
High-Throughput Drug Library Screening in Primary KMT2A-Rearranged Infant ALL Cells Favors the Identification of Drug Candidates That Activate P53 Signaling

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Cell Line Cultures
2.3. High-Throughput Drug Library Screening and Viability Assays
2.4. Western Blotting
2.5. Cloning of the P53 Wild-Type and P53 Mutant into an Inducible Vector
2.6. Transfection, Lentivirus Production, and Transduction
2.7. Apoptosis Assay
3. Results
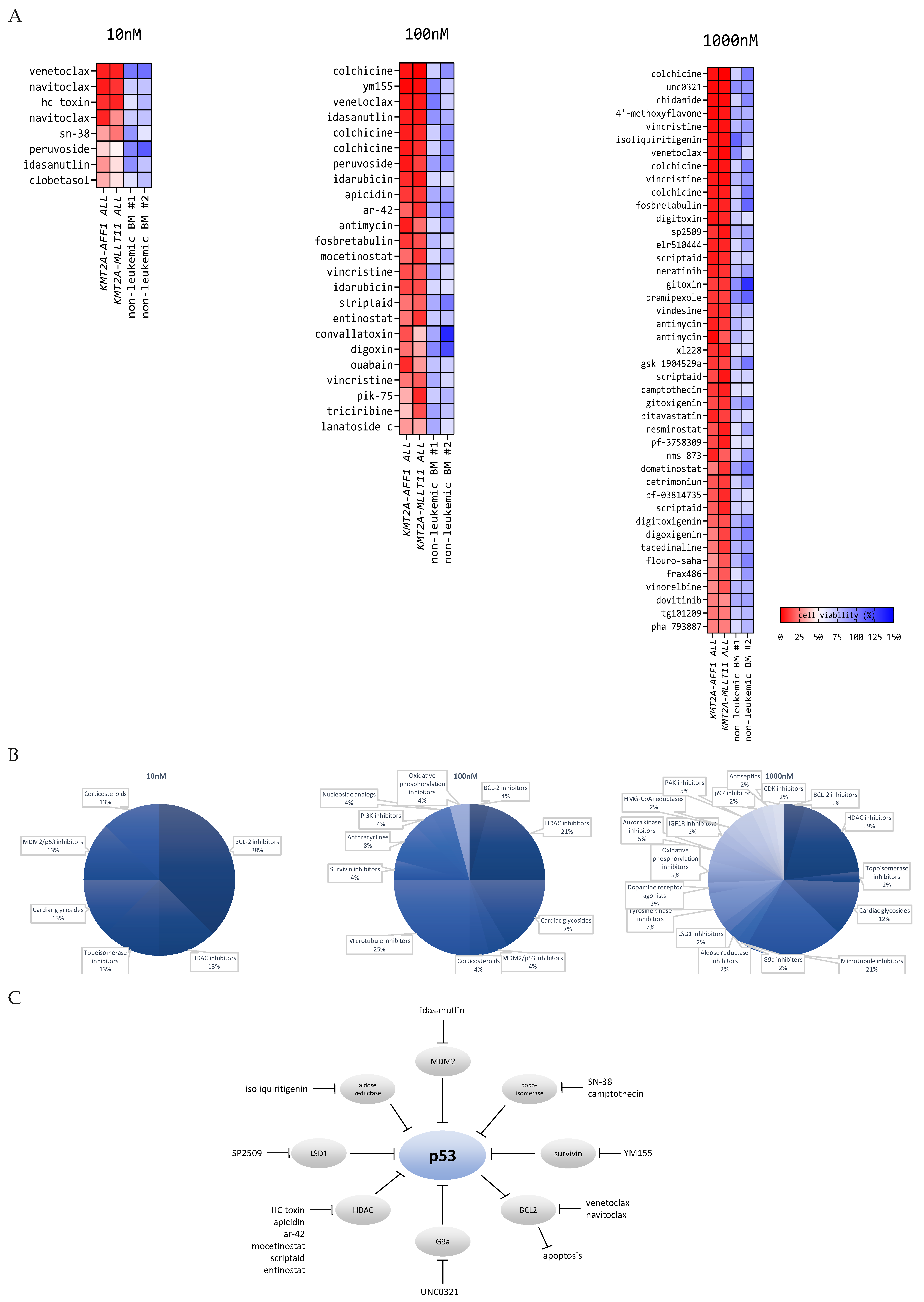
3.1. Drug Library Screens on Primary KMT2A-Rearranged Infant ALL Cells Reveal Attractive Candidate Drugs Favoring P53 Activation
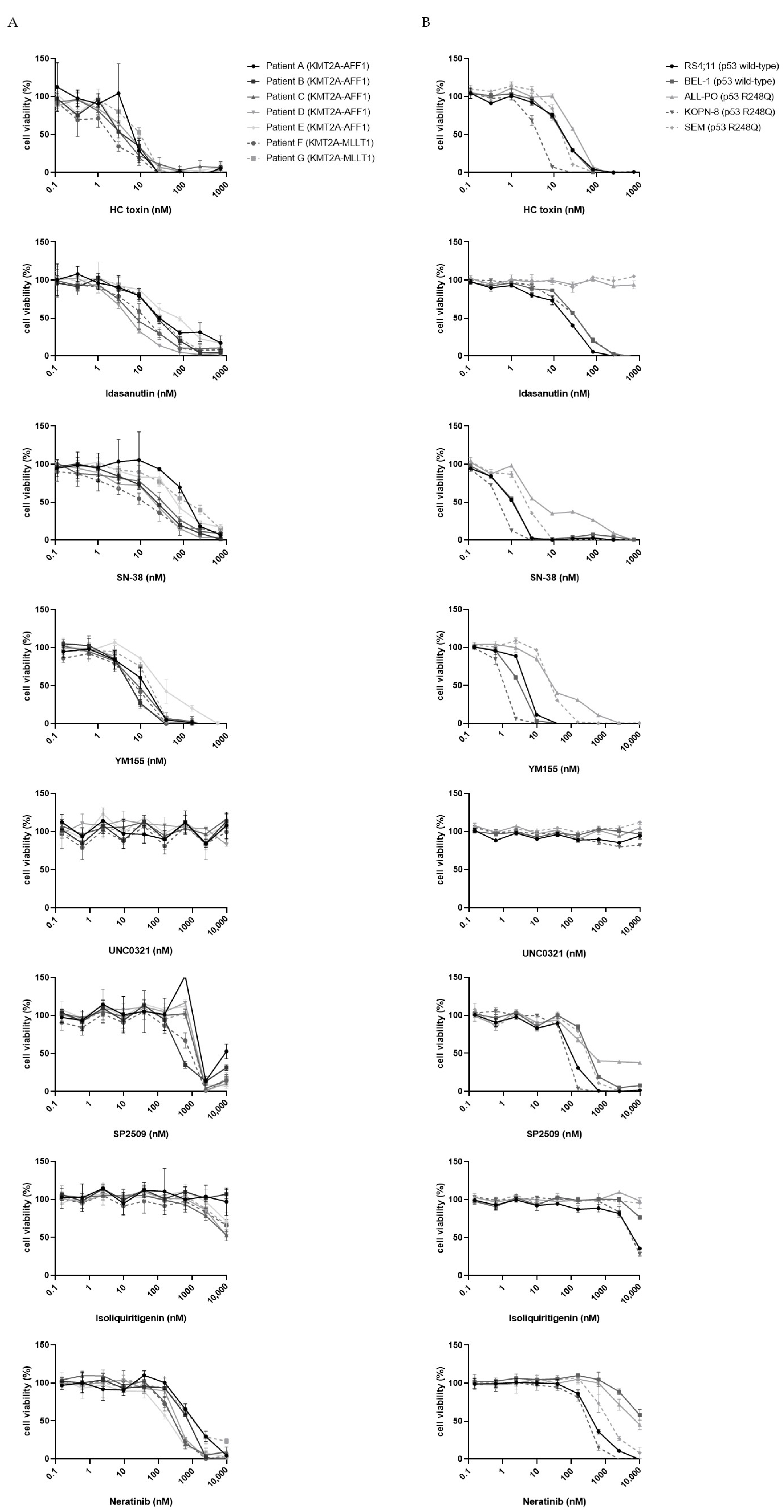
3.2. Validation of Candidate Drugs on Additional Primary KMT2A-Rearranged Infant ALL Samples and KMT2A-Rearranged ALL Cell Lines
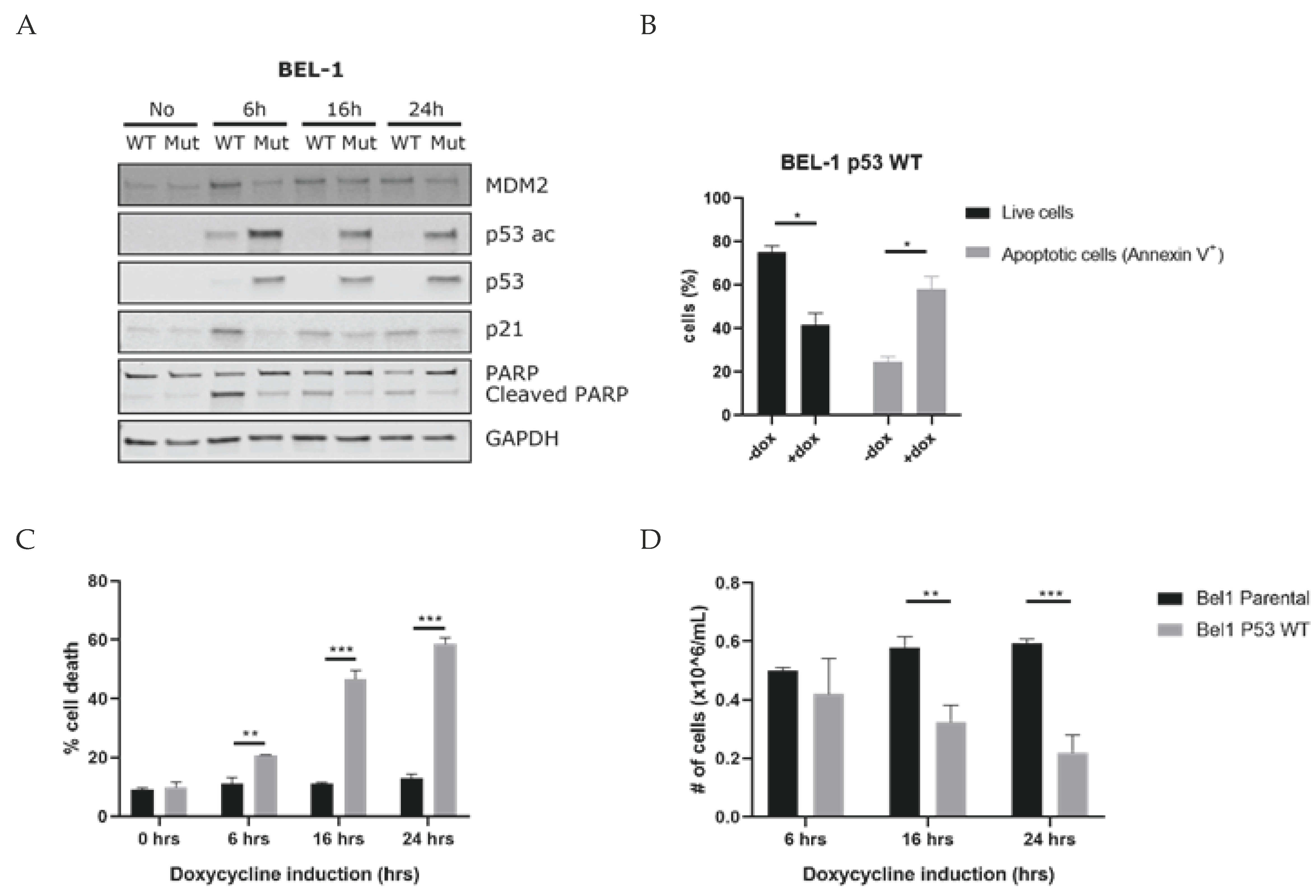
3.3. Selected Drug Candidates Affect P53 Signaling in KMT2A-Rearranged ALL Cells
3.4. Induction of P53 Leads to Apoptosis of KMT2A-Rearranged ALL Cells
3.5. MDM2 Inhibitors Are Effective against KMT2A-Rearranged ALL Cells Carrying Wild-Type P53
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Pieters, R.; Schrappe, M.; De Lorenzo, P.; Hann, I.; De Rossi, G.; Felice, M.; Hovi, L.; LeBlanc, T.; Szczepanski, T.; Ferster, A.; et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): An observational study and a multicentre randomised trial. Lancet 2007, 370, 240–250. [Google Scholar] [CrossRef]
- Pieters, R.; De Lorenzo, P.; Ancliffe, P.; Aversa, L.A.; Brethon, B.; Biondi, A.; Campbell, M.; Escherich, G.; Ferster, A.; Gardner, R.A.; et al. Outcome of Infants Younger Than 1 Year with Acute Lymphoblastic Leukemia Treated with the Interfant-06 Protocol: Results from an International Phase III Randomized Study. J. Clin. Oncol. 2019, 37, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W.; Schneider, P.; Hagelstein, J.A.P.; Van Der Linden, M.H.; Stumpel, D.J.P.M.; De Menezes, R.X.; De Lorenzo, P.; Valsecchi, M.; Maria, G.; Pieters, R. Gene expression profiling–based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 2010, 115, 2835–2844. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; Boer, M.D.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2001, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Stumpel, D.J.P.M.; Schneider, P.; Van Roon, E.H.J.; Boer, J.M.; De Lorenzo, P.; Valsecchi, M.G.; De Menezes, R.X.; Pieters, R.; Stam, R.W. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood 2009, 114, 5490–5498. [Google Scholar] [CrossRef]
- Schafer, E.; Irizarry, R.; Negi, S.; McIntyre, E.; Small, D.; Figueroa, M.E.; Melnick, A.; Brown, P. Promoter hypermethylation in MLL-r infant acute lymphoblastic leukemia: Biology and therapeutic targeting. Blood 2010, 115, 4798–4809. [Google Scholar] [CrossRef] [Green Version]
- Tejedor, J.R.; Bueno, C.; Vinyoles, M.; Petazzi, P.; Agraz-Doblas, A.; Cobo, I.; Torres-Ruiz, R.; Bayón, G.F.; Pérez, R.F.; López-Tamargo, S.; et al. Integrative methylome-transcriptome analysis unravels cancer cell vulnerabilities in infant MLL-rearranged B cell acute lymphoblastic leukemia. J. Clin. Investig. 2021, 131, e138833. [Google Scholar] [CrossRef]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Stam, R.W. Unravelling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 2019, 104, 1176. [Google Scholar] [CrossRef]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.; Feng, Z.; Punt, N.; Kedaigle, A.; Bullinger, L.; et al. MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug. Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Mansinho, A.; Boni, V.; Miguel, M.; Calvo, E. New designs in early clinical drug development. Ann. Oncol. 2019, 30, 1460–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V.G. Update on raloxifene: Role in reducing the risk of invasive breast cancer in postmenopausal women. Breast Cancer Dove Med. Press 2011, 3, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Kerstjens, M.; Castro, P.G.; Pinhanços, S.; Schneider, P.; Wander, P.; Pieters, R.; Stam, R. Irinotecan Induces Disease Remission in Xenograft Mouse Models of Pediatric MLL-Rearranged Acute Lymphoblastic Leukemia. Biomedicines 2021, 9, 711. [Google Scholar] [CrossRef] [PubMed]
- Kaspers, G.J.; Veerman, A.J.; Pieters, R.; Broekema, G.J.; Huismans, D.R.; Kazemier, K.M.; Loonen, A.H.; Rottier, M.A.; Van Zantwijk, C.H.; Hählen, K.; et al. Mononuclear cells contaminating acute lymphoblastic leukaemic samples tested for cellular drug resistance using the methyl-thiazol-tetrazolium assay. Br. J. Cancer 1994, 70, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Wander, P.; Arentsen-Peters, S.T.; Pinhanҫos, S.S.; Koopmans, B.; Dolman, M.M.; Ariese, R.; Bos, F.L.; Castro, P.G.; Jones, L.; Schneider, P.; et al. High-throughput drug screening reveals Pyrvinium pamoate as effective candidate against pediatric MLL-rearranged acute myeloid leukemia. Transl. Oncol. 2021, 14, 101048. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2017, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Benito, J.M.; Godfrey, L.; Kojima, K.; Hogdal, L.; Wunderlich, M.; Geng, H.; Marzo, I.; Harutyunyan, K.G.; Golfman, L.; North, P.; et al. MLL-Rearranged Acute Lymphoblastic Leukemias Activate BCL-2 through H3K79 Methylation and Are Sensitive to the BCL-2-Specific Antagonist ABT-199. Cell Rep. 2015, 13, 2715–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaw, S.L.; Suryani, S.; Evans, K.; Richmond, J.; Robbins, A.; Kurmasheva, R.T.; Billups, C.A.; Erickson, S.W.; Guo, Y.; Houghton, P.J.; et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood 2016, 128, 1382–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpel, D.J.P.M.; Schneider, P.; Seslija, L.; Osaki, H.; Williams, O.; Pieters, R.; Stam, R.W. Connectivity mapping identifies HDAC inhibitors for the treatment of t(4;11)-positive infant acute lymphoblastic leukemia. Leukemia 2011, 26, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido Castro, P.; Van Roon, E.H.J.; Pinhanços, S.S.; Trentin, L.; Schneider, P.; Kerstjens, M.; Stam, R.W. The HDAC inhibitor panobinostat (LBH589) exerts in vivo anti-leukaemic activity against MLL-rearranged acute lymphoblastic leukaemia and involves the RNF20/RNF40/WAC-H2B ubiquitination axis. Leukemia 2018, 32, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Katryniok, C.; Scholz, B.; Merkens, J.; Löscher, D.; Marschalek, R.; Steinhilber, D. Inhibition of class I HDACs abrogates the dominant effect of MLL-AF4 by activation of wild-type MLL. Oncogenesis 2014, 3, e127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnemann, J.; Marx, C.; Becker, S.; Wittig, S.; Palani, C.D.; Krämer, O.H.; Beck, J.F. p53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. Br. J. Cancer 2014, 110, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeba, Y.; Kumai, T.; Matsumoto, N.; Nakaya, S.; Tsuzuki, Y.; Yanagida, Y.; Kobayashi, S. Irinotecan Activates p53 With Its Active Metabolite, Resulting in Human Hepatocellular Carcinoma Apoptosis. J. Pharmacol. Sci. 2007, 104, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Tyner, J.W.; Jemal, A.M.; Thayer, M.; Druker, B.J.; Chang, B.H. Targeting survivin and p53 in pediatric acute lymphoblastic leukemia. Leukemia 2011, 26, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dorsey, J.; Chuikov, S.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S. G9a and Glp Methylate Lysine 373 in the Tumor Suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Sengupta, R.; Espejo, A.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Kuo, P.-L.; Lin, C.-C. Isoliquiritigenin induces apoptosis and cell cycle arrest through p53-dependent pathway in Hep G2 cells. Life Sci. 2005, 77, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J.D. HC-toxin. Phytochemistry 2006, 67, 1406–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a Potent and Selective p53–MDM2 Inhibitor in Clinical Development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Villalona-Calero, M.A. Irinotecan: Mechanisms of tumor resistance and novel strategies for modulating its activity. Ann. Oncol. 2002, 13, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Hennig, D.; Schäfer, C.; Wirth, M.; Marx, C.; Heinzel, T.; Krämer, O.H. Survivin and YM155: How faithful is the liaison? Biochim. Biophys. Acta 2014, 1845, 202–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wigle, T.J.; Wasney, G.A.; Dong, A.; Senisterra, G.; Chau, I.; Siarheyeva, A.; et al. Protein Lysine Methyltransferase G9a Inhibitors: Design, Synthesis, and Structure Activity Relationships of 2,4-Diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 2010, 53, 5844–5857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskus, W.; Sharma, S.; Shah, B.K.; Portier, B.P.; Devaraj, S.G.T.; Liu, K.; Iyer, S.P.; Bearss, D.; Bhalla, K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014, 28, 2155–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.-X.; Wen, Y.; He, J.-L.; Huang, S.-Z.; Gao, F.; Guo, C.-J.; Liu, Q.-Q.; Zheng, S.-W.; Gong, D.-Y.; Li, Y.-Z.; et al. Isoliquiritigenin, an Orally Available Natural FLT3 Inhibitor from Licorice, Exhibits Selective Anti-Acute Myeloid Leukemia Efficacy In Vitro and In Vivo. Mol. Pharmacol. 2019, 96, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Mendoza, M.; E González-González, M.; Barrera, D.; Díaz, L.; García-Becerra, R. Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: Preclinical and clinical evidence. Am. J. Cancer Res. 2015, 5, 2531–2561. [Google Scholar] [PubMed]
- Tchelebi, L.; Ashamalla, H.; Graves, P.R. Mutant p53 and the Response to Chemotherapy and Radiation. Subcell. Biochem. 2014, 85, 133–159. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Wiederschain, D.; Kawai, H.; Gu, J.; Shilatifard, A.; Yuan, Z.-M. Molecular Basis of p53 Functional Inactivation by the Leukemic Protein MLL-ELL. Mol. Cell. Biol. 2003, 23, 4230–4246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederschain, D.; Kawai, H.; Shilatifard, A.; Yuan, Z.-M. Multiple Mixed Lineage Leukemia (MLL) Fusion Proteins Suppress p53-mediated Response to DNA Damage. J. Biol. Chem. 2005, 280, 24315–24321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, K.; Mitani, K.; Yamagata, T.; Kurokawa, M.; Kanda, Y.; Yazaki, Y.; Hirai, H. Transcriptional Inhibition of p53 by the MLL/MEN Chimeric Protein Found in Myeloid Leukemia. Blood 1999, 93, 3216–3224. [Google Scholar] [CrossRef] [PubMed]
- Richmond, J.; Carol, H.; Evans, K.; High, L.; Mendomo, A.; Robbins, A.; Meyer, C.; Venn, N.C.; Marschalek, R.; Henderson, M.; et al. Effective Targeting of the P53–MDM2 Axis in Preclinical Models of Infant MLL-Rearranged Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2015, 21, 1395–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wander, P.; Arentsen-Peters, S.T.C.J.M.; Vrenken, K.S.; Pinhanҫos, S.M.; Koopmans, B.; Dolman, M.E.M.; Jones, L.; Garrido Castro, P.; Schneider, P.; Kerstjens, M.; et al. High-Throughput Drug Library Screening in Primary KMT2A-Rearranged Infant ALL Cells Favors the Identification of Drug Candidates That Activate P53 Signaling. Biomedicines 2022, 10, 638. https://doi.org/10.3390/biomedicines10030638
Wander P, Arentsen-Peters STCJM, Vrenken KS, Pinhanҫos SM, Koopmans B, Dolman MEM, Jones L, Garrido Castro P, Schneider P, Kerstjens M, et al. High-Throughput Drug Library Screening in Primary KMT2A-Rearranged Infant ALL Cells Favors the Identification of Drug Candidates That Activate P53 Signaling. Biomedicines. 2022; 10(3):638. https://doi.org/10.3390/biomedicines10030638
Chicago/Turabian StyleWander, Priscilla, Susan T. C. J. M. Arentsen-Peters, Kirsten S. Vrenken, Sandra Mimoso Pinhanҫos, Bianca Koopmans, M. Emmy M. Dolman, Luke Jones, Patricia Garrido Castro, Pauline Schneider, Mark Kerstjens, and et al. 2022. "High-Throughput Drug Library Screening in Primary KMT2A-Rearranged Infant ALL Cells Favors the Identification of Drug Candidates That Activate P53 Signaling" Biomedicines 10, no. 3: 638. https://doi.org/10.3390/biomedicines10030638
APA StyleWander, P., Arentsen-Peters, S. T. C. J. M., Vrenken, K. S., Pinhanҫos, S. M., Koopmans, B., Dolman, M. E. M., Jones, L., Garrido Castro, P., Schneider, P., Kerstjens, M., Molenaar, J. J., Pieters, R., Zwaan, C. M., & Stam, R. W. (2022). High-Throughput Drug Library Screening in Primary KMT2A-Rearranged Infant ALL Cells Favors the Identification of Drug Candidates That Activate P53 Signaling. Biomedicines, 10(3), 638. https://doi.org/10.3390/biomedicines10030638