
Stathmins and Motor Neuron Diseases: Pathophysiology and Therapeutic Targets

 , ,
, , 
{kind=link}
Abstract
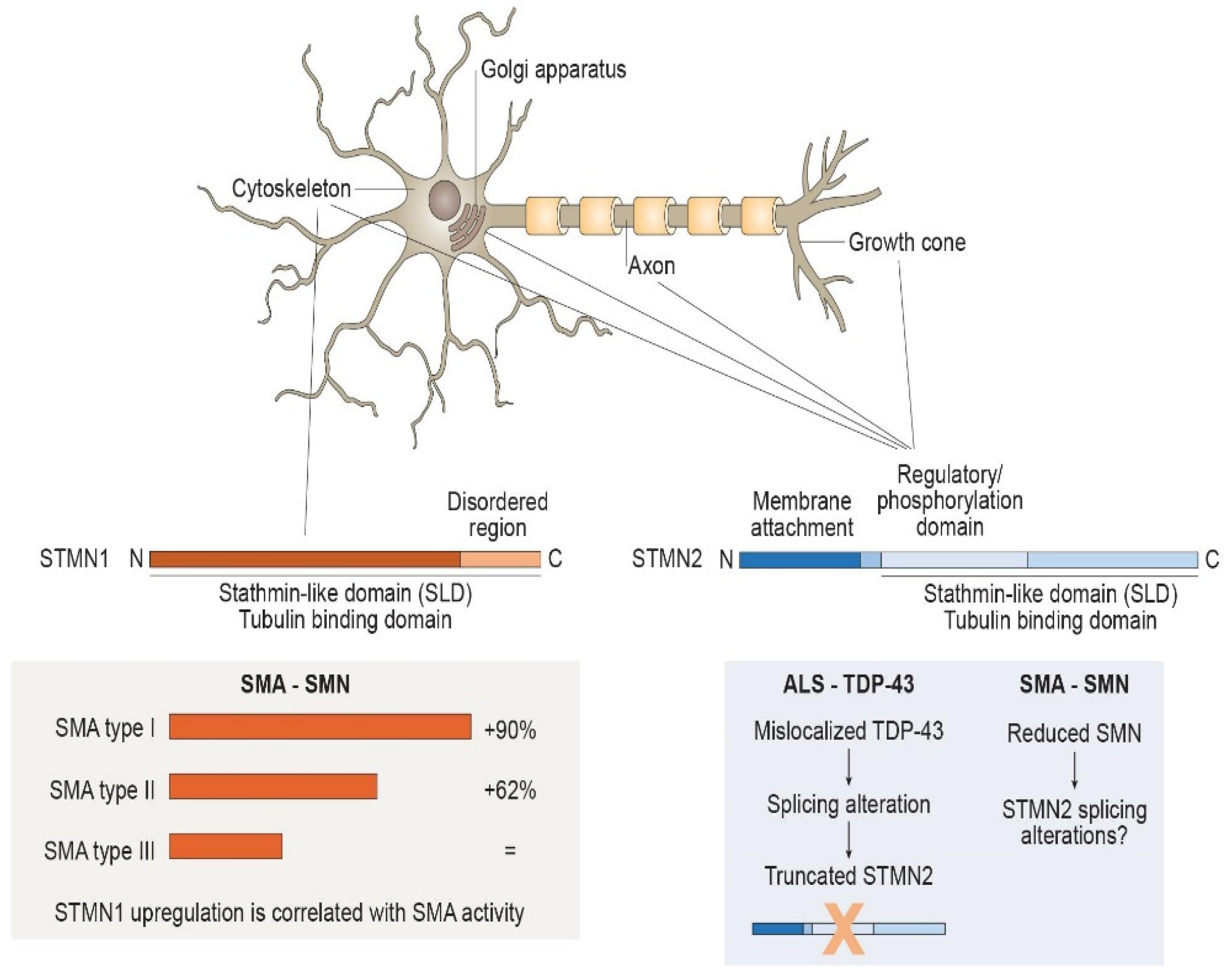
:1. Stathmins Are Relevant for Axonal Stability
2. Stathmins in Disorders Affecting MNs
2.1. Stathmin and Vulnerable MNs
2.2. Stathmins in Spinal Muscular Atrophy
2.3. Stathmins in Amyotrophic Lateral Sclerosis
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kapitein, L.C.; Hoogenraad, C.C. Building the Neuronal Microtubule Cytoskeleton. Neuron 2015, 87, 492–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauvin, S.; Sobel, A. Neuronal stathmins: A family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Prog. Neurobiol. 2015, 126, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.I.; Atweh, G.F. The role of stathmin in the regulation of the cell cycle. J. Cell. Biochem. 2004, 93, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Sobel, A.; Boutterin, M.C.; Beretta, L.; Chneiweiss, H.; Doye, V.; Peyro-Saint-Paul, H. Intracellular Substrates for Extracellular Signaling. Characterization of a ubiquitous, neuron-enriched phosphoprotein (stathmin). J. Biol. Chem. 1989, 264, 3765–3772. [Google Scholar] [CrossRef]
- Curmi, P.A.; Gavet, O.; Charbaut, E.; Ozon, S.; Lachkar-Colmerauer, S.; Manceau, V.; Siavoshian, S.; Maucuer, A.; Sobel, A. Stathmin and its Phosphoprotein Family. General Properties, Biochemical and Functional Interaction with Tubulin. Cell Struct. Funct. 1999, 24, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozon, S.; Byk, T.; Sobel, A. SCLIP: A novel SCG10-like protein of the stathmin family expressed in the nervous system. J. Neurochem. 1998, 70, 2386–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmetz, M.O.; Jahnke, W.; Towbin, H.; García-Echeverría, C.; Voshol, H.; Müller, D.; Van Oostrum, J. Phosphorylation disrupts the central helix in Op18/stathmin and suppresses binding to tubulin. EMBO Rep. 2001, 2, 505–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamed, Z.; López-Erauskin, J.; Baughn, M.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Lutjens, R.; Osen-Sand, A.; Catsicas, S.; Grenningloh, G. Differential distribution of stathmin and SCG10 in developing neurons in culture. J. Neurosci. Res. 1997, 50, 1000–1009. [Google Scholar] [CrossRef]
- Westerlund, N.; Zdrojewska, J.; Courtney, M.J.; Coffey, E.T. Superior cervical ganglion-10 protein as a molecular effector of c-Jun N-terminal kinase 1: Implications for the therapeutic targeting of Jun N-terminal kinase in nerve regeneration. Expert Opin. Ther. Targets 2008, 12, 31–43. [Google Scholar] [CrossRef]
- Shin, J.E.; Geisler, S.; DiAntonio, A. Dynamic regulation of SCG10 in regenerating axons after injury. Exp. Neurol. 2014, 252, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “Dying-Back” Phenomenon of Motor Neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.E.; Lytle, N.K.; Zuniga, A.; Goldstein, L.S.B. The Microtubule Regulatory Protein Stathmin Is Required to Maintain the Integrity of Axonal Microtubules in Drosophila. PLoS ONE 2013, 8, e68324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, E.R.; Heerssen, H.M.; Wright, C.M.; Davis, G.W.; DiAntonio, A. Stathmin is Required for Stability of the Drosophila Neuromuscular Junction. J. Neurosci. 2011, 31, 15026–15034. [Google Scholar] [CrossRef] [PubMed]
- Klim, J.R.; Williams, L.A.; Limone, F.; San Juan, I.G.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Bellouze, S.; Baillat, G.; Buttigieg, D.; De La Grange, P.; Rabouille, C.; Haase, G. Stathmin 1/2-triggered microtubule loss mediates Golgi fragmentation in mutant SOD1 motor neurons. Mol. Neurodegener. 2016, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Comley, L.H.; Nijssen, J.; Frost-Nylen, J.; Hedlund, E. Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J. Comp. Neurol. 2016, 524, 1424–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, K.K.Y.; Gibbs, R.M.; Feng, Z.; Ko, C.-P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012, 21, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, M.E.; Bieniek, K.F.; Greenberg, M.B.; DeJesus-Hernandez, M.; Rutherford, N.J.; Van Blitterswijk, M.; Niemantsverdriet, E.; Ash, P.E.; Gendron, T.F.; Kouri, N.; et al. Progressive amnestic dementia, hippocampal sclerosis, and mutation in C9ORF72. Acta Neuropathol. 2013, 126, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, L.M.; Comley, L.H.; Thomson, D.; Parkinson, N.; Talbot, K.; Gillingwater, T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008, 17, 949–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gizzi, M.; DiRocco, A.; Sivak, M.; Cohen, B. Ocular motor function in motor neuron disease. Neurology 1992, 42, 1037. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Sakakihara, Y.; Uchiyama, Y.; Nara, A.; Nagata, T.; Nitta, H.; Ishimoto, K.; Oka, A.; Horio, K.; Yanagisawa, M. New ocular movement detector system as a communication tool in ventilator-assisted Werdnig-Hoffmann disease. Dev. Med. Child Neurol. 2000, 42, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, E.; Karlsson, M.; Osborn, T.; Ludwig, W.; Isacson, O. Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain 2010, 133, 2313–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achsel, T.; Barabino, S.; Cozzolino, M.; Carrì, M.T. The intriguing case of motor neuron disease: ALS and SMA come closer. Biochem. Soc. Trans. 2013, 41, 1593–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockington, A.; Ning, K.; Heath, P.R.; Wood, E.; Kirby, J.; Fusi, N.; Lawrence, N.; Wharton, S.B.; Ince, P.G.; Shaw, P.J. Unravelling the enigma of selective vulnerability in neurodegeneration: Motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013, 125, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comley, L.; Allodi, I.; Nichterwitz, S.; Nizzardo, M.; Simone, C.; Corti, S.; Hedlund, E. Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience 2015, 291, 216–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizzardo, M.; Taiana, M.; Rizzo, F.; Benitez, J.A.; Nijssen, J.; Allodi, I.; Melzi, V.; Bresolin, N.; Comi, G.P.; Hedlund, E.; et al. Synaptotagmin 13 is neuroprotective across motor neuron diseases. Acta Neuropathol. 2020, 139, 837–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, R.A.; Kaifer, K.A.; Osman, E.; Carella, F.; Tiberi, A.; Ross, J.; Pennetta, G.; Lorson, C.L.; Murray, L.M. Comparison of independent screens on differentially vulnerable motor neurons reveals alpha-synuclein as a common modifier in motor neuron diseases. PLoS Genet. 2017, 13, e1006680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allodi, I.; Comley, L.; Nichterwitz, S.; Nizzardo, M.; Simone, C.; Benitez, J.A.; Cao, M.; Corti, S.; Hedlund, E. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 2016, 6, 25960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.-L.; Lin, Y.-T.; Ting, C.-H.; Lin-Chao, S.; Li, H.; Hsieh-Li, H.M. Stathmin, a microtubule-destabilizing protein, is dysregulated in spinal muscular atrophy. Hum. Mol. Genet. 2010, 19, 1766–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.-L.; Ting, C.-H.; Liu, H.-C.; Li, H.; Lin-Chao, S. Decreased stathmin expression ameliorates neuromuscular defects but fails to prolong survival in a mouse model of spinal muscular atrophy. Neurobiol. Dis. 2013, 52, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bsibsi, M.; Bajramovic, J.J.; Vogt, M.H.J.; van Duijvenvoorden, E.; Baghat, A.; Persoon-Deen, C.; Tielen, F.; Verbeek, R.; Huitinga, I.; Ryffel, B.; et al. The Microtubule Regulator Stathmin Is an Endogenous Protein Agonist for TLR3. J. Immunol. 2010, 184, 6929–6937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, F.; Nizzardo, M.; Vashisht, S.; Molteni, E.; Melzi, V.; Taiana, M.; Salani, S.; Santonicola, P.; Di Schiavi, E.; Bucchia, M.; et al. Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain 2019, 142, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Corti, S.; Nizzardo, M.; Simone, C.; Falcone, M.; Nardini, M.; Ronchi, D.; Donadoni, C.; Salani, S.; Riboldi, G.; Magri, F.; et al. Genetic Correction of Human Induced Pluripotent Stem Cells from Patients with Spinal Muscular Atrophy. Sci. Transl. Med. 2012, 4, 165ra162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalón, E.; Kline, R.A.; Smith, C.; Lorson, Z.C.; Osman, E.; O’Day, S.; Murray, L.M.; Lorson, C.L. AAV9-Stathmin1 gene delivery improves disease phenotype in an intermediate mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2019, 28, 3742–3754. [Google Scholar] [CrossRef] [PubMed]
- Tararuk, T.; Östman, N.; Li, W.; Björkblom, B.; Padzik, A.; Zdrojewska, J.; Hongisto, V.; Herdegen, T.; Konopka, W.; Courtney, M.; et al. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J. Cell Biol. 2006, 173, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, J.D. Stathmin-2: Adding another piece to the puzzle of TDP-43 proteinopathies and neurodegeneration. J. Clin. Investig. 2020, 130, 5677–5680. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Glass, J.D. Axonal Degeneration in Motor Neuron Disease. Neurodegener. Dis. 2007, 4, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal Transport of TDP-43 mRNA Granules Is Impaired by ALS-Causing Mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.B.; Lee, V.M.-Y.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2011, 13, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, M.; Humphrey, J.; Pickles, S.; Brown, A.-L.; Hill, S.E.; Kachergus, J.M.; Shi, J.; Heckman, M.G.; Spiegel, M.R.; Cook, C.; et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Investig. 2020, 130, 6080–6092. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, F.; Anderton, R.S.; Mastaglia, F.L.; Flynn, L.L.; Winter, S.J.; James, I.; Bedlack, R.; Hodgetts, S.; Fletcher, S.; Wilton, S.D.; et al. Novel STMN2 Variant Linked to Amyotrophic Lateral Sclerosis Risk and Clinical Phenotype. Front. Aging Neurosci. 2021, 13, 658226. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wang, M.; Song, W.-M.; Shen, Q.; McKenzie, A.; Choi, I.; Zhou, X.; Pan, P.-Y.; Yue, Z.; et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun. 2019, 10, 5234. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, D.; Faravelli, I.; Meneri, M.; Saccomanno, D.; Govoni, A.; Magri, F.; Ricci, G.; Siciliano, G.; Comi, G.P.; Corti, S. Diagnostic and prognostic value of CSF neurofilaments in a cohort of patients with motor neuron disease: A cross-sectional study. J. Cell. Mol. Med. 2021, 25, 3765–3771. [Google Scholar] [CrossRef] [PubMed]
- Zeugin, D.; Ionta, S. Anatomo-Functional Origins of the Cortical Silent Period: Spotlight on the Basal Ganglia. Brain Sci. 2021, 11, 705. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagliardi, D.; Pagliari, E.; Meneri, M.; Melzi, V.; Rizzo, F.; Comi, G.P.; Corti, S.; Taiana, M.; Nizzardo, M. Stathmins and Motor Neuron Diseases: Pathophysiology and Therapeutic Targets. Biomedicines 2022, 10, 711. https://doi.org/10.3390/biomedicines10030711
Gagliardi D, Pagliari E, Meneri M, Melzi V, Rizzo F, Comi GP, Corti S, Taiana M, Nizzardo M. Stathmins and Motor Neuron Diseases: Pathophysiology and Therapeutic Targets. Biomedicines. 2022; 10(3):711. https://doi.org/10.3390/biomedicines10030711
Chicago/Turabian StyleGagliardi, Delia, Elisa Pagliari, Megi Meneri, Valentina Melzi, Federica Rizzo, Giacomo Pietro Comi, Stefania Corti, Michela Taiana, and Monica Nizzardo. 2022. "Stathmins and Motor Neuron Diseases: Pathophysiology and Therapeutic Targets" Biomedicines 10, no. 3: 711. https://doi.org/10.3390/biomedicines10030711
APA StyleGagliardi, D., Pagliari, E., Meneri, M., Melzi, V., Rizzo, F., Comi, G. P., Corti, S., Taiana, M., & Nizzardo, M. (2022). Stathmins and Motor Neuron Diseases: Pathophysiology and Therapeutic Targets. Biomedicines, 10(3), 711. https://doi.org/10.3390/biomedicines10030711