
Periodontal Disease Augments Cardiovascular Disease Risk Biomarkers in Rheumatoid Arthritis

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Rheumatoid Arthritis Group
2.2. Periodontal Disease Group
2.3. Periodontal Examination
- Bleeding on probing (BOP)
- ΣPPD Total
- ΣPPD Disease
- Adjusted PPD Total
- Adjusted PPD Diseased sites
- Σ Marginal bone loss (MBL)
- Adjusted ΣMBL
2.4. Anthropometric Measures
2.5. Glycated Hemoglobin (HbA1c)
2.6. Proteomic Profiling
2.7. Protein–Protein Interaction (PPI) Network Analysis
2.8. Statistical Analyses
3. Results
3.1. Characteristics of Study Groups
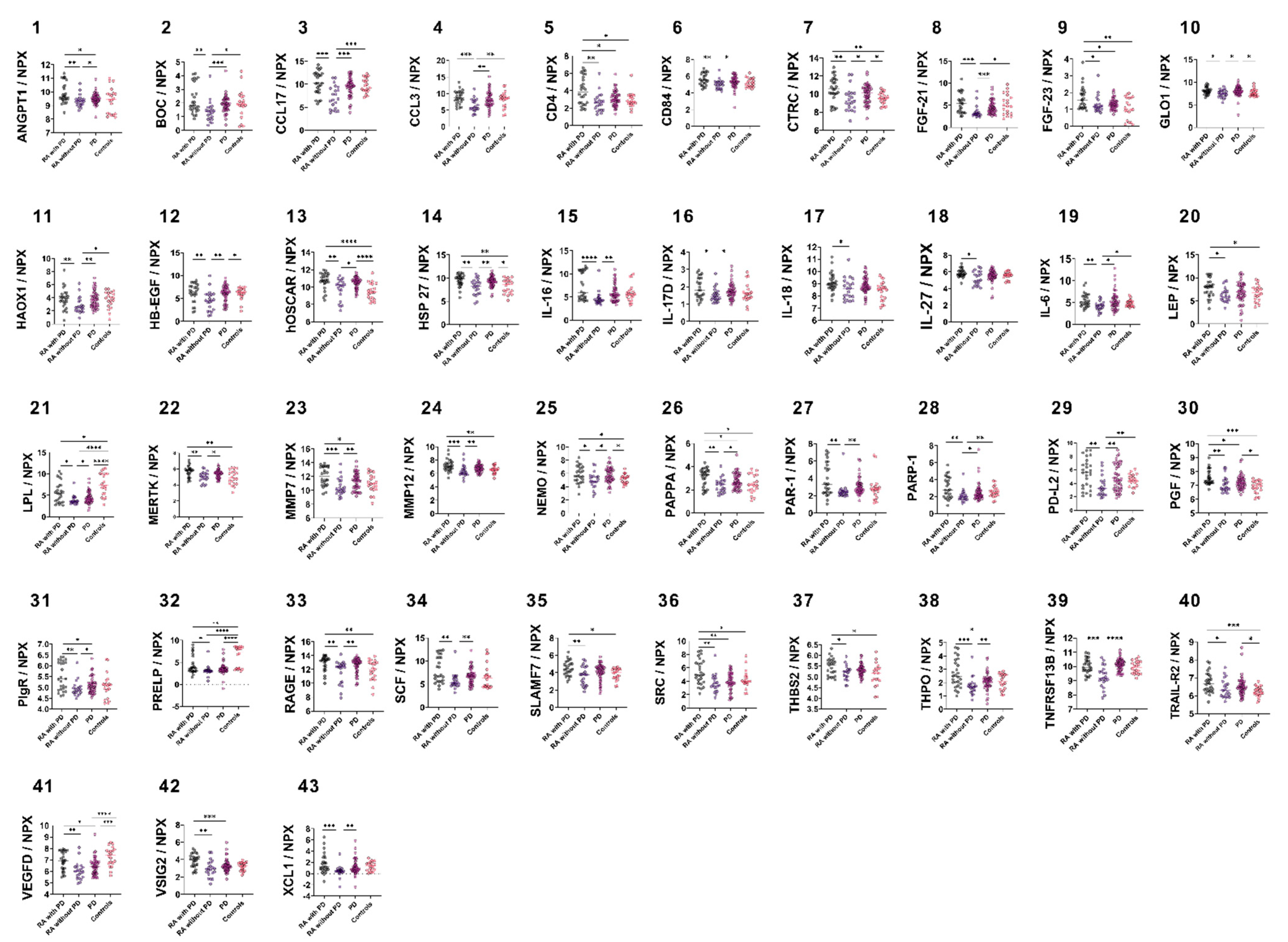
3.2. Group-Wise Biomarker Distribution
3.3. Correlation of CVD Biomarkers with Periodontal Parameters
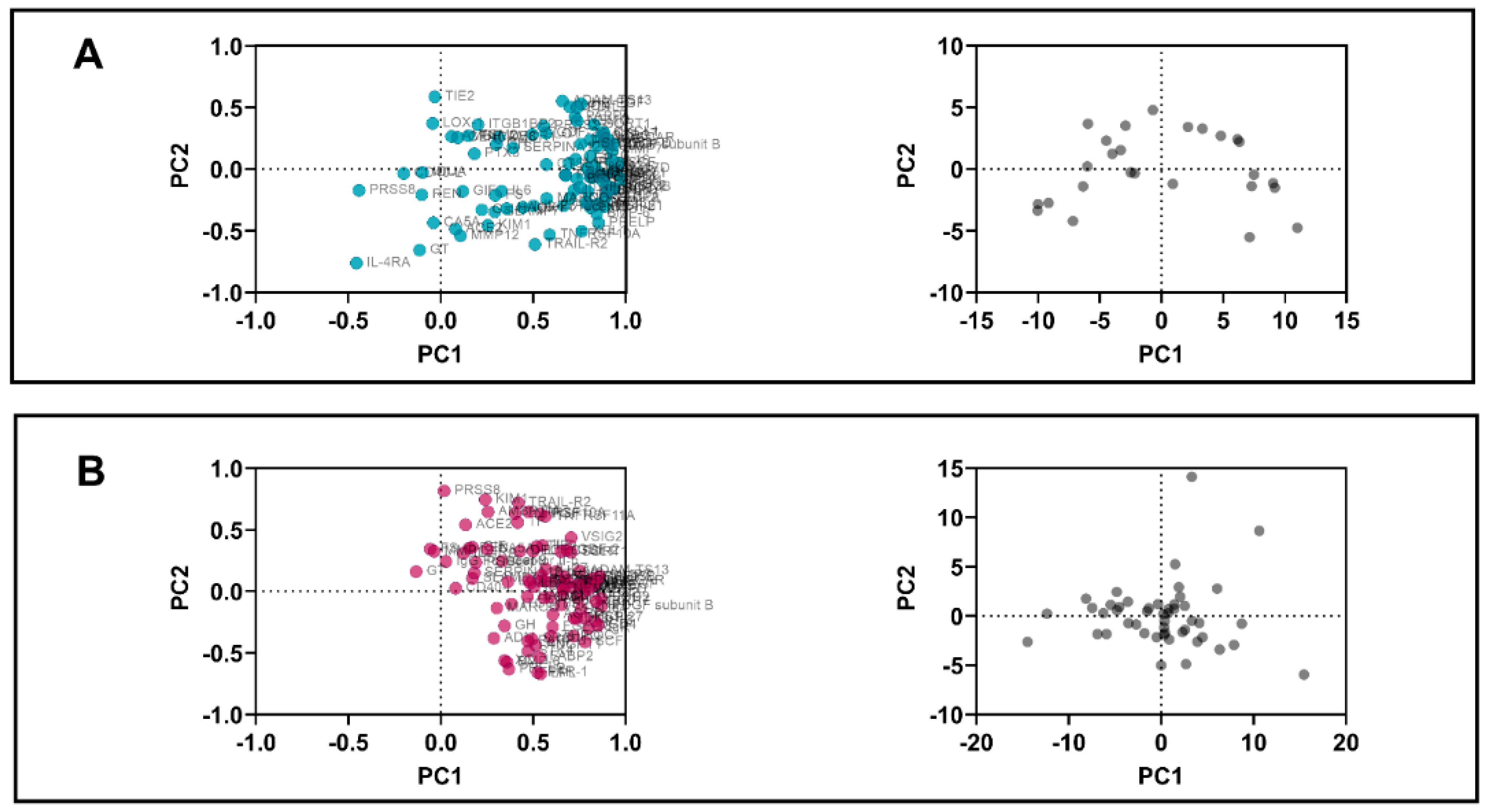
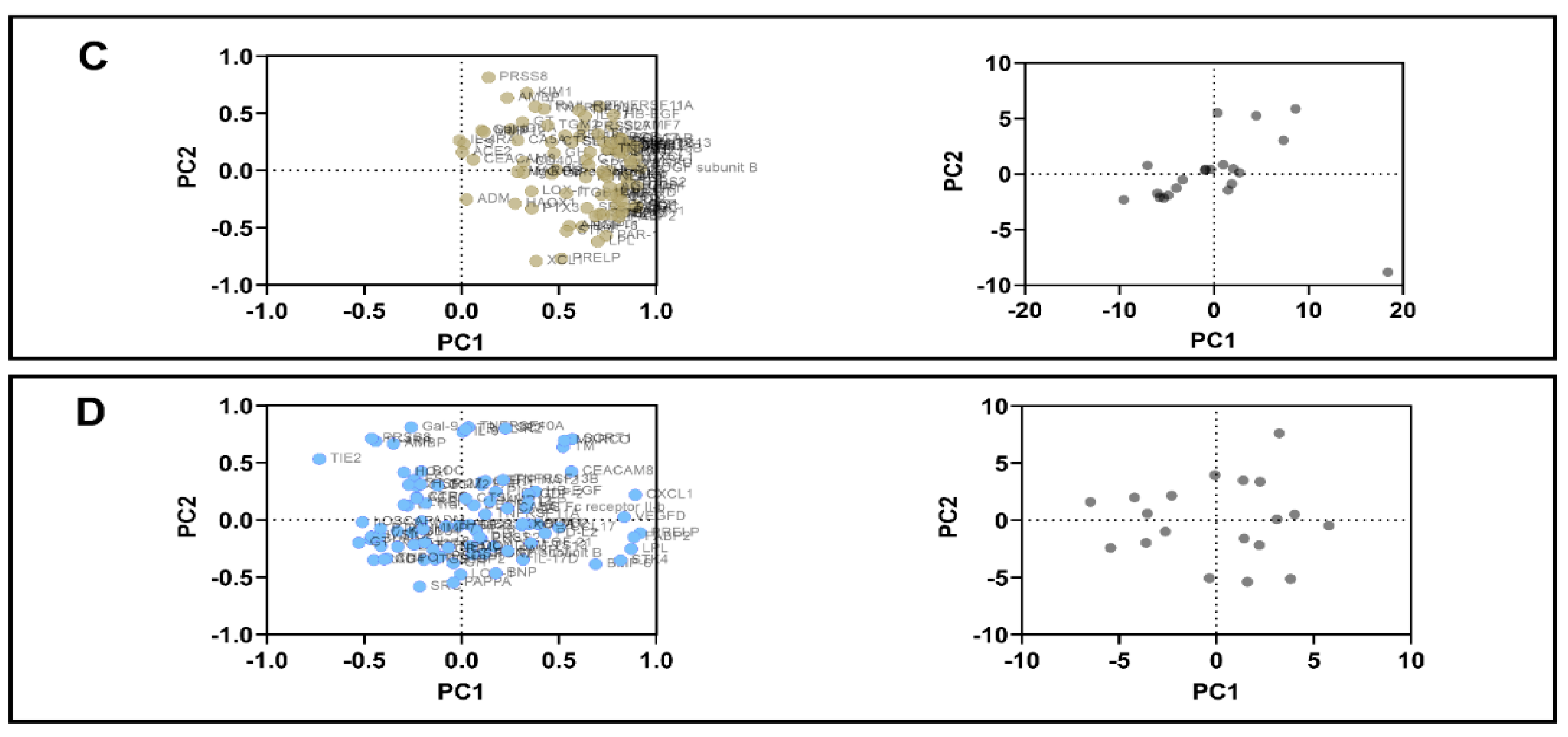
3.4. PCA
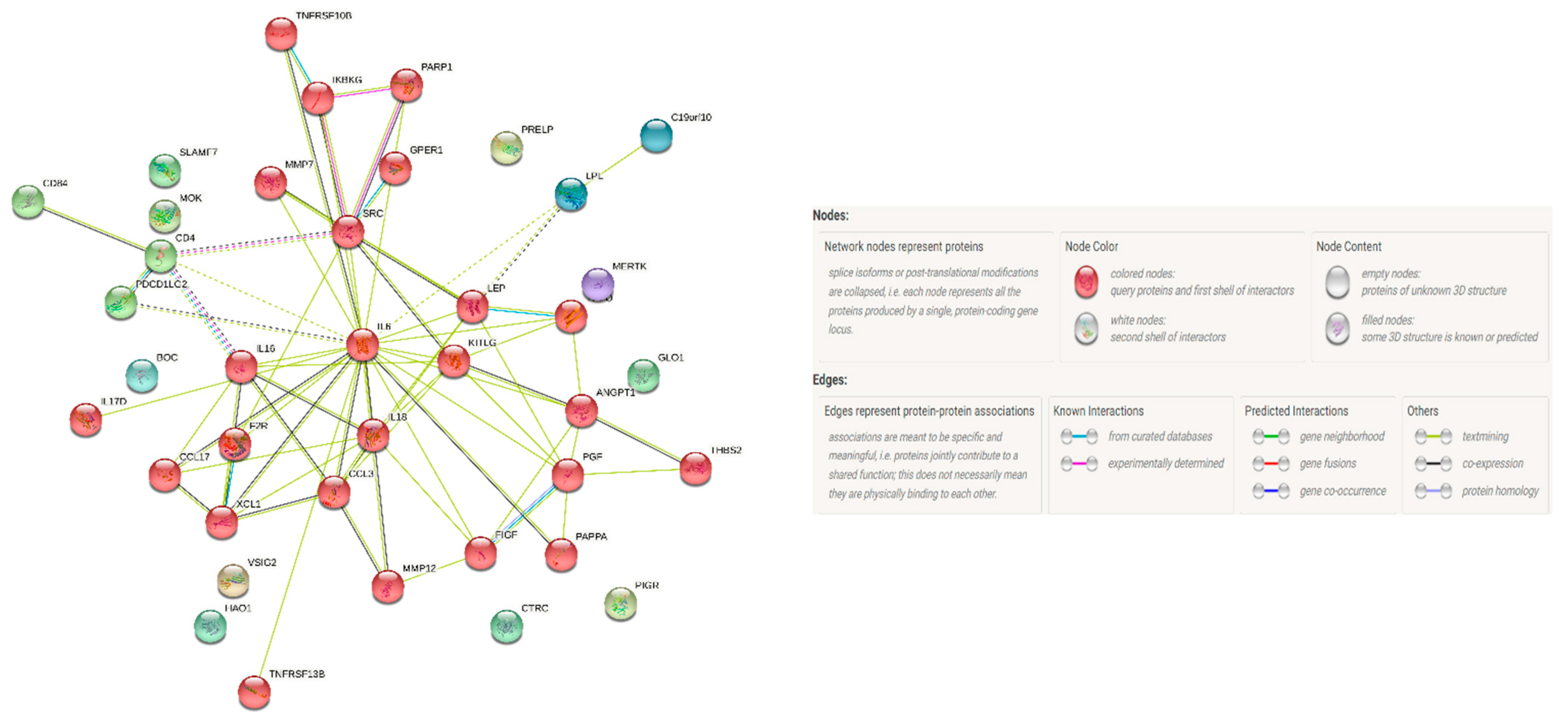
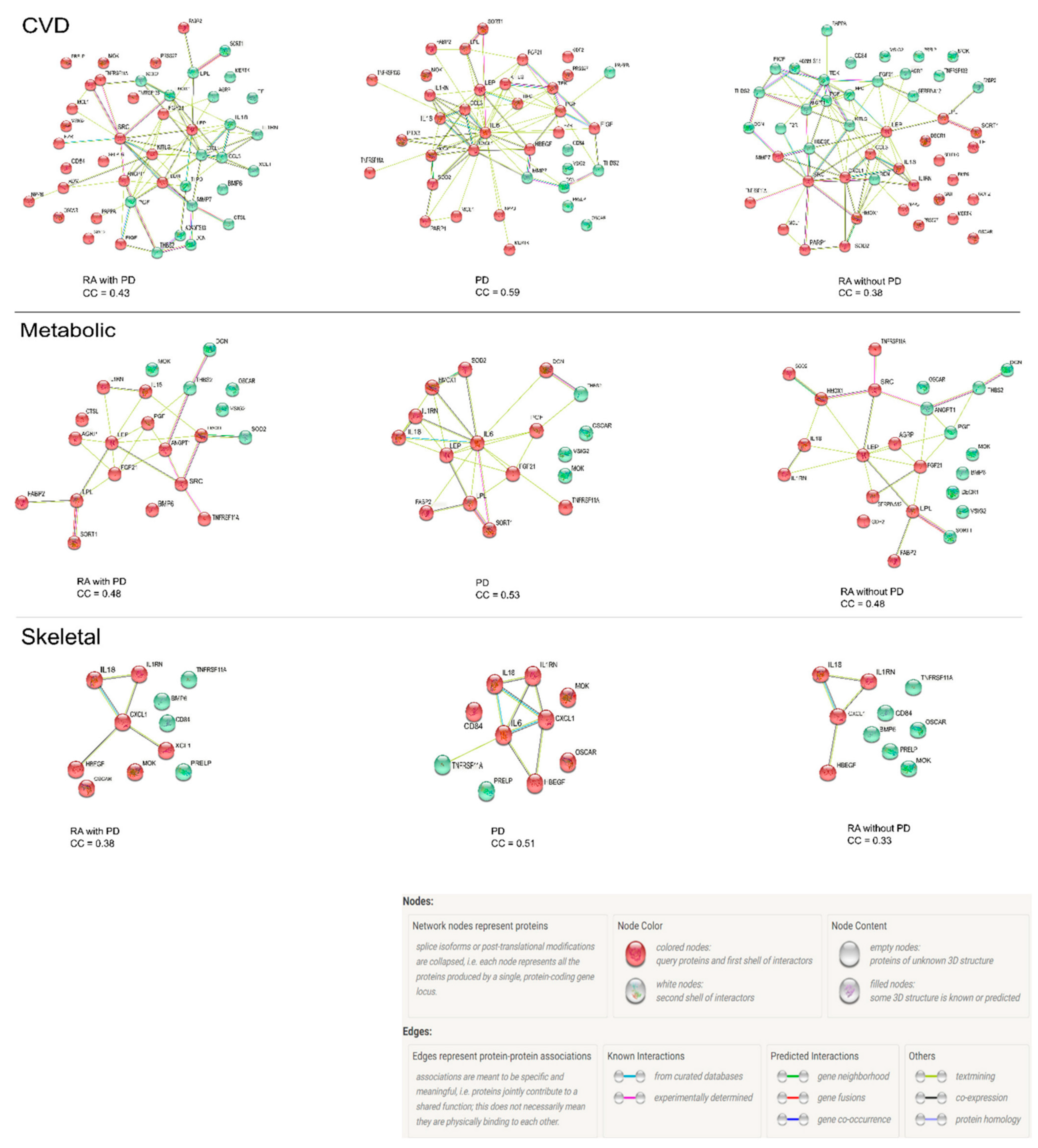
3.5. Protein–Protein Interaction Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hajishengallis, G.; Moutsopoulos, N.M.; Hajishengallis, E.; Chavakis, T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin. Immunol. 2016, 28, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lin, L.; Wu, H.; Yan, L.; Wang, H.; Yang, H.; Li, H. Global, Regional, and National Death, and Disability-Adjusted Life-Years (DALYs) for Cardiovascular Disease in 2017 and Trends and Risk Analysis from 1990 to 2017 Using the Global Burden of Disease Study and Implications for Prevention. Public Health Front. 2021, 9, 559751. [Google Scholar] [CrossRef]
- Gallino, A.; Aboyans, V.; Diehm, C.; Cosentino, F.; Stricker, H.; Falk, E.; Schouten, O.; Lekakis, J.; Amann-Vesti, B.; Siclari, F.; et al. Non-coronary atherosclerosis. Eur. Heart J. 2014, 35, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Kobiyama, K.; Ley, K. Atherosclerosis. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef]
- Beukers, N.G.; van der Heijden, G.J.; van Wijk, A.J.; Loos, B.G. Periodontitis is an independent risk indicator for atherosclerotic cardiovascular diseases among 60174 participants in a large dental school in the Netherlands. J. Epidemiol. Community Health 2017, 71, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Makkar, H.; Reynolds, M.A.; Wadhawan, A.; Dagdag, A.; Merchant, A.T.; Postolache, T.T. Periodontal, metabolic, and cardiovascular disease: Exploring the role of inflammation and mental health. Pteridines 2018, 29, 124–163. [Google Scholar] [CrossRef]
- Aviña-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Care Res. 2008, 59, 1690–1697. [Google Scholar] [CrossRef]
- Santos, M.J.; Cordeiro, A.C.; Gil, V.M. Biomarkers of Cardiometabolic Risk, Inflammation and Disease; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Sergeant, ESG, 2018. Epitools Epidemiological Calculators. Ausvet. Available online: http://epitools.ausvet.com (accessed on 20 May 2021).
- Potikuri, D.; Dannana, K.C.; Kanchinadam, S.; Agrawal, S.; Kancharla, A.; Rajasekhar, L.; Pothuraju, S.; Gumdal, N. Periodontal disease is significantly higher in non-smoking treatment-naive rheumatoid arthritis patients: Results from a case-control study. Ann. Rheum. Dis. 2012, 71, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Panezai, J.; Ali, A.; Ghaffar, A.; Benchimol, D.; Altamash, M.; Klinge, B.; Engström, P.E.; Larsson, A. Upregulation of circulating inflammatory biomarkers under the influence of periodontal disease in rheumatoid arthritis patients. Cytokine 2020, 131, 155117. [Google Scholar] [CrossRef] [PubMed]
- Panezai, J.; Ghaffar, A.; Altamash, M.; Sundqvist, K.G.; Engström, P.E.; Larsson, A. Correlation of serum cytokines, chemokines, growth factors and enzymes with periodontal disease parameters. PLoS ONE 2017, 12, e0188945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, R.R.; Rohlfing, C.L.; Sacks, D.B. National Glycohemoglobin Standardization Program (NGSP) Steering Committee Status of hemoglobin A1c measurement and goals for improvement: From chaos to order for improving diabetes care. Clin. Chem. 2011, 57, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Bjorkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Rennel Dickens, E.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021, customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R Stat. Soc. Ser. B Stat. Methodol. 1995, 1, 289–300. [Google Scholar] [CrossRef]
- Lipsky, M.S.; Su, S.; Crespo, C.J.; Hung, M. Men and Oral Health: A Review of Sex and Gender Differences. Am. J. Men’s Health 2021, 15, 15579883211016361. [Google Scholar] [CrossRef]
- Fransen, J.; Kazemi-Bajestani, S.M.; Bredie, S.J.; Popa, C.D. Rheumatoid Arthritis Disadvantages Younger Patients for Cardiovascular Diseases: A Meta-Analysis. PLoS ONE 2016, 11, e0157360. [Google Scholar] [CrossRef]
- Lindström, J.; Tuomilehto, J. The diabetes risk score: A practical tool to predict type 2 diabetes risk. Diabetes Care 2003, 26, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Thalhamer, T.; McGrath, M.A.; Harnett, M.M. MAPKs and their relevance to arthritis and inflammation. Rheumatology 2008, 47, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Braz, N.F.; Pinto, M.R.; Vieira, É.L.; Souza, A.J.; Teixeira, A.L.; Simoes-e-Silva, A.C.; Kakehasi, A.M. Renin–angiotensin system molecules are associated with subclinical atherosclerosis and disease activity in rheumatoid arthritis. Mod. Rheumatol. 2021, 31, 119–126. [Google Scholar] [CrossRef] [PubMed]
- CieŚlik, P.; Hrycek, A. Long pentraxin 3 (PTX3) in the light of its structure, mechanism of action and clinical implications. Autoimmunity 2012, 45, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Ito, H.; Akiyoshi, M.; Kume, N.; Yoshitomi, H.; Mitsuoka, H.; Tanida, S.; Murata, K.; Shibuya, H.; Kasahara, T.; et al. Lectin-like oxidized low-density lipoprotein receptor 1 signal is a potent biomarker and therapeutic target for human rheumatoid arthritis. Arthritis Rheum. 2012, 64, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Barreto, J.; Karathanasis, S.K.; Remaley, A.; Sposito, A.C. Role of LOX-1 (lectin-like oxidized low-density lipoprotein receptor 1) as a cardiovascular risk predictor: Mechanistic insight and potential clinical use. Arter. Thromb. Vasc. Biol. 2021, 41, 153–166. [Google Scholar] [CrossRef]
- Pradeep, A.R.; Roopa, Y.; Swati, P.P. Interleukin-4, a T-helper 2 cell cytokine, is associated with the remission of periodontal disease. J. Periodontal Res. 2008, 43, 712–716. [Google Scholar] [CrossRef]
- Dufour, A.; Bellac, C.L.; Eckhard, U.; Solis, N.; Klein, T.; Kappelhoff, R.; Fortelny, N.; Jobin, P.; Rozmus, J.; Mark, J.; et al. C-terminal truncation of IFN-γ inhibits proinflammatory macrophage responses and is deficient in autoimmune disease. Nature Commun. 2018, 9, 2416. [Google Scholar] [CrossRef]
- Czaya, B.; Faul, C. The role of fibroblast growth factor 23 in inflammation and anemia. Int. J. Mol. Sci. 2019, 20, 4195. [Google Scholar] [CrossRef] [Green Version]
- Rossini, M.; Viapiana, O.; Adami, S.; Fracassi, E.; Idolazzi, L.; Dartizio, C.; Povino, M.R.; Orsolini, G.; Gatti, D. In patients with rheumatoid arthritis Dickkopf-1 serum levels are correlated with parathyroid hormone, bone erosions and bone mineral density. Clin. Exp. Rheumatol. 2015, 33, 77–83. [Google Scholar]
- Park, Y.W.; Kang, Y.M.; Butterfield, J.; Detmar, M.; Goronzy, J.J.; Weyand, C.M. Thrombospondin 2 functions as an endogenous regulator of angiogenesis and inflammation in rheumatoid arthritis. Am. J. Pathol. 2004, 165, 2087–2098. [Google Scholar] [CrossRef] [Green Version]
- Wannamethee, S.G.; Tchernova, J.; Whincup, P.; Lowe, G.D.; Kelly, A.; Rumley, A.; Wallace, A.M.; Sattar, N. Plasma leptin: Associations with metabolic, inflammatory and haemostatic risk factors for cardiovascular disease. Atherosclerosis 2007, 191, 418–426. [Google Scholar] [CrossRef]
- Abe, T.; AlSarhan, M.; Benakanakere, M.R.; Maekawa, T.; Kinane, D.F.; Cancro, M.P.; Korostoff, J.M.; Hajishengallis, G. The B cell-stimulatory cytokines BLyS and APRIL are elevated in human periodontitis and are required for B cell-dependent bone loss in experimental murine periodontitis. J. Immunol. 2015, 195, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Chang, Y.; Wei, W. The role of BAFF in the progression of rheumatoid arthritis. Cytokine. 2015, 76, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Al Shahi, H.; Shimada, K.; Miyauchi, K.; Yoshihara, T.; Sai, E.; Shiozawa, T.; Naito, R.; Aikawa, T.; Ouchi, S.; Kadoguchi, T.; et al. Elevated circulating levels of inflammatory markers in patients with acute coronary syndrome. Int. J. Vasc. Med. 2015, 2015, 805375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarese, G.; Currò, M.; Isola, G.; Caccamo, D.; Vecchio, M.; Giunta, M.L.; Ramaglia, L.; Cordasco, G.; Williams, R.C.; Ientile, R. Transglutaminase 2 up-regulation is associated with RANKL/OPG pathway in cultured HPDL cells and THP-1-differentiated macrophages. Amino Acids 2015, 47, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO Hub, Web Presence Working Group. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Song, I.S.; Han, K.; Park, Y.M.; Ji, S.; Jun, S.H.; Ryu, J.J.; Park, J.B. Severe periodontitis is associated with insulin resistance in non-abdominal obese adults. J. Clin. Endocrinol. Metab. 2016, 101, 4251–4259. [Google Scholar] [CrossRef] [PubMed]
- Llorente, I.; García-Castañeda, N.; Valero, C.; González-Álvaro, I.; Castañeda, S. Osteoporosis in rheumatoid arthritis: Dangerous liaisons. Front. Med. 2020, 7, 802. [Google Scholar] [CrossRef]
- Avouac, J.; Elhai, M.; Forien, M.; Sellam, J.; Eymard, F.; Molto, A.; Banal, F.; Damiano, J.; Dieudé, P.; Larger, E.; et al. Influence of inflammatory and non-inflammatory rheumatic disorders on the clinical and biological profile of type-2 diabetes. Rheumatology 2021, 60, 3598–3606. [Google Scholar] [CrossRef]
- Hong, S.J.; Yang, B.E.; Yoo, D.M.; Kim, S.J.; Choi, H.G.; Byun, S.H. Analysis of the relationship between periodontitis and osteoporosis/fractures: A cross-sectional study. BMC Oral Health 2021, 21, 125. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Disease Groups | Control | p-Value | ||
|---|---|---|---|---|---|
| RA with PD (n = 26) | RA without PD (n = 21) | PD (n = 51) | (n = 20) | ||
| Age (years) a | 48.5 (8.8) | 43.1 (13.3) | 47 (9.5) | 43 (6.3) | 0.11 |
| Female sex, n (%) b | 21 (81) | 20 (95) | 33 (65) | 8 (40) | <0.001 |
| Clinical Status, n b | |||||
| – Hypertension | 9 | 7 | 8 | - | 0.14 |
| – Diabetes | 2 | 6 | 10 | - | |
| BOP c | 23 (57) | 43 (60) | 77 (56) | 15 (32) | <0.0001 |
| PPD Total c | 301(81) | 276 (86) | 384 (113) | 191 (24) | <0.0001 |
| PPD Disease c | 107.5 (104) | 0 (2.5) | 229 (136) | 0 (5) | <0.0001 |
| Adjusted PPD Total c | 11.6 (2.9) | 10.8 (2.8) | 15.5 (4.2) | 6.8 (1) | <0.0001 |
| Adjusted PPD Disease c | 8 (4.3) | 0 (0) | 10.4 (4) | 0 (0) | <0.0001 |
| ∑MBL c | 27.4 (10.8) d | 13.5 (12.9) e | 34.2 (15.4) | 8.8 (17.5) f | <0.0001 |
| Adjusted ∑MBL c | 4.57 (1) | 3.02 (0.9) | 5.24 (2) | 2.88 (0.8) | <0.0001 |
| Body mass index (kg/m2) c | 24.2 (5) | 24.1 (6.2) | 25.2 (4) | 23.9 (4.6) | 0.35 |
| Waist circumference (cm) c | 102 (30) | 97 (23) | 109 (19) | 86 (17) | <0.0001 |
| HbA1c % c | 5.0 (1) | 5.0 (2) | 5.7 (1.2) | 4.5 (0.8) | <0.0001 |
| Periodontal Pocketing and Inflammation | Marginal Bone Loss | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BOP | PPD Total | PPD Disease | Adj. PPD Total | Adj. PPD Disease | ∑MBL | Adj. MBL | ||||||||
| Analyte | r | Analyte | r | Analyte | r | Analyte | r | Analyte | r | Analyte | r | Analyte | r | |
| RA with PD | ACE-2 | −0.42 | LOX-1 | 0.41 | PTX3 | 0.44 | ANGPT1 PGF | 0.47 0.45 | LEP TNFRSF13B IL-27 | 0.48 0.48 0.46 | ||||
| PD | CXCL1 SRC XCL1 | −0.31 −0.32 −0.36 | IL-4RA | −0.29 | IL-4RA MERTK SRC | −0.37 −0.28 −0.29 | IL-4RA MMP-12 SRC | −0.33 −0.31 −0.38 | MMP-12 SRC | −0.34 −0.42 | ADAM-TS13 | −0.29 | ||
| RA without PD | FGF-23 | −0.52 | Dkk-1 THBS2 | 0.47 0.49 | CD40-L TGM2 | −0.47 −0.45 | ||||||||
| RA with PD | PD | RA without PD | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Variable | PC1 | PC2 | Variable | PC1 | PC2 | Variable | PC1 | PC2 | |
| C22D4 | 0.98 | −0.08 | PDGF subunit B | 0.87 | −0.12 | PDGF subunit B | 0.92 | 0.02 | |
| SCF | 0.96 | 0.05 | SOD2 | 0.87 | −0.05 | CD84 | 0.90 | −0.14 | |
| IL-17D | 0.95 | 0.00 | MMP7 | 0.86 | 0.12 | SCF | 0.90 | −0.31 | |
| PAR-1 | 0.93 | −0.16 | CD4 | 0.85 | −0.28 | BOC | 0.90 | −0.32 | |
| BOC | 0.93 | −0.16 | hOSCAR | 0.84 | 0.08 | CXCL1 | 0.90 | 0.11 | |
| PIgR | 0.93 | −0.02 | CCL17 | 0.84 | −0.08 | PD-L2 | 0.89 | 0.21 | |
| VEGFD | 0.93 | 0.20 | IL16 | 0.83 | −0.24 | MERTK | 0.87 | 0.22 | |
| IL16 | 0.93 | 0.07 | HB-EGF | 0.83 | 0.05 | VEGFD | 0.87 | 0.06 | |
| MMP7 | 0.92 | 0.15 | CCL3 | 0.83 | 0.11 | VSIG2 | 0.87 | 0.20 | |
| SPON2 | 0.91 | −0.14 | PIgR | 0.80 | −0.30 | MMP7 | 0.86 | 0.09 | |
| PDGF subunit B | 0.91 | 0.19 | VEGFD | 0.80 | 0.01 | THBS2 | 0.86 | −0.11 | |
| THPO | 0.91 | −0.18 | RAGE | 0.79 | 0.07 | BNP | 0.85 | −0.06 | |
| CD84 | 0.90 | 0.21 | SCF | 0.78 | −0.41 | PIgR | 0.84 | −0.33 | |
| hOSCAR | 0.90 | 0.25 | HSP 27 | 0.78 | −0.21 | PARP-1 | 0.82 | −0.36 | |
| LPL | 0.90 | −0.18 | IL-17D | 0.77 | 0.04 | HO-1 | 0.82 | 0.16 | |
| FABP2 | 0.89 | −0.23 | THBS2 | 0.77 | −0.02 | hOSCAR | 0.82 | 0.27 | |
| FGF-21 | 0.89 | −0.30 | CD84 | 0.76 | −0.14 | CD4 | 0.82 | −0.26 | |
| THBS2 | 0.88 | −0.06 | ADAM-TS13 | 0.75 | 0.17 | CCL17 | 0.82 | 0.28 | |
| CCL17 | 0.88 | 0.28 | PD-L2 | 0.75 | 0.05 | IL-17D | 0.82 | −0.20 | |
| CXCL1 | 0.88 | 0.30 | FGF-21 | 0.74 | −0.06 | DECR1 | 0.81 | −0.17 | |
| PARP-1 | 0.88 | −0.10 | HO-1 | 0.73 | −0.22 | FABP2 | 0.81 | −0.41 | |
| CTRC | 0.87 | 0.26 | Dkk-1 | 0.72 | −0.22 | GDF-2 | 0.81 | 0.15 | |
| ANGPT1 | 0.86 | −0.04 | BOC | 0.72 | −0.36 | RAGE | 0.80 | 0.17 | |
| PRELP | 0.85 | −0.44 | PAPPA | 0.71 | 0.03 | SORT1 | 0.80 | 0.20 | |
| MERTK | 0.85 | −0.06 | VSIG2 | 0.70 | 0.44 | PGF | 0.80 | 0.28 | |
| TM | 0.85 | −0.28 | SORT1 | 0.70 | 0.32 | ADAM-TS13 | 0.80 | 0.23 | |
| CCL3 | 0.84 | 0.18 | MERTK | 0.69 | 0.07 | CCL3 | 0.80 | −0.23 | |
| BMP-6 | 0.84 | −0.36 | GDF-2 | 0.68 | 0.33 | FGF-21 | 0.80 | −0.31 | |
| SOD2 | 0.83 | 0.02 | TNFRSF13B | 0.67 | 0.11 | HB-EGF | 0.78 | 0.48 | |
| SORT1 | 0.83 | 0.36 | CXCL1 | 0.66 | 0.07 | THPO | 0.78 | −0.39 | |
| STK4 | 0.81 | −0.07 | GLO1 | 0.66 | 0.03 | SLAMF7 | 0.77 | 0.39 | |
| VSIG2 | 0.81 | −0.04 | NEMO | 0.66 | 0.02 | LEP | 0.77 | 0.24 | |
| SRC | 0.81 | 0.10 | CTRC | 0.66 | −0.02 | Dkk-1 | 0.77 | −0.22 | |
| RAGE | 0.81 | 0.24 | SPON2 | 0.65 | −0.11 | TF | 0.76 | −0.01 | |
| PGF | 0.81 | −0.31 | CTSL1 | 0.65 | 0.32 | AGRP | 0.76 | −0.14 | |
| HO-1 | 0.81 | −0.06 | IL-1ra | 0.63 | 0.16 | IL-1ra | 0.76 | 0.18 | |
| FGF-23 | 0.80 | −0.28 | AGRP | 0.61 | −0.19 | NEMO | 0.75 | −0.06 | |
| IL18 | 0.79 | −0.18 | FGF-23 | 0.60 | −0.29 | TNFRSF13B | 0.74 | 0.19 | |
| Dkk-1 | 0.79 | 0.01 | IL1RL2 | 0.60 | 0.08 | PAR-1 | 0.74 | −0.57 | |
| AGRP | 0.78 | −0.27 | THPO | 0.60 | −0.37 | PSGL-1 | 0.73 | −0.04 | |
| XCL1 | 0.76 | −0.50 | DCN | 0.59 | −0.05 | IL16 | 0.73 | −0.39 | |
| HB-EGF | 0.76 | 0.53 | IL-27 | 0.57 | 0.18 | DCN | 0.73 | 0.25 | |
| HSP 27 | 0.76 | 0.20 | TNFRSF11A | 0.56 | 0.61 | HSP 27 | 0.72 | −0.01 | |
| TNFRSF13B | 0.75 | −0.15 | PTX3 | 0.56 | 0.08 | PAPPA | 0.72 | −0.22 | |
| PD-L2 | 0.74 | 0.50 | BNP | 0.56 | −0.05 | SOD2 | 0.71 | −0.38 | |
| NEMO | 0.74 | 0.39 | IL6 | 0.55 | 0.37 | TNFRSF11A | 0.71 | 0.56 | |
| BNP | 0.74 | −0.08 | TGM2 | 0.54 | 0.13 | TM | 0.70 | 0.32 | |
| LEP | 0.73 | 0.08 | PGF | 0.54 | 0.63 | LPL | 0.70 | −0.62 | |
| PAPPA | 0.73 | 0.42 | LPL | 0.54 | −0.67 | FGF-23 | 0.69 | −0.40 | |
| IL-1ra | 0.72 | −0.17 | PRSS27 | 0.54 | 0.08 | GLO1 | 0.66 | 0.16 | |
| TNFRSF11A | 0.70 | −0.24 | FABP2 | 0.53 | −0.53 | SPON2 | 0.65 | 0.05 | |
| DCN | 0.70 | 0.50 | PAR-1 | 0.52 | −0.66 | SRC | 0.64 | −0.33 | |
| IL1RL2 | 0.69 | 0.04 | TIE2 | 0.52 | 0.36 | CTRC | 0.64 | 0.10 | |
| PSGL-1 | 0.68 | −0.05 | ANGPT1 | 0.51 | −0.44 | IL1RL2 | 0.64 | −0.06 | |
| TF | 0.67 | −0.05 | IDUA | 0.50 | 0.04 | IL-27 | 0.63 | 0.47 | |
| ADM | 0.66 | −0.30 | IL18 | 0.50 | 0.33 | PRSS27 | 0.62 | 0.38 | |
| ADAM-TS13 | 0.66 | 0.55 | SRC | 0.49 | −0.39 | BMP-6 | 0.62 | −0.49 | |
| TNFRSF10A | 0.59 | −0.53 | LEP | 0.48 | 0.07 | IL18 | 0.61 | 0.30 | |
| MARCO | 0.57 | −0.24 | TM | 0.47 | 0.64 | TIE2 | 0.60 | 0.52 | |
| CTSL1 | 0.57 | 0.04 | CEACAM8 | 0.47 | 0.09 | SERPINA12 | 0.57 | 0.25 | |
| GDF-2 | 0.57 | 0.29 | STK4 | 0.47 | −0.49 | ANGPT1 | 0.55 | −0.48 | |
| PRSS27 | 0.56 | 0.35 | PARP-1 | 0.47 | −0.40 | STK4 | 0.54 | −0.53 | |
| TRAIL-R2 | 0.51 | −0.61 | HAOX1 | 0.47 | −0.04 | ITGB1BP2 | 0.54 | −0.20 | |
| IL-27 | 0.50 | −0.30 | DECR1 | 0.43 | 0.33 | REN | 0.53 | 0.31 | |
| GLO1 | 0.50 | 0.28 | TRAIL-R2 | 0.42 | 0.72 | PRELP | 0.51 | −0.77 | |
| IgG Fc receptor II-b | 0.44 | −0.31 | TF | 0.42 | 0.56 | IL6 | 0.49 | 0.01 | |
| SERPINA12 | 0.39 | 0.17 | TNFRSF10A | 0.40 | 0.63 | GH | 0.47 | 0.15 | |
| HAOX1 | 0.36 | −0.32 | ITGB1BP2 | 0.38 | −0.10 | GIF | 0.46 | −0.03 | |
| IL6 | 0.33 | −0.18 | PRELP | 0.37 | −0.63 | CTSL1 | 0.46 | 0.25 | |
| DECR1 | 0.31 | 0.26 | LOX-1 | 0.36 | 0.08 | TGM2 | 0.44 | 0.39 | |
| GH | 0.30 | 0.20 | BMP-6 | 0.36 | −0.58 | TNFRSF10A | 0.42 | 0.54 | |
| FS | 0.29 | −0.21 | XCL1 | 0.34 | −0.56 | XCL1 | 0.38 | −0.79 | |
| SLAMF7 | 0.29 | −0.35 | GH | 0.34 | −0.28 | TRAIL-R2 | 0.38 | 0.56 | |
| KIM1 | 0.25 | −0.46 | Gal-9 | 0.33 | 0.24 | PTX3 | 0.36 | −0.33 | |
| Gal-9 | 0.22 | −0.33 | MARCO | 0.30 | −0.14 | LOX-1 | 0.36 | −0.18 | |
| ITGB1BP2 | 0.20 | 0.36 | ADM | 0.29 | −0.38 | KIM1 | 0.34 | 0.68 | |
| PTX3 | 0.18 | 0.12 | AMBP | 0.26 | 0.64 | CD40-L | 0.32 | 0.07 | |
| TGM2 | 0.15 | 0.27 | CA5A | 0.25 | 0.34 | IgG Fc receptor II-b | 0.32 | −0.02 | |
| GIF | 0.12 | −0.18 | KIM1 | 0.24 | 0.74 | GT | 0.31 | 0.42 | |
| MMP12 | 0.11 | −0.54 | PSGL-1 | 0.19 | 0.23 | CA5A | 0.29 | 0.26 | |
| CEACAM8 | 0.09 | 0.25 | SERPINA12 | 0.18 | 0.15 | MARCO | 0.29 | −0.01 | |
| ACE2 | 0.08 | −0.49 | SLAMF7 | 0.17 | 0.10 | HAOX1 | 0.27 | −0.29 | |
| AMBP | 0.06 | 0.26 | GIF | 0.17 | 0.36 | IDUA | 0.25 | 0.36 | |
| TIE2 | −0.03 | 0.58 | REN | 0.15 | 0.35 | AMBP | 0.23 | 0.63 | |
| CA5A | −0.04 | −0.44 | ACE2 | 0.13 | 0.54 | PRSS8 | 0.14 | 0.81 | |
| LOX-1 | −0.04 | 0.37 | IL-4RA | 0.12 | 0.31 | MMP12 | 0.11 | 0.34 | |
| IDUA | −0.10 | −0.03 | CD40-L | 0.08 | 0.02 | Gal-9 | 0.10 | 0.35 | |
| REN | −0.10 | −0.21 | IgG Fc receptor II-b | 0.03 | 0.24 | CEACAM8 | 0.06 | 0.09 | |
| GT | −0.11 | −0.66 | PRSS8 | 0.02 | 0.81 | ADM | 0.03 | −0.25 | |
| CD40-L | −0.20 | −0.04 | MMP12 | −0.04 | 0.32 | FS | 0.01 | 0.23 | |
| PRSS8 | −0.44 | −0.17 | FS | −0.06 | 0.34 | ACE2 | 0.00 | 0.16 | |
| IL-4RA | −0.46 | −0.76 | GT | −0.13 | 0.16 | IL-4RA | −0.01 | 0.26 | |
| Proportion of variance | 47.0% | 8.9% | 32.9% | 11.6% | 43.2% | 11.3% | |||
| Cumulative proportion of variance | 47.0% | 55.9% | 32.9% | 44.5% | 43.2% | 54.5% | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panezai, J.; Ghaffar, A.; Altamash, M.; Åberg, M.; Van Dyke, T.E.; Larsson, A.; Engström, P.-E. Periodontal Disease Augments Cardiovascular Disease Risk Biomarkers in Rheumatoid Arthritis. Biomedicines 2022, 10, 714. https://doi.org/10.3390/biomedicines10030714
Panezai J, Ghaffar A, Altamash M, Åberg M, Van Dyke TE, Larsson A, Engström P-E. Periodontal Disease Augments Cardiovascular Disease Risk Biomarkers in Rheumatoid Arthritis. Biomedicines. 2022; 10(3):714. https://doi.org/10.3390/biomedicines10030714
Chicago/Turabian StylePanezai, Jeneen, Ambereen Ghaffar, Mohammad Altamash, Mikael Åberg, Thomas E. Van Dyke, Anders Larsson, and Per-Erik Engström. 2022. "Periodontal Disease Augments Cardiovascular Disease Risk Biomarkers in Rheumatoid Arthritis" Biomedicines 10, no. 3: 714. https://doi.org/10.3390/biomedicines10030714
APA StylePanezai, J., Ghaffar, A., Altamash, M., Åberg, M., Van Dyke, T. E., Larsson, A., & Engström, P. -E. (2022). Periodontal Disease Augments Cardiovascular Disease Risk Biomarkers in Rheumatoid Arthritis. Biomedicines, 10(3), 714. https://doi.org/10.3390/biomedicines10030714