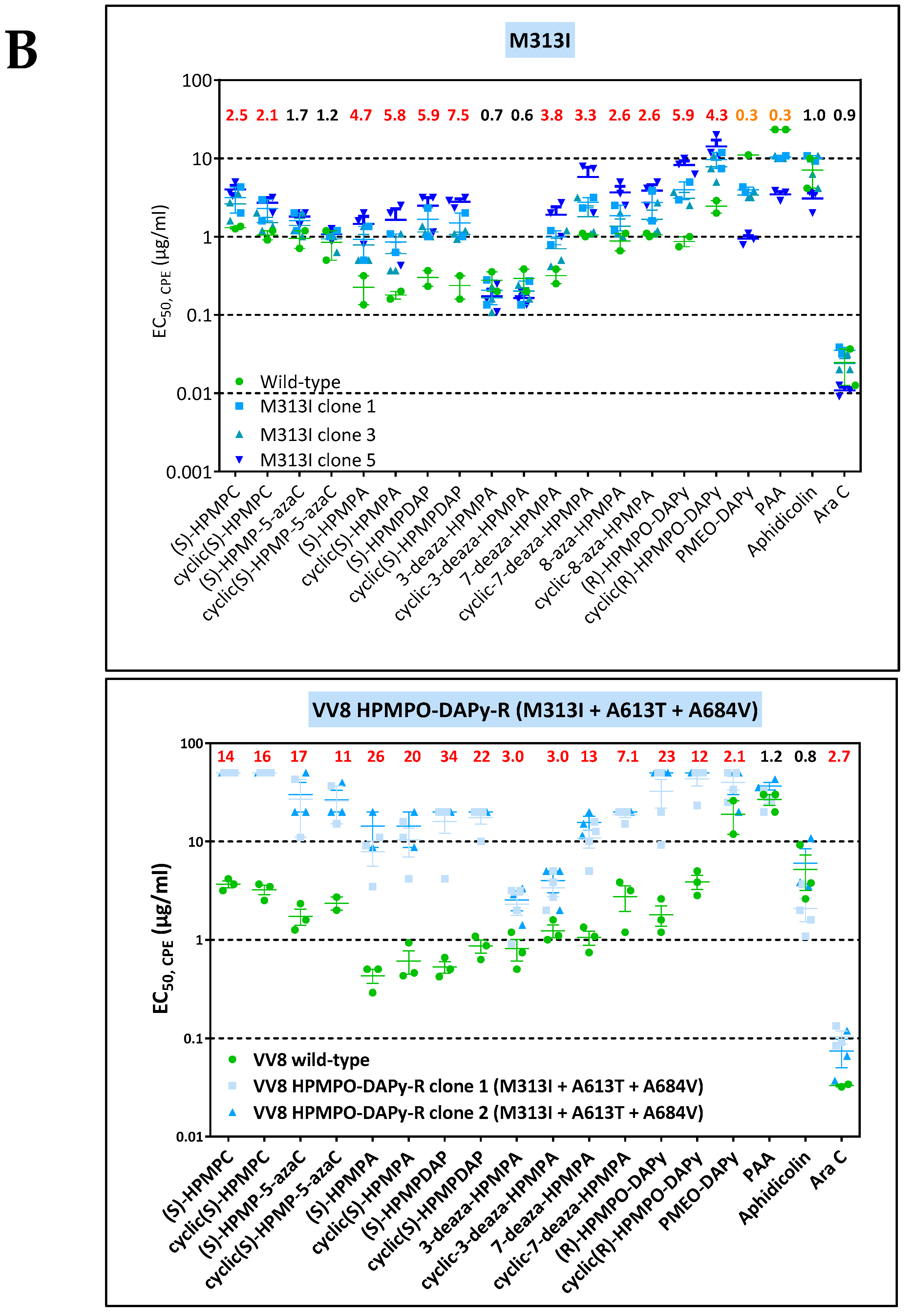
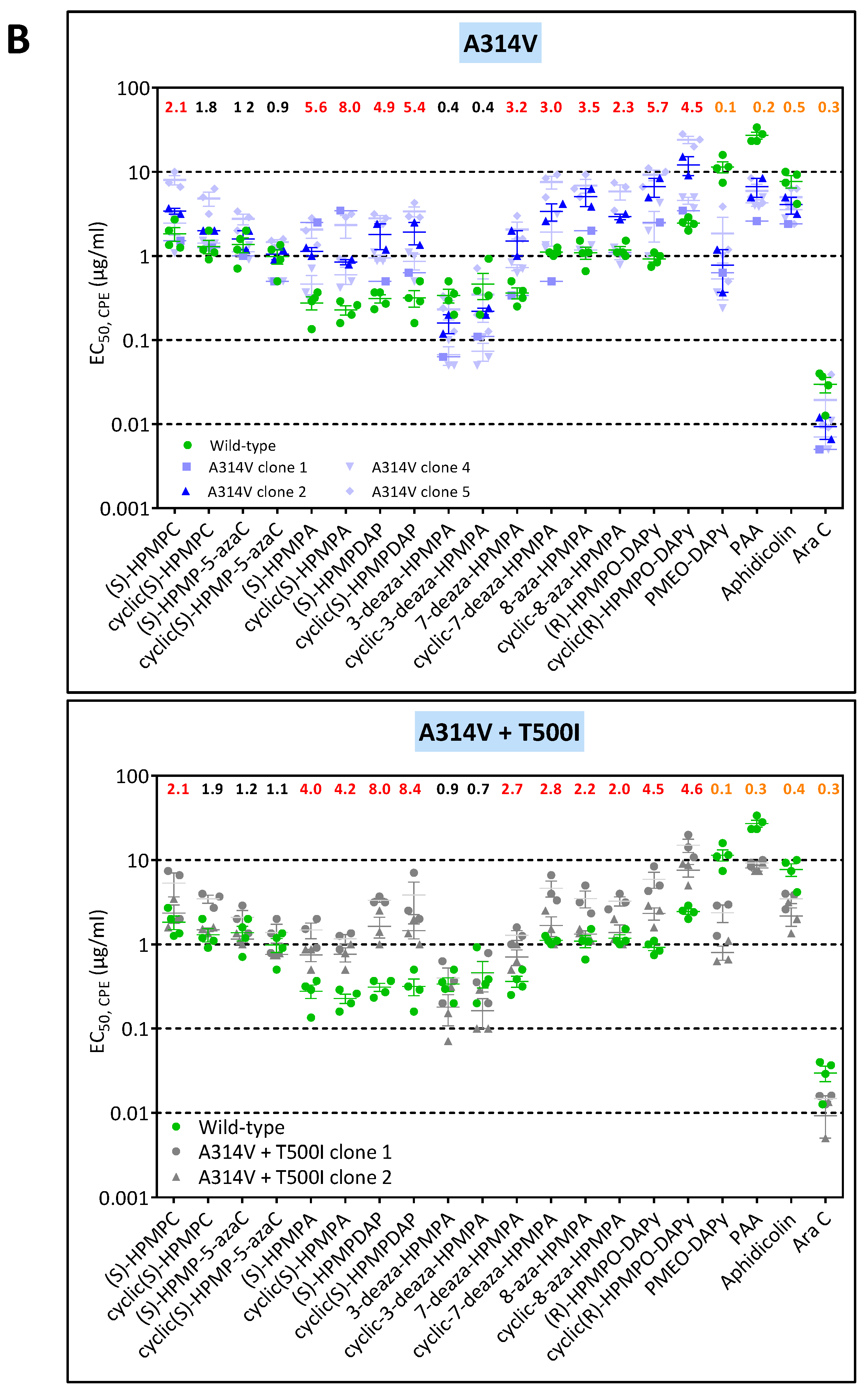
3.1.3. Genotypic Characterization of VACV DNA Pol Mutants Arising Following Four Independent Selection Procedures with HPMPC, HPMPDAP, and HPMPO-DAPy
3.2. Chronological Analysis of Emergence of DNA pol VACV Mutants Resistant to HPMPC, HPMPDAP, and HPMPO-DAPy
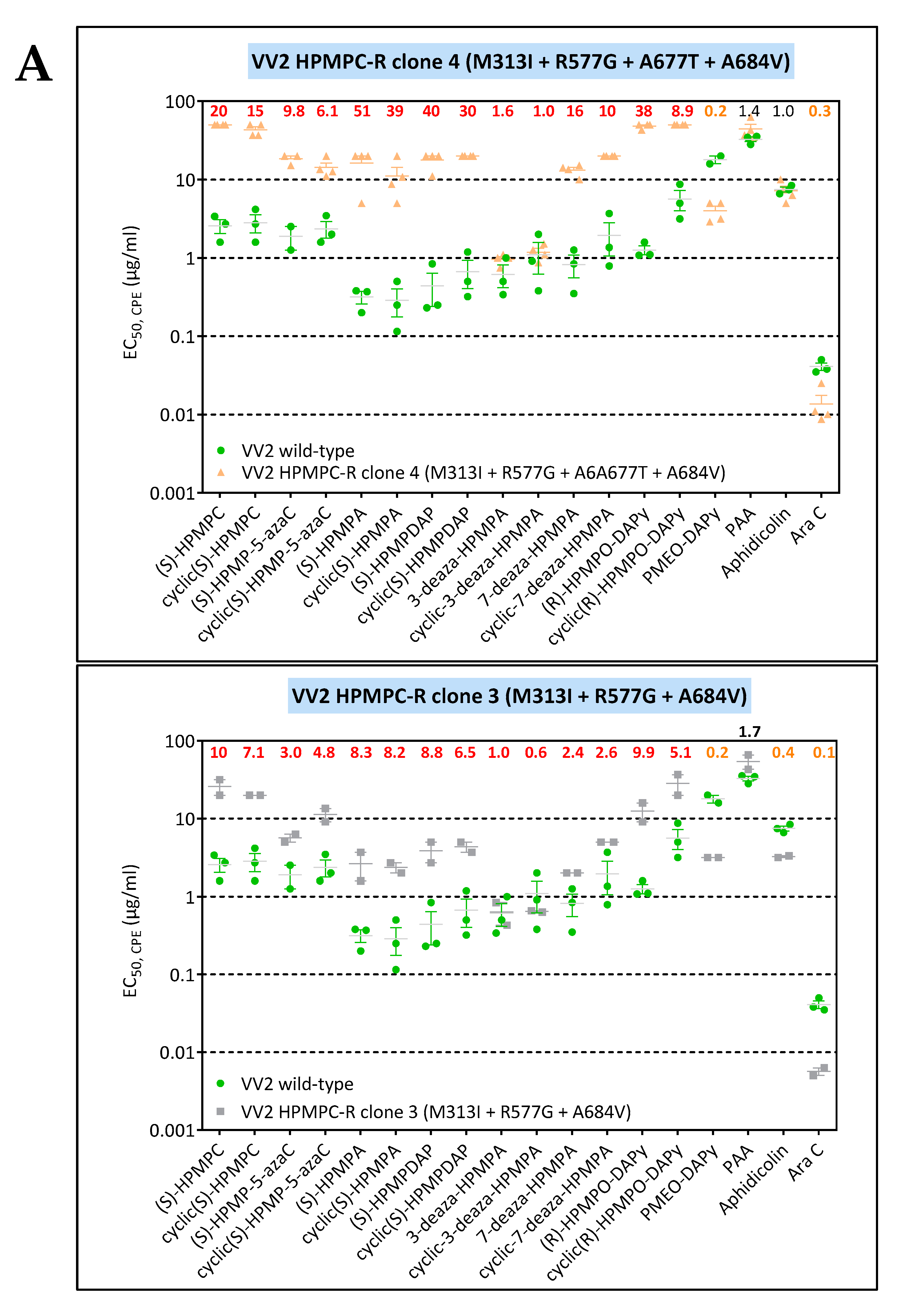
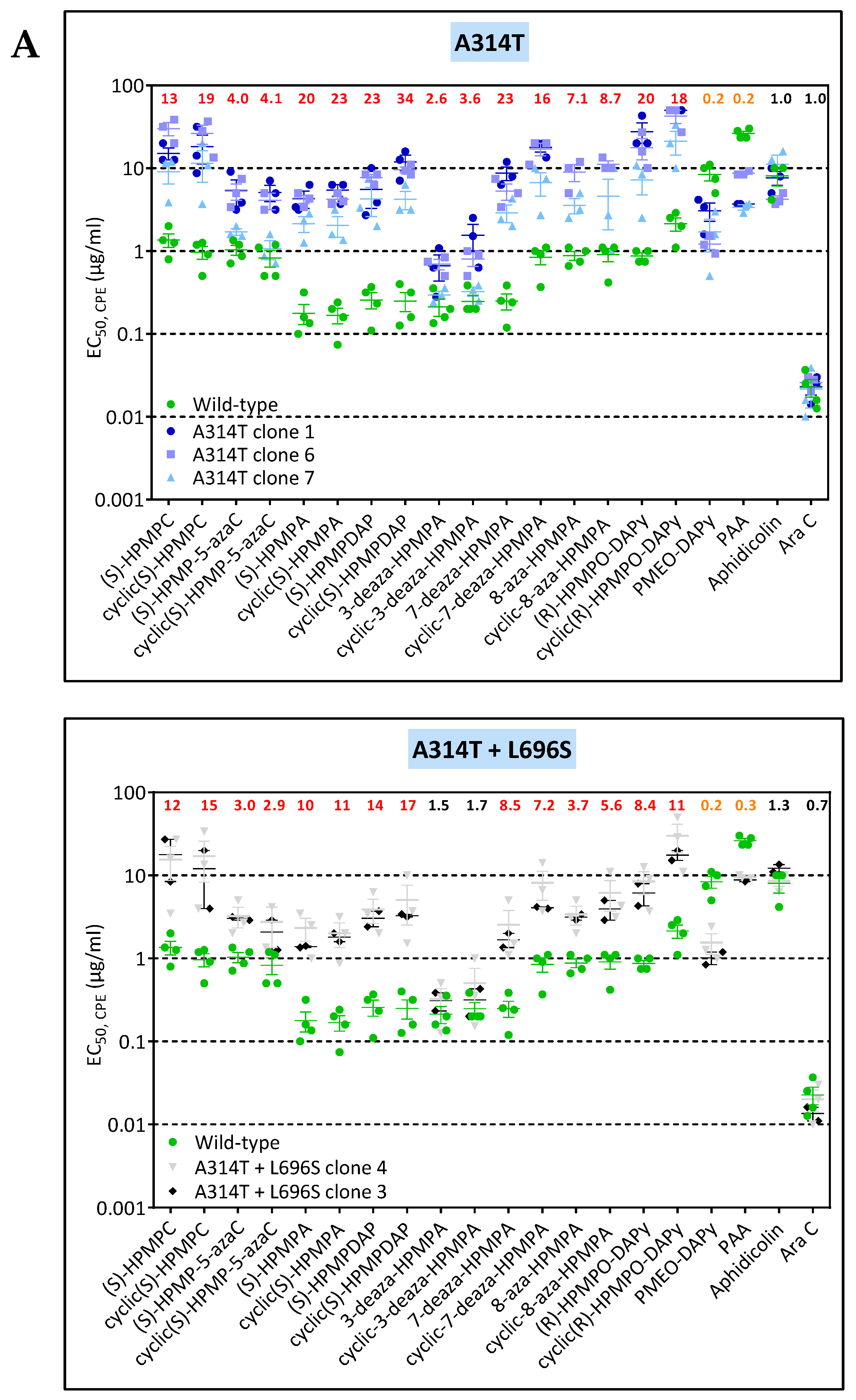
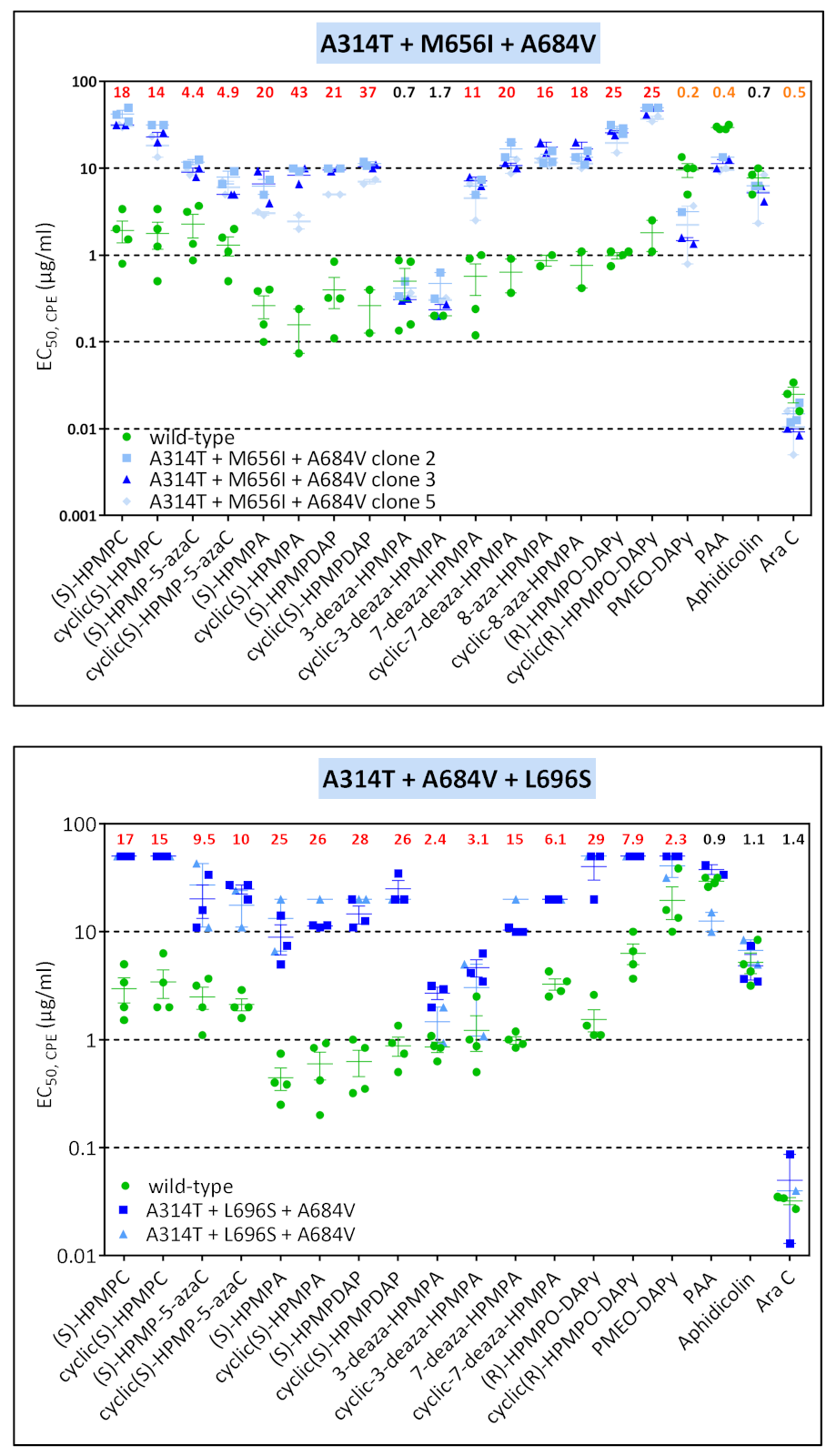
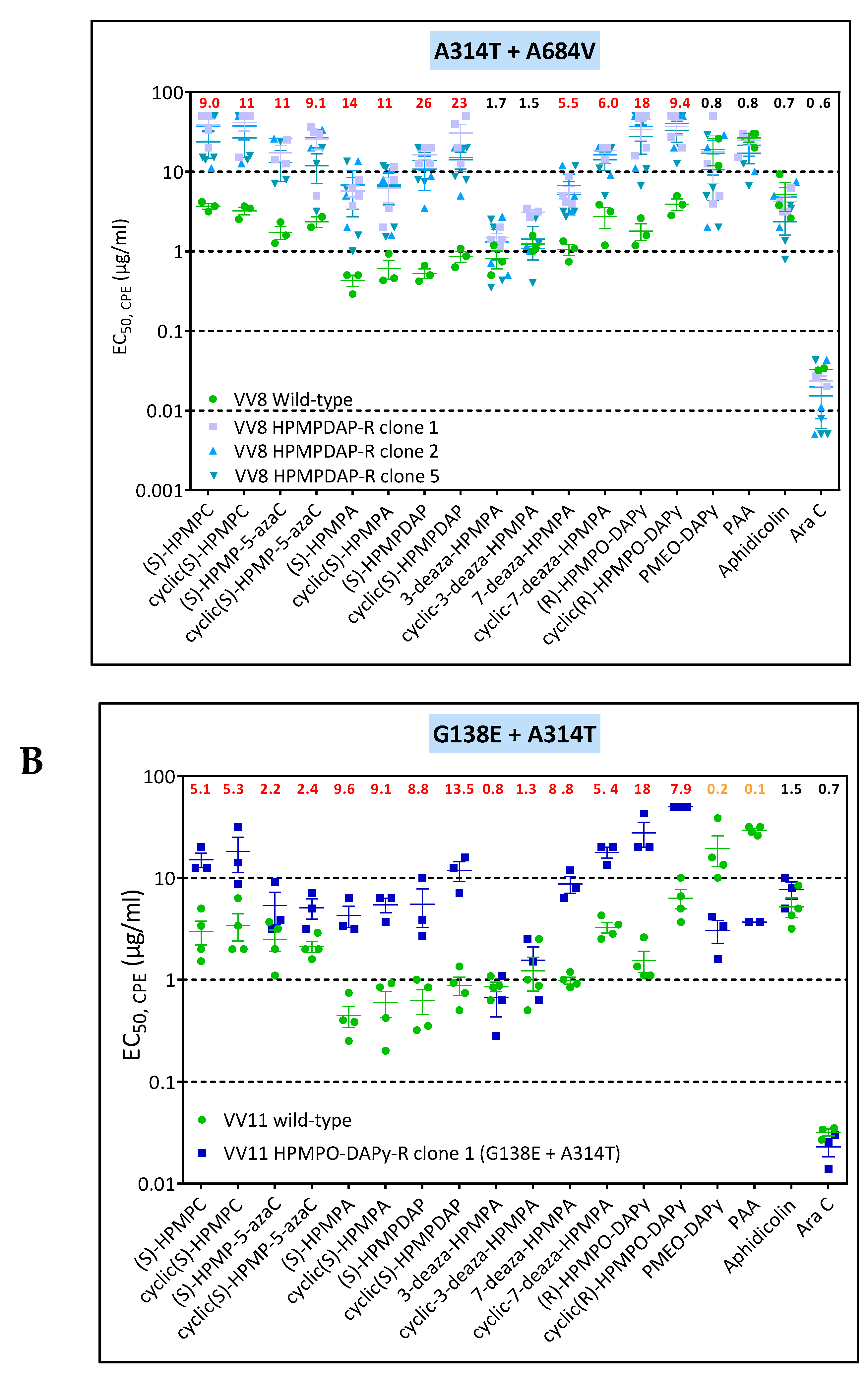
3.3. Phenotypic Characterization of VACV Viral Clones Selected under Pressure of HPMPC, HPMPDAP, and HPMPO-DAPy
3.3.1. VACV Clones Emerging under HPMPC Pressure
3.3.2. VACV Clones Emerging under HPMPDAP Pressure
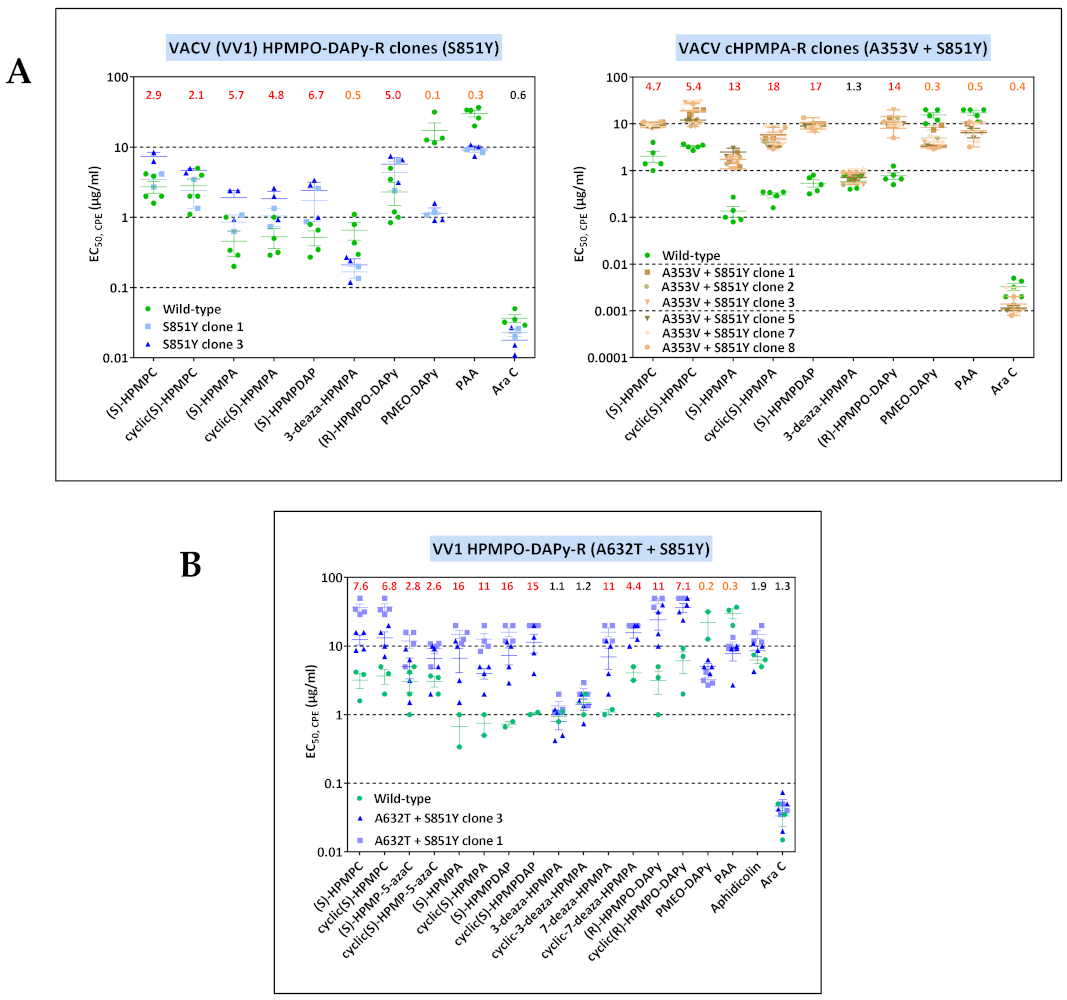
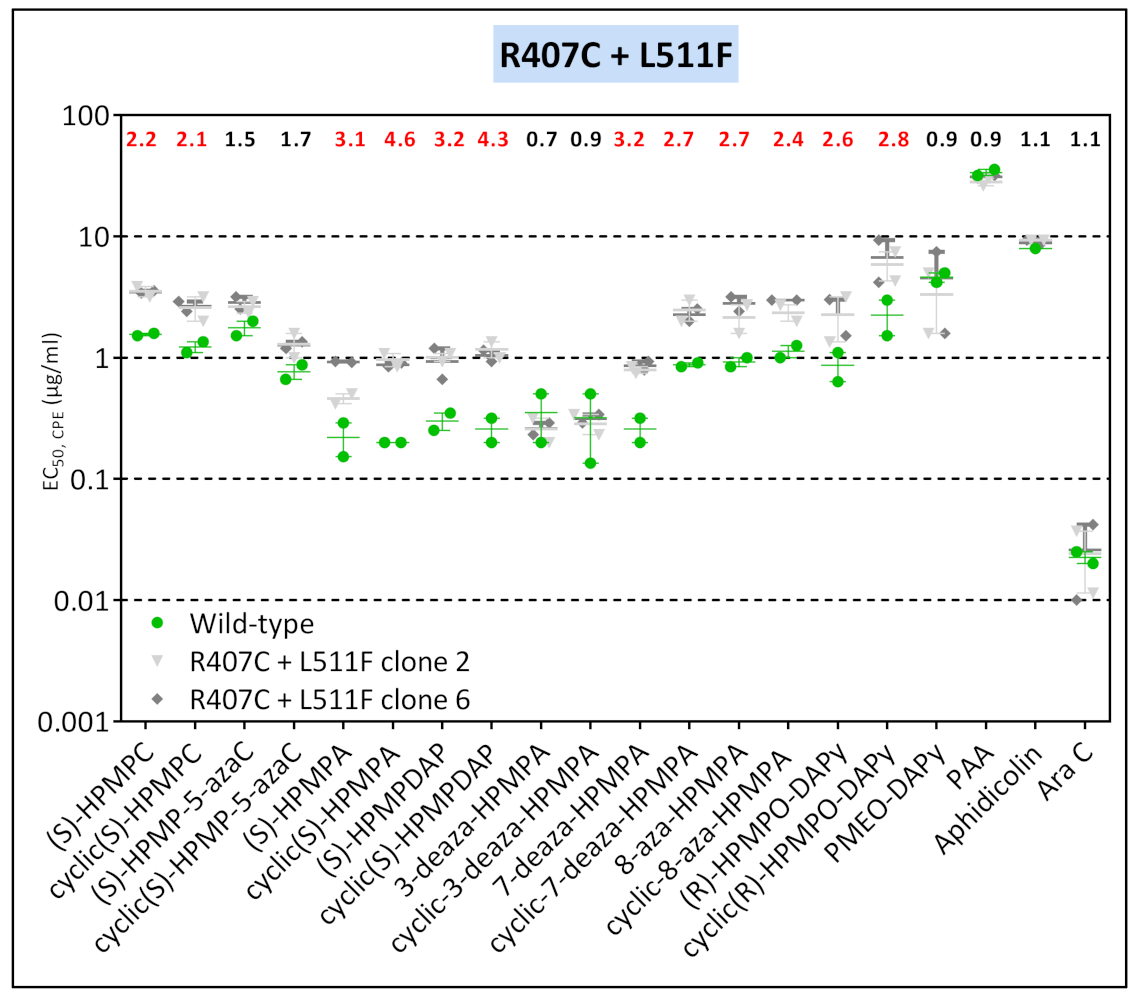
3.3.3. VACV Clones Emerging under HPMPO-DAPy Pressure
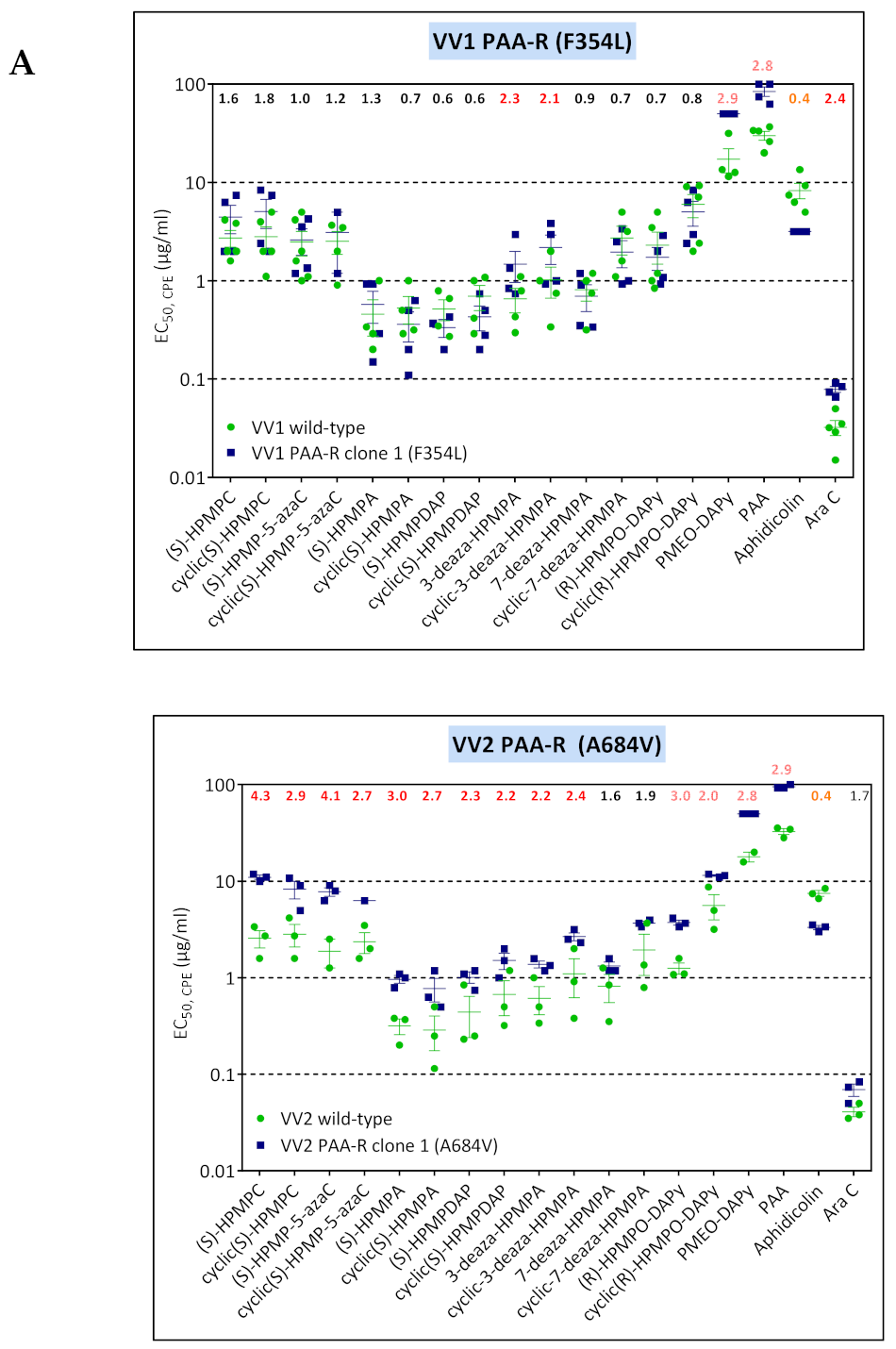
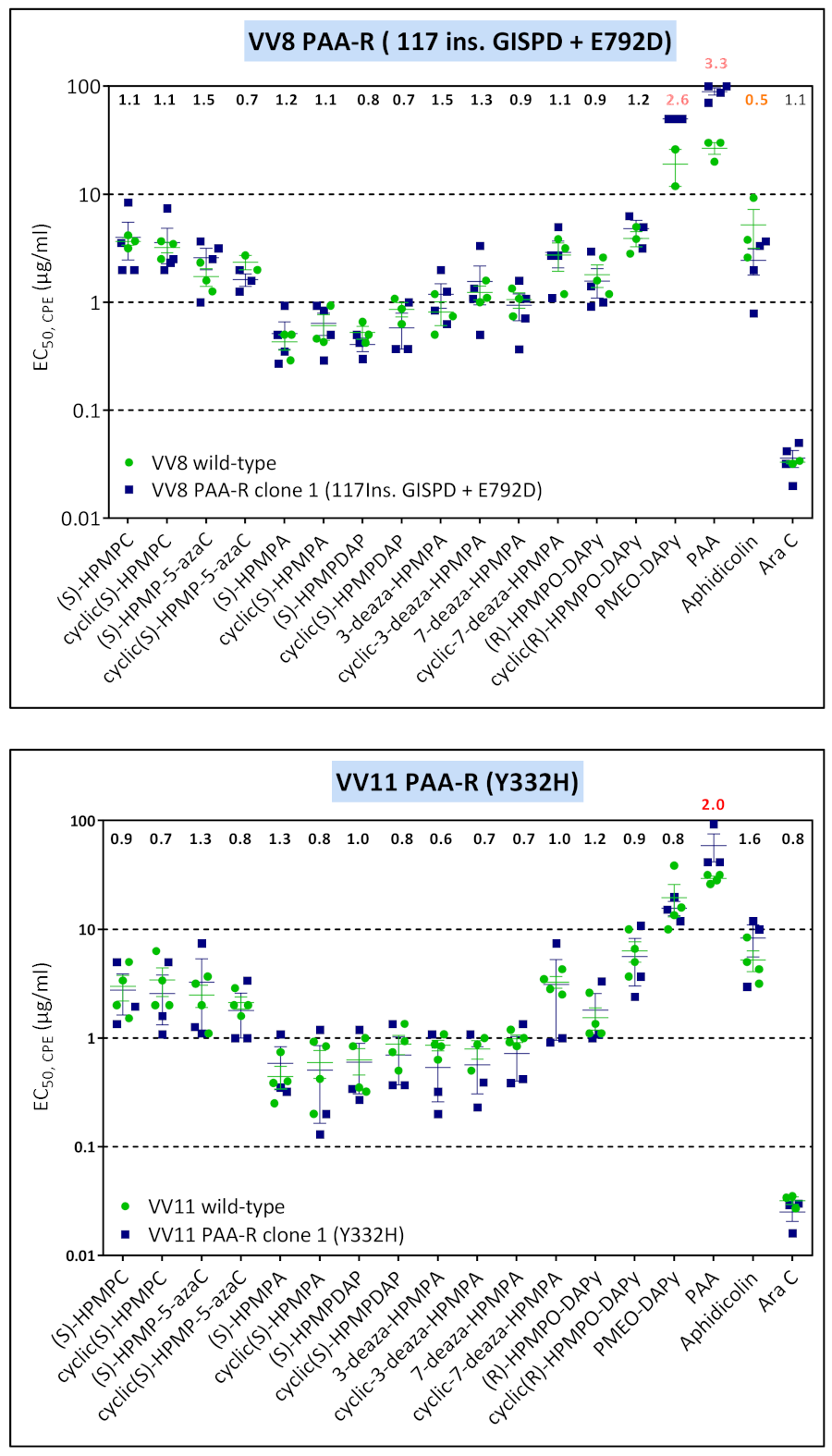
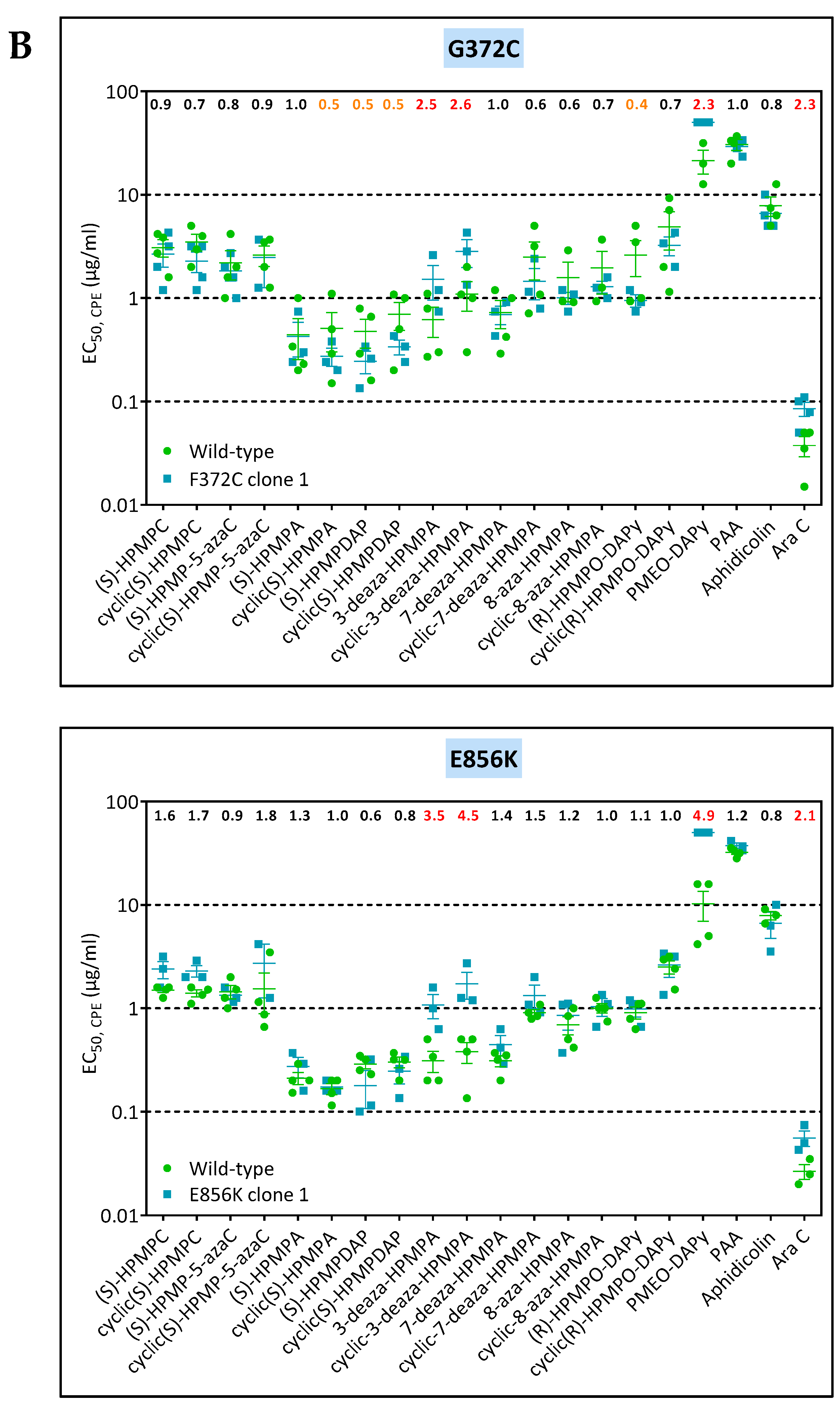
3.4. Characterization of Drug-Resistance VACV against PAA and PMEO-DAPy
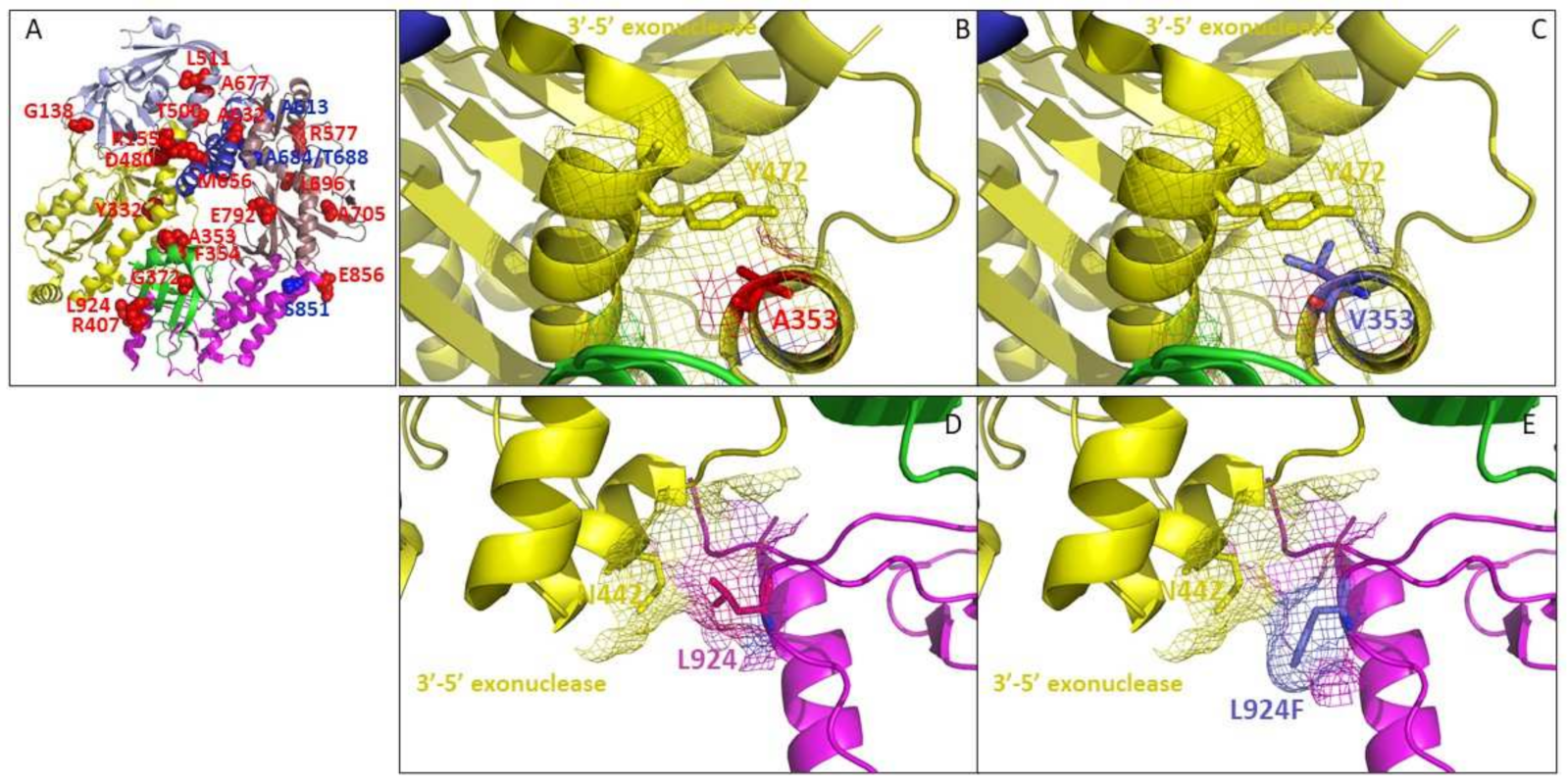
3.5. Topological Distribution of Drug-Ressitance Mutations in Poxvirus DNA Polymerase
3.5.1. A353V and L924F May Affect the 3′-5′ Exonuclease Domain of Poxvirus E9 Protein
3.5.2. The A632T, M656I, and A677T Might Alter the Positioning of the Fingers Domain to Avoid Incorporation of ANPs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foster, S.A.; Parker, S.; Lanier, R. The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak. Viruses 2017, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenner, F. Smallpox: Emergence, global spread, and eradication. Hist. Philos. Life Sci. 1993, 15, 397–420. [Google Scholar] [PubMed]
- Breman, J.G.; Arita, I. The Confirmation and Maintenance of Smallpox Eradication. N. Engl. J. Med. 1980, 303, 1263–1273. [Google Scholar] [CrossRef]
- Melamed, S.; Israely, T.; Paran, N. Challenges and Achievements in Prevention and Treatment of Smallpox. Vaccines 2018, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Grosenbach, D.W.; Honeychurch, K.; Rose, E.A.; Chinsangaram, J.; Frimm, A.; Maiti, B.; Lovejoy, C.; Meara, I.; Long, P.; Hruby, D.E. Oral Tecovirimat for the Treatment of Smallpox. N. Engl. J. Med. 2018, 379, 44–53. [Google Scholar] [CrossRef]
- Yang, G.; Pevear, D.C.; Davies, M.H.; Collett, M.S.; Bailey, T.; Rippen, S.; Barone, L.; Burns, C.; Rhodes, G.; Tohan, S.; et al. An Orally Bioavailable Antipoxvirus Compound (ST-246) Inhibits Extracellular Virus Formation and Protects Mice from Lethal Orthopoxvirus Challenge. J. Virol. 2005, 79, 13139–13149. [Google Scholar] [CrossRef] [Green Version]
- Duraffour, S.; Lorenzo, M.M.; Zöller, G.; Topalis, D.; Grosenbach, D.; Hruby, D.E.; Andrei, G.; Blasco, R.; Meyer, H.; Snoeck, R. ST-246 is a key antiviral to inhibit the viral F13L phospholipase, one of the essential proteins for orthopoxvirus wrapping. J. Antimicrob. Chemother. 2015, 70, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Chan-Tack, K.; Harrington, P.; Bensman, T.; Choi, S.-Y.; Donaldson, E.; O’Rear, J.; McMillan, D.; Myers, L.; Seaton, M.; Ghantous, H.; et al. Benefit-risk assessment for brincidofovir for the treatment of smallpox: U.S. Food and Drug Administration’s Evaluation. Antivir. Res. 2021, 195, 105182. [Google Scholar] [CrossRef]
- Alvarez-Cardona, J.J.; Whited, L.K.; Chemaly, R.F. Brincidofovir: Understanding its unique profile and potential role against ade-novirus and other viral infections. Future Microbiol. 2020, 15, 389–400. [Google Scholar]
- Delaune, D.; Iseni, F. Drug Development against Smallpox: Present and Future. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Kabuga, A.I.; El Zowalaty, M.E. A review of the monkeypox virus and a recent outbreak of skin rash disease in Nigeria. J. Med. Virol. 2019, 91, 533–540. [Google Scholar]
- Guagliardo, S.A.J.; Monroe, B.; Moundjoa, C.; Athanase, A.; Okpu, G.; Burgado, J.; Townsend, M.B.; Satheshkumar, P.S.; Epperson, S.; Doty, J.B.; et al. Asymptomatic Orthopoxvirus Circulation in Humans in the Wake of a Monkeypox Outbreak among Chimpanzees in Cameroon. Am. J. Trop. Med. Hyg. 2020, 102, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, M.G.; Doty, J.B.; Mccollum, A.M.; Olson, V.A.; Nakazawa, Y. Monkeypox re-emergence in Africa: A call to expand the concept and practice of One Health. Expert Rev. Anti-Infect. Ther. 2018, 17, 129–139. [Google Scholar] [CrossRef]
- Di Giulio, D.B.; Eckburg, P.B. Human monkeypox: An emerging zoonosis. Lancet Infect. Dis. 2004, 4, 15–25. [Google Scholar]
- Shisler, J.L. Immune Evasion Strategies of Molluscum Contagiosum Virus. Adv. Virus Res. 2015, 92, 201–252. [Google Scholar] [CrossRef]
- Romero, R.M.; Navarrete-Dechent, C.; Downey, C. Molluscum contagiosum: An update and review of new perspectives in etiology, diagnosis, and treatment. Clin. Cosmet. Investig. Dermatol. 2019, 12, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Abrahao, J.S.; Campos, R.K.; Trindade Gde, S.; Guimaraes da Fonseca, F.; Ferreira, P.C.; Kroon, E.G. Outbreak of severe zoonotic vaccinia virus infection, Southeastern Brazil. Emerg. Infect. Dis. 2015, 21, 695–698. [Google Scholar]
- Lu, B.; Cui, L.-B.; Gu, M.-H.; Shi, C.; Sun, C.-W.; Zhao, K.-C.; Bi, J.; Tan, Z.-M.; Guo, X.-L.; Huo, X.; et al. Outbreak of Vaccinia Virus Infection from Occupational Exposure, China, 2017. Emerg. Infect. Dis. 2019, 25, 1192–1195. [Google Scholar] [CrossRef] [Green Version]
- Lima, M.T.; Oliveira, G.; Afonso, J.A.B.; Souto, R.J.C.; De Mendonça, C.L.; Dantas, A.F.M.; Abrahao, J.S.; Kroon, E.G. An Update on the Known Host Range of the Brazilian Vaccinia Virus: An Outbreak in Buffalo Calves. Front. Microbiol. 2019, 9, 3327. [Google Scholar] [CrossRef] [Green Version]
- Silva, N.I.O.; De Oliveira, J.S.; Kroon, E.G.; Trindade, G.D.S.; Drumond, B.P. Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses. Viruses 2020, 13, 43. [Google Scholar] [CrossRef]
- Wollenberg, A.; Vogel, S.; Sã¡rdy, M.; Glos, K.; Korting, H.; Ruzicka, T. The Munich Outbreak of Cutaneous Cowpox Infection: Transmission by Infected Pet Rats. Acta Derm. Venereol. 2012, 92, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Snoeck, R.; Holý, A.; Dewolf-Peeters, C.; Oord, J.V.D.; De Clercq, E.; Andrei, G. Antivaccinia Activities of Acyclic Nucleoside Phosphonate Derivatives in Epithelial Cells and Organotypic Cultures. Antimicrob. Agents Chemother. 2002, 46, 3356–3361. [Google Scholar] [CrossRef] [Green Version]
- Keith, K.A.; Wan, W.B.; Ciesla, S.L.; Beadle, J.R.; Hostetler, K.Y.; Kern, E.R. Inhibitory Activity of Alkoxyalkyl and Alkyl Esters of Cidofovir and Cyclic Cidofovir against Orthopoxvirus Replication In Vitro. Antimicrob. Agents Chemother. 2004, 48, 1869–1871. [Google Scholar] [CrossRef] [Green Version]
- Hocková, D.; Holý, A.; Masojídková, M.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. 5-Substituted-2,4-diamino-6-[2-(phosphonomethoxy)ethoxy]pyrimidines-acycli c nucleoside phosphonate analogues with an-tiviral activity. J. Med. Chem. 2003, 46, 5064–5073. [Google Scholar]
- De Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar]
- Duraffour, S.; Snoeck, R.; Krecmerová, M.; Oord, J.V.D.; De Vos, R.; Holý, A.; Crance, J.-M.; Garin, D.; De Clercq, E.; Andrei, G. Activities of Several Classes of Acyclic Nucleoside Phosphonates against Camelpox Virus Replication in Different Cell Culture Models. Antimicrob. Agents Chemother. 2007, 51, 4410–4419. [Google Scholar] [CrossRef] [Green Version]
- Dal Pozzo, F.; Andrei, G.; Holy, A.; Van Den Oord, J.; Scagliarini, A.; De Clercq, E.; Snoeck, R. Activities of acyclic nucleoside phosphonates against Orf virus in human and ovine cell monolayers and organotypic ovine raft cultures. Antimicrob Agents Chemother. 2005, 49, 4843–4852. [Google Scholar] [PubMed] [Green Version]
- Lebeau, I.; Andrei, G.; Dal Pozzo, F.; Beadle, J.R.; Hostetler, K.Y.; De Clercq, E.; Van Den Oord, J.; Snoeck, R. Activities of alkoxyalkyl esters of cidofovir (CDV), cyclic CDV, and (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine against orthopoxviruses in cell monolayers and in organotypic cultures. Antimicrob Agents Chemother. 2006, 50, 2525–2529. [Google Scholar] [PubMed] [Green Version]
- Dal Pozzo, F.; Andrei, G.; Lebeau, I.; Beadle, J.R.; Hostetler, K.Y.; De Clercq, E.; Snoeck, R. In vitro evaluation of the anti-orf virus activity of alkoxyalkyl esters of CDV, cCDV and (S)-HPMPA. Antiviral Res. 2007, 75, 52–57. [Google Scholar] [PubMed]
- Hostetler, K.Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antivir. Res. 2009, 82, A84–A98. [Google Scholar] [CrossRef] [Green Version]
- Magee, W.C.; Aldern, K.A.; Hostetler, K.Y.; Evans, D.H. Cidofovir and (S)-9-[3-Hydroxy-(2-Phosphonomethoxy)Propyl]Adenine Are Highly Effective Inhibitors of Vaccinia Virus DNA Polymerase When Incorporated into the Template Strand. Antimicrob. Agents Chemother. 2008, 52, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Zahn, K.; Tchesnokov, E.P.; Götte, M.; Doublié, S. Phosphonoformic Acid Inhibits Viral Replication by Trapping the Closed Form of the DNA Polymerase. J. Biol. Chem. 2011, 286, 25246–25255. [Google Scholar] [CrossRef] [Green Version]
- Czarnecki, M.W.; Traktman, P. The vaccinia virus DNA polymerase and its processivity factor. Virus Res. 2017, 234, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Andrei, G.; Snoeck, R. Cidofovir Activity against Poxvirus Infections. Viruses 2010, 2, 2803–2830. [Google Scholar] [CrossRef] [Green Version]
- Sèle, C.; Gabel, F.; Gutsche, I.; Ivanov, I.; Burmeister, W.P.; Iseni, F.; Tarbouriech, N. Low-Resolution Structure of Vaccinia Virus DNA Replication Machinery. J. Virol. 2012, 87, 1679–1689. [Google Scholar] [CrossRef] [Green Version]
- Andrei, G.; Gammon, D.B.; Fiten, P.; De Clercq, E.; Opdenakker, G.; Snoeck, R.; Evans, D.H. Cidofovir resistance in vaccinia virus is linked to diminished virulence in mice. J. Virol. 2006, 80, 9391–9401. [Google Scholar]
- Farlow, J.; Ichou, M.A.; Huggins, J.; Ibrahim, S. Comparative whole genome sequence analysis of wild-type and cidofovir-resistant monkeypoxvirus. Virol. J. 2010, 7, 110. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.N.; Obraztsova, M.; Kern, E.R.; Quenelle, D.C.; Keith, K.; Prichard, M.N.; Luo, M.; Moyer, R.W. Isolation and characterization of cidofovir resistant vaccinia viruses. Virol. J. 2008, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Kornbluth, R.S.; Smee, D.F.; Sidwell, R.W.; Snarsky, V.; Evans, D.H.; Hostetler, K.Y. Mutations in the E9L polymerase gene of cidofo-vir-resistant vaccinia virus strain WR are associated with the drug resistance phenotype. Antimicrob. Agents Chemother. 2006, 50, 4038–4043. [Google Scholar]
- Gammon, D.B.; Snoeck, R.; Fiten, P.; Krecmerová, M.; Holyý, A.; De Clercq, E.; Opdenakker, G.; Evans, D.; Andrei, G. Mechanism of Antiviral Drug Resistance of Vaccinia Virus: Identification of Residues in the Viral DNA Polymerase Conferring Differential Resistance to Antipoxvirus Drugs. J. Virol. 2008, 82, 12520–12534. [Google Scholar] [CrossRef] [Green Version]
- Duraffour, S.; Andrei, G.; Topalis, D.; Krečmerová, M.; Crance, J.-M.; Garin, D.; Snoeck, R. Mutations Conferring Resistance to Viral DNA Polymerase Inhibitors in Camelpox Virus Give Different Drug-Susceptibility Profiles in Vaccinia Virus. J. Virol. 2012, 86, 7310–7325. [Google Scholar] [CrossRef] [Green Version]
- Taddie, J.A.; Traktman, P. Genetic characterization of the vaccinia virus DNA polymerase: Identification of point mutations conferring altered drug sensitivities and reduced fidelity. J. Virol. 1991, 65, 869–879. [Google Scholar]
- Taddie, J.A.; Traktman, P. Genetic characterization of the vaccinia virus DNA polymerase: Cytosine arabinoside resistance re-quires a variable lesion conferring phosphonoacetate resistance in conjunction with an invariant mutation localized to the 3′-5′ exonuclease domain. J. Virol. 1993, 67, 4323–4336. [Google Scholar]
- Tarbouriech, N.; Ducournau, C.; Hutin, S.; Mas, P.J.; Man, P.; Forest, E.; Hart, D.J.; Peyrefitte, C.N.; Burmeister, W.P.; Iseni, F. The vaccinia virus DNA polymerase structure provides insights into the mode of processivity factor binding. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- DeFilippes, F.M. Site of the base change in the vaccinia virus DNA polymerase gene which confers aphidicolin resistance. J. Virol. 1989, 63, 4060–4063. [Google Scholar]
- Magee, W.C.; Hostetler, K.; Evans, D.H. Mechanism of Inhibition of Vaccinia Virus DNA Polymerase by Cidofovir Diphosphate. Antimicrob. Agents Chemother. 2005, 49, 3153–3162. [Google Scholar] [CrossRef] [Green Version]
- Gammon, D.B.; Evans, D.H. The 3′-to-5′ Exonuclease Activity of Vaccinia Virus DNA Polymerase Is Essential and Plays a Role in Promoting Virus Genetic Recombination. J. Virol. 2009, 83, 4236–4250. [Google Scholar] [CrossRef] [Green Version]
- Balzarini, J.; Pannecouque, C.; Naesens, L.; Andrei, G.; Snoeck, R.; De Clercq, E.; Hockova, D.; Holy, A. 6-[2-(Phosphonomethoxy)alkoxy]-2,4-diaminopyrimidines: A New Class of Acyclic Pyrimidine Nucleoside Phosphonates with Antiviral Activity. Nucleotides Nucleic Acids 2004, 23, 1321–1327. [Google Scholar]
- Andrei, G.; Fiten, P.; Froeyen, M.; De Clercq, E.; Opdenakker, G.; Snoeck, R. DNA polymerase mutations in drug-resistant herpes simplex virus mutants determine in vivo neurovirulence and drug-enzyme interactions. Antivir. Ther. 2007, 12, 719–732. [Google Scholar]
- Andrei, G.; Topalis, D.; Fiten, P.; McGuigan, C.; Balzarini, J.; Opdenakker, G.; Snoeck, R. In Vitro-Selected Drug-Resistant Varicella-Zoster Virus Mutants in the Thymidine Kinase and DNA Polymerase Genes Yield Novel Phenotype-Genotype Associations and Highlight Differences between Antiherpesvirus Drugs. J. Virol. 2011, 86, 2641–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Amino Acid Changes in Viral DNA Polymerase | |||
|---|---|---|---|---|
| Known Natural Occurring Genetic Polymorphisms | Known Drug-Resistant Mutations (Dark Blue) and Novel Mutations (Yellow) | |||
| HPMPA | T845M, G857R, 936–938 ANV | A314T | — | — |
| cHPMPA | T845M, G857R, 936–938 ANV | — | A353V | S851Y |
| cHPMPC | T845M, G857R, 936–938 NΔG | A314T | — | — |
| Drug | Amino Acid Changes in Viral DNA Polymerase | |||||
|---|---|---|---|---|---|---|
| Known Natural Occurring Genetic Polymorphisms | Known Drug-R Mutations (Dark Blue) and Novel Mutations (Yellow) | |||||
| HPMPC (clones 1, 2, 4, 5, 6, 7) | — | A314V | — | A684V | — | — |
| HPMPDAP (clones 1, 2, 3) | — | — | Ins408 (amino acids KDIICKVIH) | — | — | L924F |
| HPMPDAP (clones 4, 5, 6, 7, 8) | — | — | — | — | — | L924F |
| HPMPO-DAPy (clones 1, 4, 5, 6, 7) | — | — | — | — | Ins851L | — |
| A. VACV Mutants Selected under Pressure of HPMPC (Cidofovir). | |||||||||||
| Virus | Clone | Amino Acid Changes in Viral DNA Polymerase | |||||||||
| Known Natural Occurring Genetic Polymorphisms | Known Drug-Resistant Mutations (Dark Blue) and Novel Mutations (Yellow) | ||||||||||
| VV1 | clone 3 | R97H, T845M, G857R, 936–938 NΔG | — | A314T | — | — | A684V | ||||
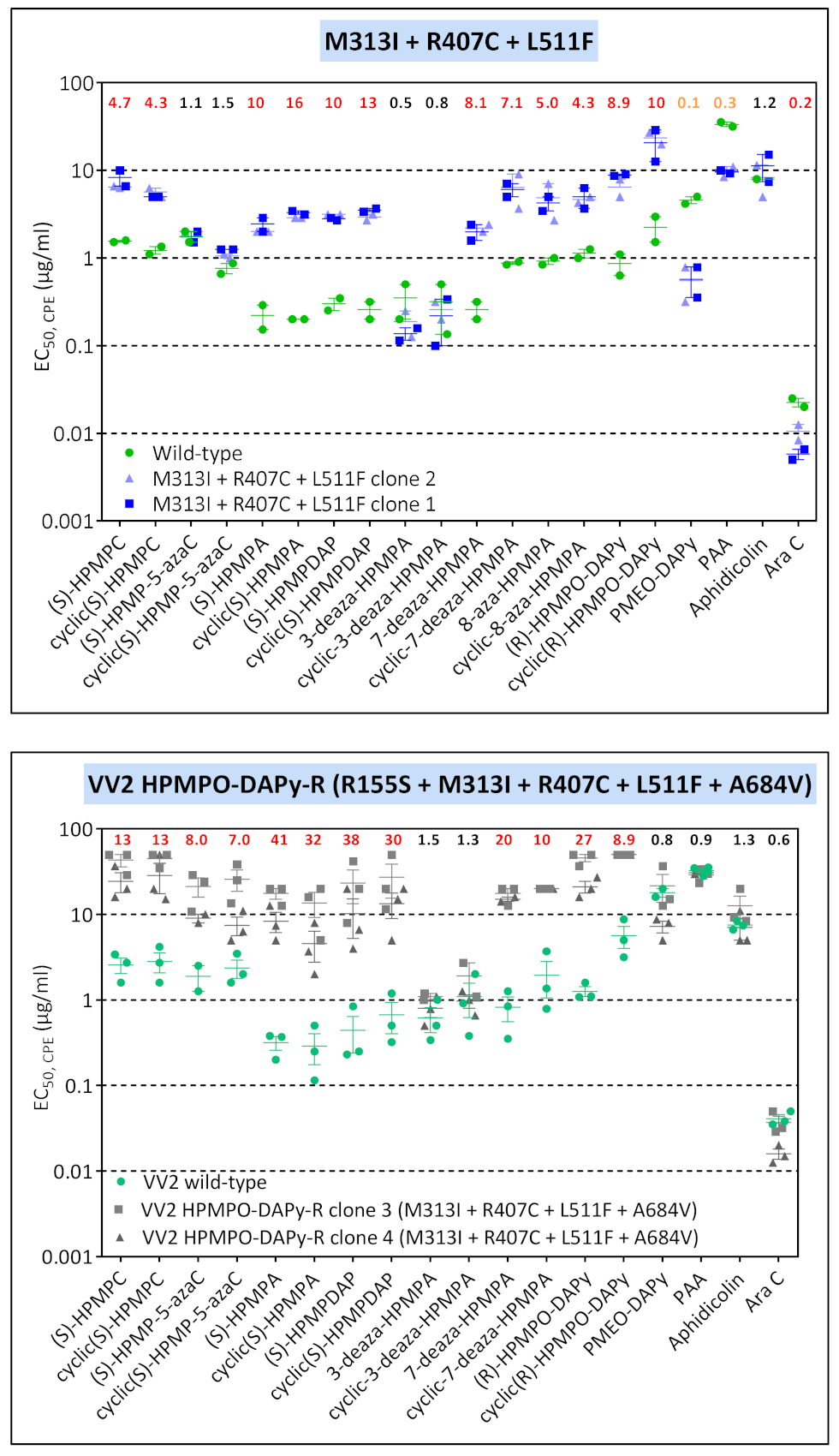
| VV2 | clone 2 | W8C, 936–938 ANV | M313I | — | R577G | A677T | A684V | ||||
| VV8 | clone 1 | R246Q, G857R, 936–938 ANV | — | A314V | L510S | — | A684V | ||||
| VV11 | clone 1 | L90S, R235L, T845M, G857R, S898T, 936–938 NΔG | — | A314T | — | M656I | A684V | ||||
| B. VACV Mutants Selected under Pressure of HPMPDAP. | |||||||||||
| Virus | Clone | Amino Acid Changes in Viral DNA Polymerase | |||||||||
| Known Natural Occurring Genetic Polymorphisms | Known Drug-Resistant Mutations (Dark Blue) and Novel Mutations (Yellow) | ||||||||||
| VV1 | clone 1 | R23G, L420S, T845M, G857R, 936–938 NΔG | A314V | — | T500I | — | T688A | — | |||
| VV2 | clone 3 | T71I, T845M, G857R, 936–938 NΔG | — | D480Y | — | A684V | — | A705T | |||
| VV8 | clone 1 | R246Q, T845M, G857R, 936–938 NΔG | A314T | — | — | A684V | — | — | |||
| VV11 | clone 1 | I68T, L420S, T845M, G857R, C865S, 936–938 NΔG | A314T | — | — | A684V | — | L696S | |||
| C. VACV Mutants Selected under Pressure of HPMPO-DAPy. | |||||||||||
| Virus | Clone | Amino Acid Changes in Viral DNA Polymerase | |||||||||
| Known Natural Occurring Genetic Polymorphisms | Known Drug-Resistant Mutations (Dark Blue) and Novel Mutations (Yellow) | ||||||||||
| VV1 | clone 3 | T845M, G857R, 936–938 NΔG | — | — | — | — | — | — | A632T | — | S851Y |
| VV2 | clone 3 | T845M, G857R, 936–938 NΔG | R155S | M313I | - | R407C | L511F | — | — | A684V | — |
| VV8 | clone 1 | R246Q, T845M, G857R, 936–938 NΔG | — | M313I | — | — | — | A613T | — | A684V | — |
| VV11 | clone 1 | T845M, G857R, 936–938 NΔG | G138E | — | A314T | — | — | — | — | — | — |
| A. VACV and CPXV Mutants Selected under Pressure of PAA. | |||||||
| Virus | Clone | Amino Acid Changes in Viral DNA Polymerase | |||||
| Known Natural Occurring Genetic Polymorphisms | Known Drug-Resistant Mutations (Dark Blue) and Novel Mutations (Yellow) | ||||||
| VV1 | clone 1 | T845M, G857R, 936–938 NΔG | — | — | F354L | — | — |
| VV2 | clone 1 | T845M, G857R, 936–938 NΔG | — | — | — | A684V | — |
| VV8 | clone 1 | R246Q, G857R | 117Ins. GISPD | — | — | — | E792D |
| VV11 | clone 1 | L420S, T845M, G857R, 936–938 NΔG | — | Y332H | — | — | — |
| B. VACV Mutants Selected under Pressure of PMEO-DAPy. | |||||||
| Virus | Clone | Amino Acid Changes in Viral DNA Polymerase | |||||
| Known Natural Occurring Genetic Polymorphisms | Novel Drug-Resistant Mutations (Yellow) | ||||||
| VV1 | clone 1 | L420, T845M, 936–938 ANV | G372C | — | |||
| VV2 | clone 1 | T845M, 936–938 NΔG | — | E856K | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrei, G.; Fiten, P.; Krečmerová, M.; Opdenakker, G.; Topalis, D.; Snoeck, R. Poxviruses Bearing DNA Polymerase Mutations Show Complex Patterns of Cross-Resistance. Biomedicines 2022, 10, 580. https://doi.org/10.3390/biomedicines10030580
Andrei G, Fiten P, Krečmerová M, Opdenakker G, Topalis D, Snoeck R. Poxviruses Bearing DNA Polymerase Mutations Show Complex Patterns of Cross-Resistance. Biomedicines. 2022; 10(3):580. https://doi.org/10.3390/biomedicines10030580
Chicago/Turabian StyleAndrei, Graciela, Pierre Fiten, Marcela Krečmerová, Ghislain Opdenakker, Dimitrios Topalis, and Robert Snoeck. 2022. "Poxviruses Bearing DNA Polymerase Mutations Show Complex Patterns of Cross-Resistance" Biomedicines 10, no. 3: 580. https://doi.org/10.3390/biomedicines10030580
APA StyleAndrei, G., Fiten, P., Krečmerová, M., Opdenakker, G., Topalis, D., & Snoeck, R. (2022). Poxviruses Bearing DNA Polymerase Mutations Show Complex Patterns of Cross-Resistance. Biomedicines, 10(3), 580. https://doi.org/10.3390/biomedicines10030580