
Anticancer Effects with Molecular Docking Confirmation of Newly Synthesized Isatin Sulfonamide Molecular Hybrid Derivatives against Hepatic Cancer Cell Lines

 ,
,  ,
, 
Abstract
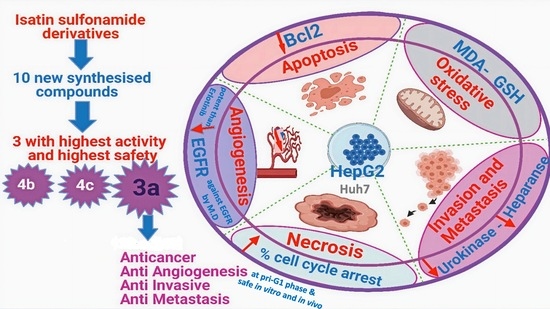
:
1. Introduction
2. Materials and Methods
2.1. Biochemical Reagents, Chemicals, Solvents
2.2. Biochemical/Molecular Assay Kits
2.3. Cell Lines
2.4. Animals
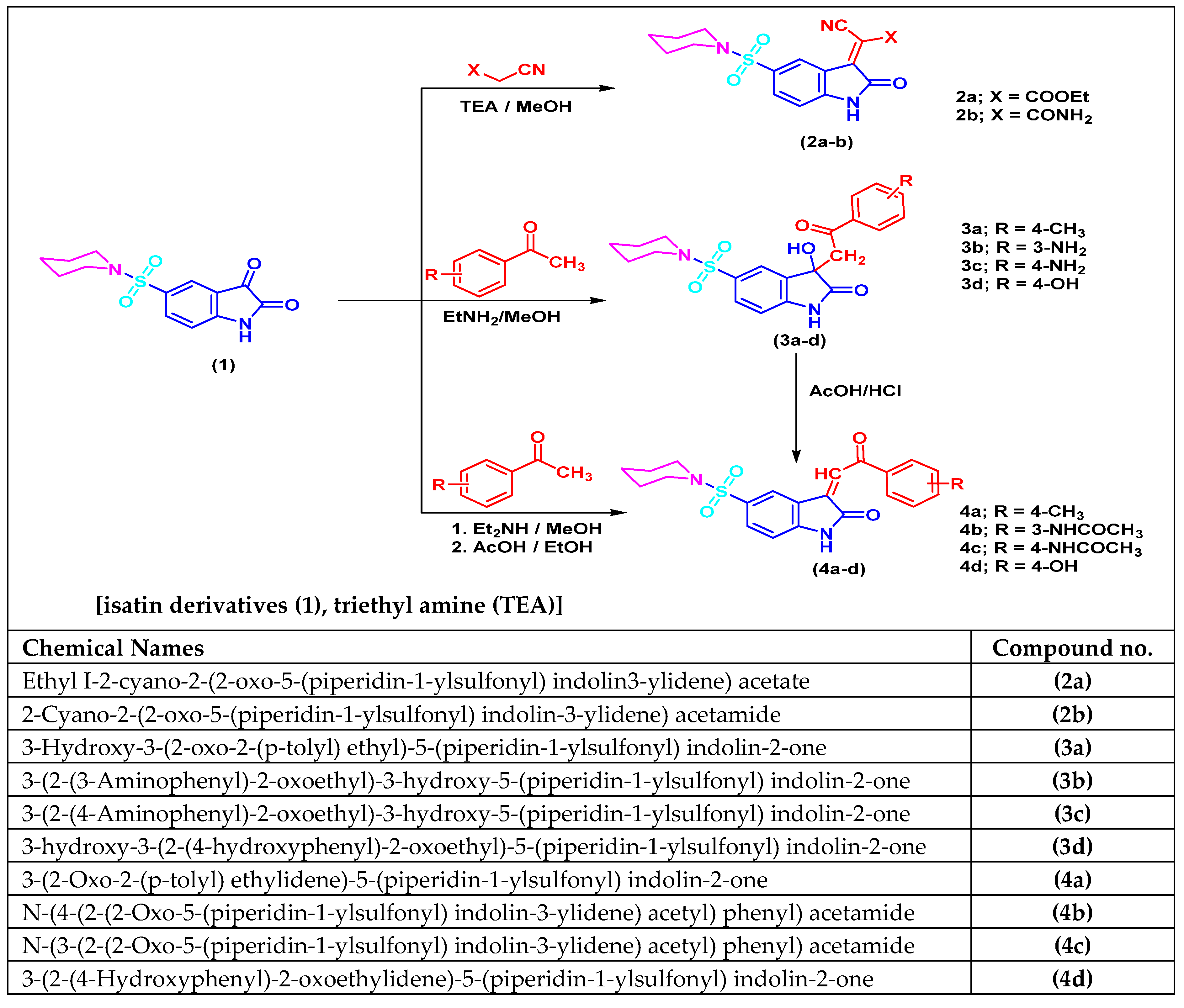
2.5. Isatin Sulfonamide Derivative Synthesis
2.6. Chemical Synthetic Pathway
2.7. Cancer Cell Proliferation Inhibition
2.8. In Vitro Safety/Cell Viability Assay towards Human Healthy RPE1 Cell Line
2.9. In Vivo Acute Oral Toxicity Study
2.10. Ethics Statement
2.11. Experimental Design
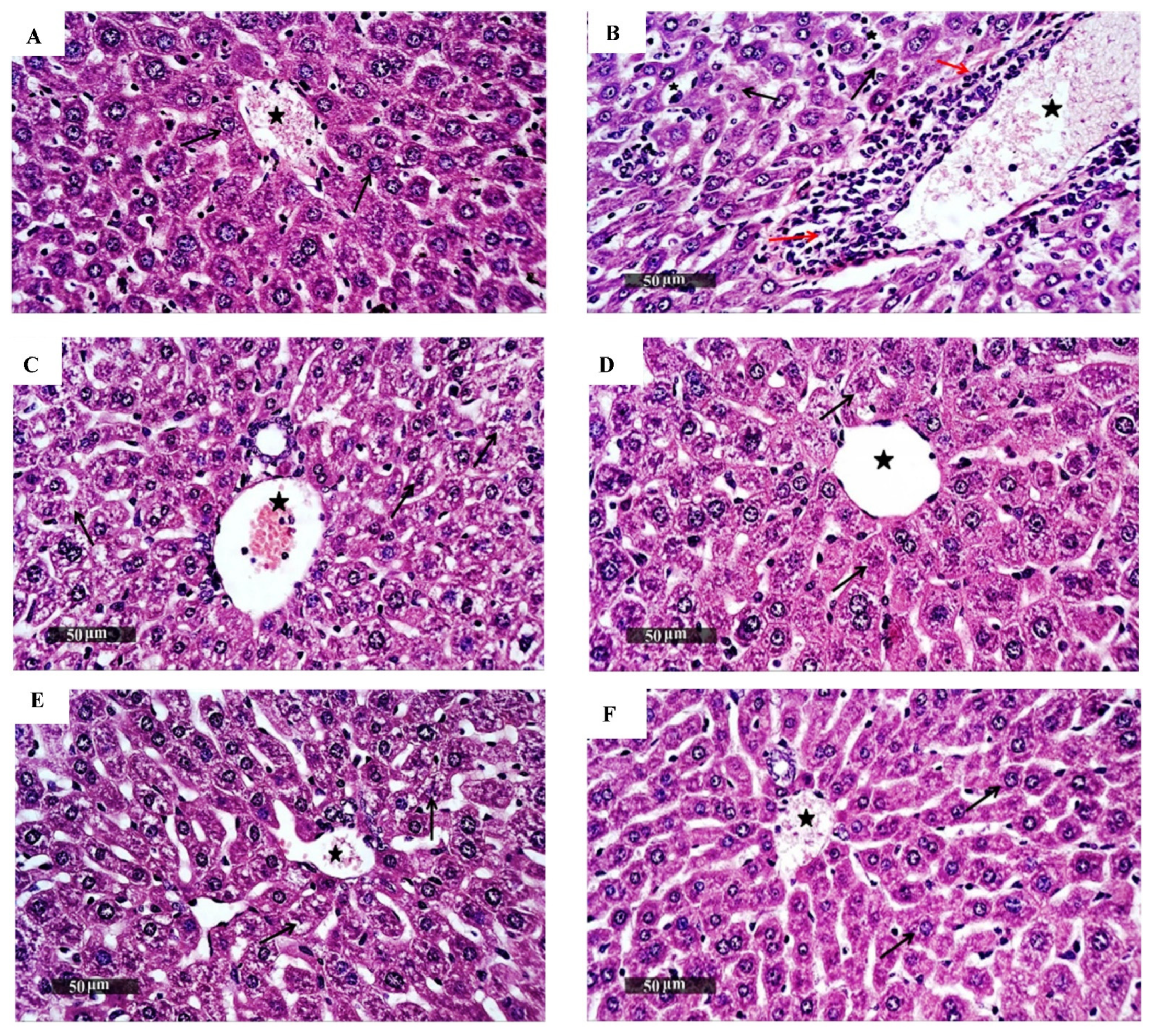
2.12. Histopathological Examination
2.13. Flow Cytometric Analysis and Apoptotic Studies
2.13.1. Cell Cycle Analysis
2.13.2. Annexin V/FITC Apoptosis Assay
2.14. Biochemical Evaluation
2.15. Molecular Docking Simulation
2.16. Statistical Analysis
3. Results
3.1. Biological Evaluation
3.1.1. Antiproliferative and Anticancer Activities of Isatin Sulfonamide Derivatives on HepG2 and Huh7 Cell Lines
3.1.2. In Vitro Safety Assay towards Human Healthy Retina Pigmented Epithelial (RPE1) Cell Line
3.1.3. In Vivo Acute Oral Toxicity Assay
3.1.4. Isatin Sulfonamide Molecular Hybrids Effect Cell Cycle Analysis
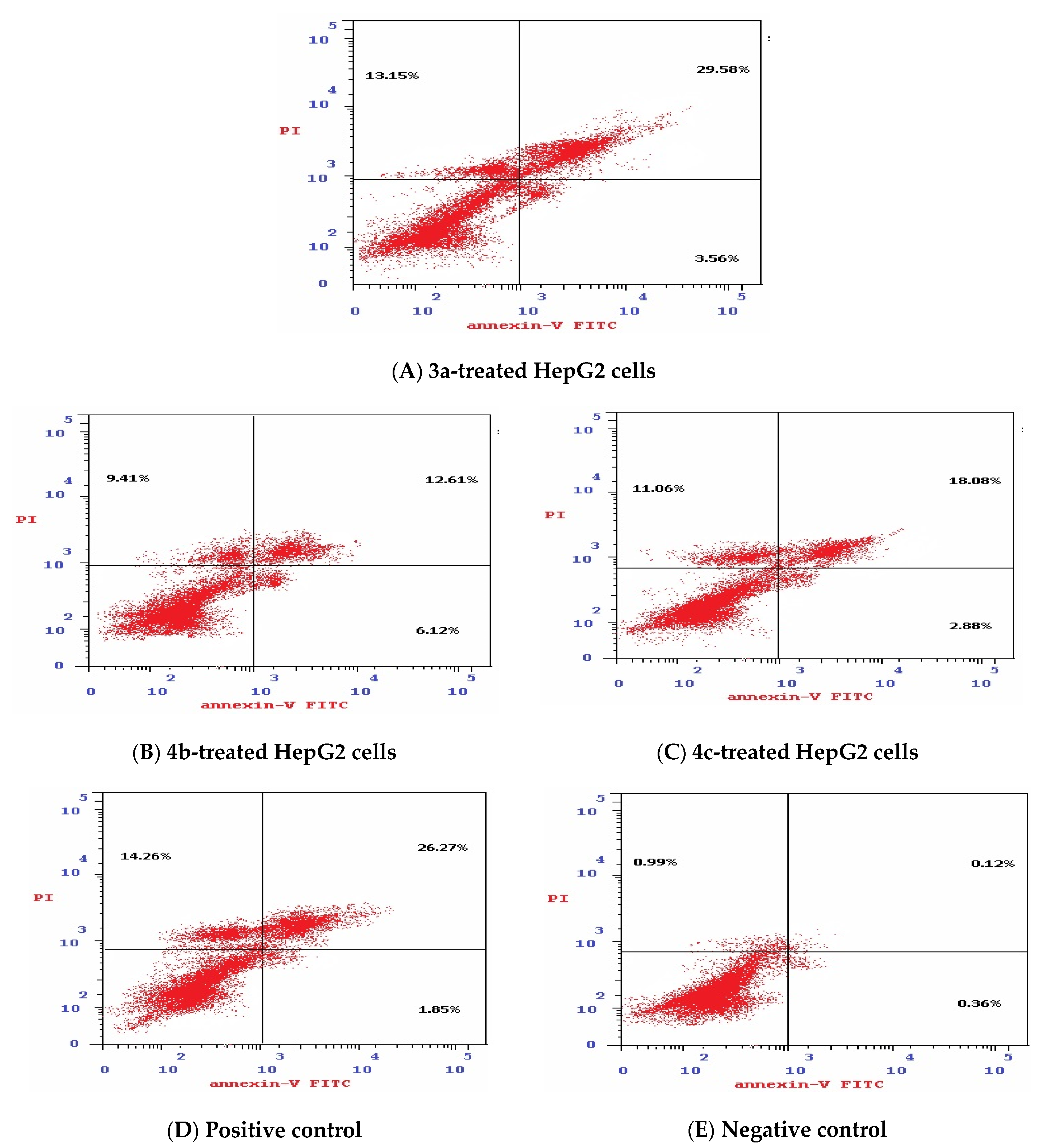
3.1.5. Effect of Isatin Sulfonamide Derivatives on Apoptotic Assay
- (1)
- Viable cells, which are negative to both probes (PI/FITC −/−),
- (2)
- Apoptotic cells, which are PI negative and Annexin positive (PI/FITC −/+),
- (3)
- Late apoptotic cells, which are both PI and Annexin positive (PI/FITC +/+),
- (4)
- Necrotic cells, which are PI positive and Annexin negative (PI/FITC+/−).
3.1.6. Effect of Isatin Sulfonamide Molecular Hybrids on Other Cancer Hallmark Markers Assay
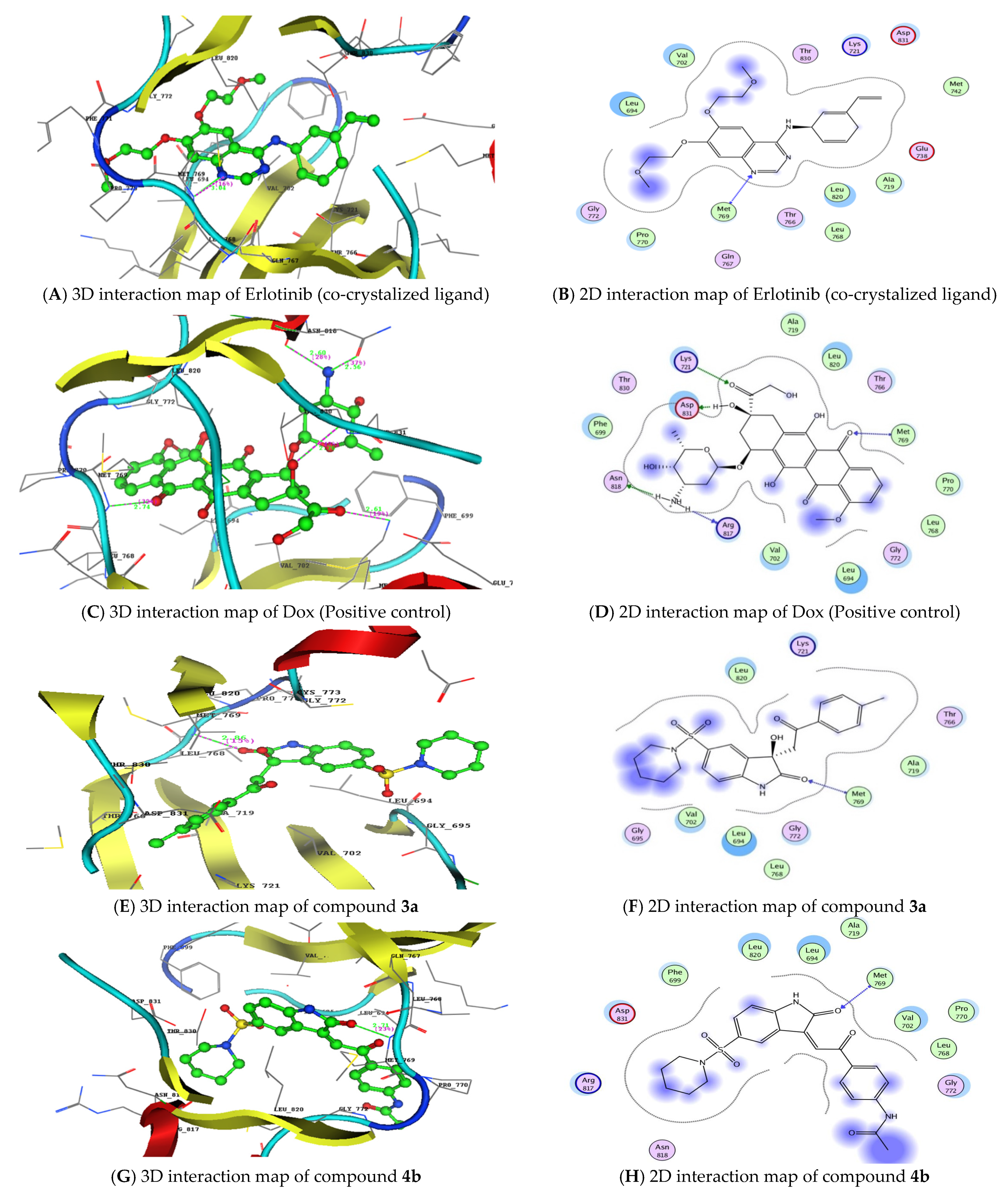
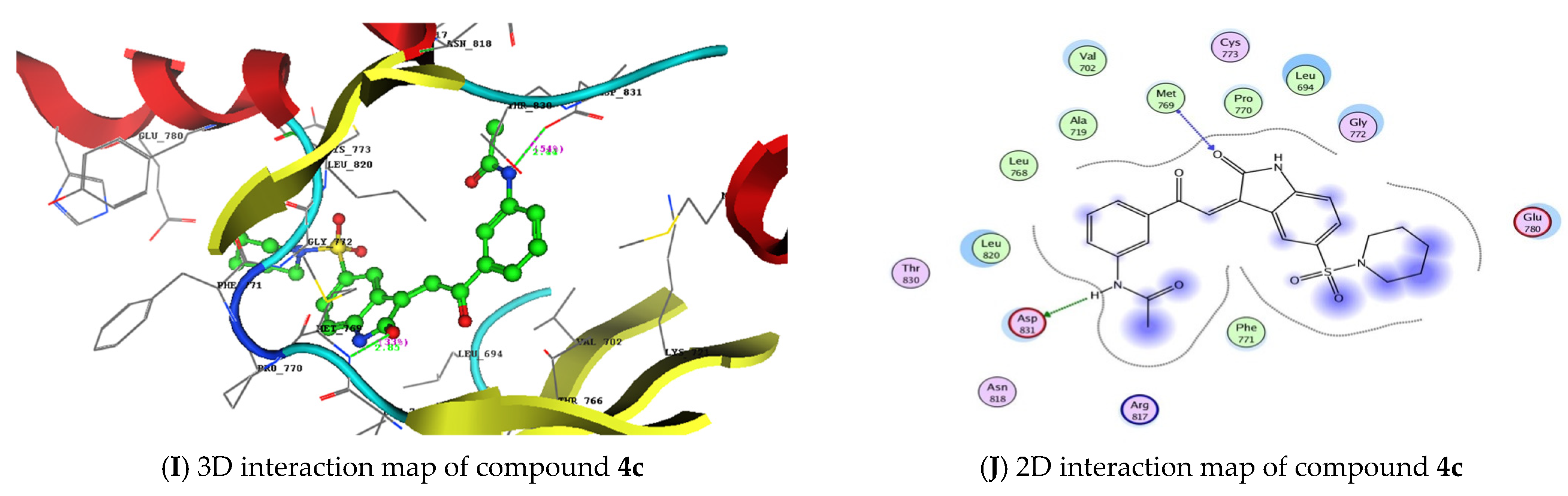
3.2. Molecular Docking Studies and Binding to EGFR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsuchiya, K. Switching from Apoptosis to Pyroptosis: Gasdermin-Elicited Inflammation and Antitumor Immunity. Int. J. Mol. Sci. 2021, 22, 426. [Google Scholar] [CrossRef] [PubMed]
- Hussien, A.G.; Borai, I.H.; Said, M.M.; Mahmoud, K.; Ali, M.M. Chemotherapeutic effect of Ulmus pumila leaves methanolic extract against N-methyl-N-nitrosourea-induced mammary carcinoma in female rats: An in vitro and in vivo study. J. Appl. Pharm. Sci. 2019, 9, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, S.; Shi, L.; Yang, Z. Discovery of novel isatin–dehydroepiandrosterone conjugates as potential anticancer agents. Bioorg. Med. Chem. Lett. 2015, 25, 4628–4631. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Bhargava, G.; Land, K.M.; Chang, K.-H.; Arora, R.; Sen, S.; Kumar, V. N-Propargylated isatin-Mannich mono-and bis-adducts: Synthesis and preliminary analysis of in vitro activity against Tritrichomonas foetus. Eur. J. Med. Chem. 2014, 74, 657–663. [Google Scholar] [CrossRef]
- Lin, H.-H.; Wu, W.-Y.; Cao, S.-L.; Liao, J.; Ma, L.; Gao, M.; Li, Z.-F.; Xu, X. Synthesis and antiproliferative evaluation of piperazine-1-carbothiohydrazide derivatives of indolin-2-one. Bioorg. Med. Chem. Lett. 2013, 23, 3304–3307. [Google Scholar] [CrossRef]
- Krause-Heuer, A.M.; Howell, N.R.; Matesic, L.; Dhand, G.; Young, E.L.; Burgess, L.; Jiang, C.D.; Lengkeek, N.A.; Fookes, C.J.R.; Pham, T.Q. A new class of fluorinated 5-pyrrolidinylsulfonyl isatin caspase inhibitors for PET imaging of apoptosis. MedChemComm 2013, 4, 347–352. [Google Scholar] [CrossRef]
- Salem, M.A.; Ragab, A.; El-Khalafawy, A.; Makhlouf, A.H.; Askar, A.A.; Ammar, Y.A. Design, synthesis, in vitro antimicrobial evaluation and molecular docking studies of indol-2-one tagged with morpholinosulfonyl moiety as DNA gyrase inhibitors. Bioorg. Chem. 2020, 96, 103619. [Google Scholar] [CrossRef] [PubMed]
- Nath, R.; Pathania, S.; Grover, G.; Akhtar, M.J. Isatin containing heterocycles for different biological activities: Analysis of structure activity relationship. J. Mol. Struct. 2020, 1222, 128900. [Google Scholar] [CrossRef]
- Wang, J.; Yun, D.; Yao, J.; Fu, W.; Huang, F.; Chen, L.; Wei, T.; Yu, C.; Xu, H.; Zhou, X. Design, synthesis and QSAR study of novel isatin analogues inspired Michael acceptor as potential anticancer compounds. Eur. J. Med. Chem. 2018, 144, 493–503. [Google Scholar] [CrossRef]
- Gupta, A.K.; Tulsyan, S.; Bharadwaj, M.; Mehrotra, R. Systematic review on cytotoxic and anticancer potential of n-substituted isatins as novel class of compounds useful in multidrug-resistant cancer therapy: In silico and in vitro analysis. Top. Curr. Chem. 2019, 377, 15. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-W.; Ohnishi, K.; Murakami, A.; Lee, J.-S.; Kundu, J.K.; Na, H.-K.; Ohigashi, H.; Surh, Y.-J. Zerumbone induces heme oxygenase-1 expression in mouse skin and cultured murine epidermal cells through activation of Nrf2. Cancer Prev. Res. 2011, 4, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, L.; Schwarz, S.; Perl, V.; Köwitsch, A.; Siewert, B.; Csuk, R. Incorporation of a Michael acceptor enhances the antitumor activity of triterpenoic acids. Eur. J. Med. Chem. 2015, 101, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar] [CrossRef]
- Fang, L.; Jumpertz, S.; Zhang, Y.; Appenroth, D.; Fleck, C.; Mohr, K.; Tränkle, C.; Decker, M. Hybrid molecules from xanomeline and tacrine: Enhanced tacrine actions on cholinesterases and muscarinic M1 receptors. J. Med. Chem. 2010, 53, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, H.A.; Eldehna, W.M.; Keeton, A.B.; Piazza, G.A.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S.; Attia, M.I. Isatin-benzoazine molecular hybrids as potential antiproliferative agents: Synthesis and in vitro pharmacological profiling. Drug Des. Dev. Ther. 2017, 11, 2333. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Zhou, M.; Zeng, C. Recent advances in isatin hybrids as potential anticancer agents. Arch. Pharm. 2020, 353, 1900367. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.; Buneeva, O.; Gnedenko, O.; Ershov, P.; Ivanov, A. Isatin, an endogenous nonpeptide biofactor: A review of its molecular targets, mechanisms of actions, and their biomedical implications. BioFactors 2018, 44, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Varun, V.; Sonam, S.; Kakkar, R. Isatin and its derivatives: A survey of recent syntheses, reactions, and applications. Medchemcomm 2019, 10, 351–368. [Google Scholar] [CrossRef]
- Singh, S.N.; Regati, S.; Paul, A.K.; Layek, M.; Jayaprakash, S.; Reddy, K.V.; Deora, G.S.; Mukherjee, S.; Pal, M. Cu-mediated 1, 3-dipolar cycloaddition of azomethine ylides with dipolarophiles: A faster access to spirooxindoles of potential pharmacological interest. Tetrahedron Lett. 2013, 54, 5448–5452. [Google Scholar] [CrossRef]
- Senwar, K.R.; Reddy, T.S.; Thummuri, D.; Sharma, P.; Naidu, V.G.M.; Srinivasulu, G.; Shankaraiah, N. Design, synthesis and apoptosis inducing effect of novel (Z)-3-(3′-methoxy-4′-(2-amino-2-oxoethoxy)-benzylidene) indolin-2-ones as potential antitumour agents. Eur. J. Med. Chem. 2016, 118, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Ammar, Y.A.; El-Sharief, A.M.S.; Belal, A.; Abbas, S.Y.; Mohamed, Y.A.; Mehany, A.B.M.; Ragab, A. Design, synthesis, antiproliferative activity, molecular docking and cell cycle analysis of some novel (morpholinosulfonyl) isatins with potential EGFR inhibitory activity. Eur. J. Med. Chem. 2018, 156, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Li, X.; Zhao, B.; Peng, W.; Li, W.; Fu, W. Discovery of aryl-piperidine derivatives as potential antipsychotic agents using molecular hybridization strategy. Eur. J. Med. Chem. 2020, 193, 112214. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Santiago, M.; Santiago, Á.; Pastor, N.; Alvarez, L.; Razo-Hernández, R.S. Isatin derivatives as DNA minor groove-binding agents: A structural and theoretical study. Struct. Chem. 2020, 31, 1289–1307. [Google Scholar] [CrossRef]
- Ali Mohamed, H.; Ammar, Y.A.; AM Elhagali, G.; Eyada, H.A.; Aboul-Magd, D.S.; Ragab, A. In Vitro Antimicrobial Evaluation, Single-Point Resistance Study, and Radiosterilization of Novel Pyrazole Incorporating Thiazol-4-one/Thiophene Derivatives as Dual DNA Gyrase and DHFR Inhibitors against MDR Pathogens. ACS omega 2022, 7, 4970–4990. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Kamiloglu, S.; Sari, G.; Ozdal, T.; Capanoglu, E. Guidelines for cell viability assays. Food Front. 2020, 1, 332–349. [Google Scholar] [CrossRef]
- Oecd, T.N. 425: Acute Oral Toxicity: Up-and-Down Procedure. OECD Guidel. Test. Chem. Sect. 2008, 4, 1–27. [Google Scholar]
- Khan, M.A.; Aljarbou, A.N.; Aldebasi, Y.H.; Alorainy, M.S.; Khan, A. Combination of glycosphingosomes and liposomal doxorubicin shows increased activity against dimethyl-α-benzanthracene-induced fibrosarcoma in mice. Int. J. Nanomed. 2015, 10, 6331. [Google Scholar] [CrossRef] [Green Version]
- Mahesh, B.U.; Shrivastava, S.; Kuncha, M.; Sahu, B.D.; Swamy, C.V.; Pragada, R.R.; Naidu, V.G.M.; Sistla, R. Ethanolic extract of Boswellia ovalifoliolata bark and leaf attenuates doxorubicin-induced cardiotoxicity in mice. Environ. Toxicol. Pharmacol. 2013, 36, 840–849. [Google Scholar] [CrossRef]
- Akhila, J.S.; Shyamjith, D.; Alwar, M.C. Acute toxicity studies and determination of median lethal dose. Curr. Sci. 2007, 917–920. [Google Scholar]
- Suvarna, K.S.; Layton, C.; Bancroft, J.D. Bancroft’s theory and practice of histological techniques E-Book; Elsevier Health Sciences, 2018; ISBN 0702068861. [Google Scholar]
- Culling, C.F.A. Handbook of Histopathological and Histochemical Techniques: Including Museum Techniques; Butterworth-Heinemann: Cambridge, UK, 2013; ISBN 1483164799. [Google Scholar]
- Karrowni, W.; Li, Y.; Jones, P.G.; Cresci, S.; Abdallah, M.S.; Lanfear, D.E.; Maddox, T.M.; McGuire, D.K.; Spertus, J.A.; Horwitz, P.A. Insulin resistance is associated with significant clinical atherosclerosis in nondiabetic patients with acute myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2245–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldehna, W.M.; Almahli, H.; Al-Ansary, G.H.; Ghabbour, H.A.; Aly, M.H.; Ismael, O.E.; Al-Dhfyan, A.; Abdel-Aziz, H.A. Synthesis and in vitro anti-proliferative activity of some novel isatins conjugated with quinazoline/phthalazine hydrazines against triple-negative breast cancer MDA-MB-231 cells as apoptosis-inducing agents. J. Enzyme Inhib. Med. Chem. 2017, 32, 600–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezki, N.; Almehmadi, M.A.; Ihmaid, S.; Shehata, A.M.; Omar, A.M.; Ahmed, H.E.A.; Aouad, M.R. Novel scaffold hopping of potent benzothiazole and isatin analogues linked to 1, 2, 3-triazole fragment that mimic quinazoline epidermal growth factor receptor inhibitors: Synthesis, antitumor and mechanistic analyses. Bioorg. Chem. 2020, 103, 104133. [Google Scholar] [CrossRef] [PubMed]
- Treger, S.; Ackerman, S.; Kaplan, V.; Ghanem, S.; Nadir, Y. Progestin type affects the increase of heparanase level and procoagulant activity mediated by the estrogen receptor. Hum. Reprod. 2021, 36, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Shamroukh, A.H.; El-Shahat, M.; Drabowicz, J.; Ali, M.M.; Rashad, A.E.; Ali, H.S. Anticancer evaluation of some newly synthesized N-nicotinonitrile derivative. Eur. J. Med. Chem. 2013, 69, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Fayed, E.A.; Ammar, Y.A.; Saleh, M.A.; Bayoumi, A.H.; Belal, A.; Mehany, A.B.M.; Ragab, A. Design, synthesis, antiproliferative evaluation, and molecular docking study of new quinoxaline derivatives as apoptotic inducers and EGFR inhibitors. J. Mol. Struct. 2021, 1236, 130317. [Google Scholar] [CrossRef]
- Beutler, E. Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 1963, 61, 882–888. [Google Scholar]
- Du, H.; Chen, B.; Jiao, N.-L.; Liu, Y.-H.; Sun, S.-Y.; Zhang, Y.-W. Elevated glutathione peroxidase 2 expression promotes cisplatin resistance in lung adenocarcinoma. Oxid. Med. Cell. Longev. 2020, 2020, 7370157. [Google Scholar] [CrossRef] [PubMed]
- Gavino, V.C.; Miller, J.S.; Ikharebha, S.O.; Milo, G.E.; Cornwell, D.G. Effect of polyunsaturated fatty acids and antioxidants on lipid peroxidation in tissue cultures. J. Lipid Res. 1981, 22, 763–769. [Google Scholar] [CrossRef]
- De Leon, J.A.D.; Borges, C.R. Evaluation of Oxidative Stress in Biological Samples Using the Thiobarbituric Acid Reactive Substances Assay. JoVE J. Vis. Exp. 2020, 159, e61122. [Google Scholar] [CrossRef]
- El-Sharief, A.M.S.; Ammar, Y.A.; Belal, A.; El-Sharief, M.A.M.S.; Mohamed, Y.A.; Mehany, A.B.M.; Ali, G.A.M.E.; Ragab, A. Design, synthesis, molecular docking and biological activity evaluation of some novel indole derivatives as potent anticancer active agents and apoptosis inducers. Bioorg. Chem. 2019, 85, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liao, X.; Yuan, L.; Wang, Y.; Zheng, Y.; Zuo, J.; Pan, Y. Visible-Light-Mediated Click Chemistry for Highly Regioselective Azide–Alkyne Cycloaddition by a Photoredox Electron-Transfer Strategy. Chem. Eur. J. 2020, 26, 5694–5700. [Google Scholar] [CrossRef] [PubMed]
- Leal-Esteban, L.C.; Fajas, L. Cell cycle regulators in cancer cell metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165715. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Li, Y.; Zhang, G. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, C.; Haribabu, J.; Jeyalakshmi, K.; Bhuvanesh, N.S.P.; Karvembu, R.; Emi, N.; Awale, S. Nickel (II) bis (isatin thiosemicarbazone) complexes induced apoptosis through mitochondrial signaling pathway and G0/G1 cell cycle arrest in IM-9 cells. J. Inorg. Biochem. 2018, 182, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.A.; Ali, A.M.; El-Sayed, W.M.; Qayed, W.S.; Farag, H.H.A.; Aboul-Fadl, T. Design and synthesis of novel isatin-based derivatives targeting cell cycle checkpoint pathways as potential anticancer agents. Bioorg. Chem. 2020, 105, 104366. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Harper, J.W. Anticancer drug targets: Cell cycle and checkpoint control. J. Clin. Investig. 1999, 104, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Zhang, L.; Zhao, X.; Zhang, Y. A hybrid chalcone combining the trimethoxyphenyl and isatinyl groups targets multiple oncogenic proteins and pathways in hepatocellular carcinoma cells. PLoS ONE 2016, 11, e0161025. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Lau, T.; Ng, I. Doxorubicin-induced apoptosis and chemosensitivity in hepatoma cell lines. Cancer Chemother. Pharmacol. 2002, 49, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fu, Y.; Zhao, Y.; Cui, S.; Wang, J.; Liu, F.; Yuan, Y.; Galons, H.; Yu, P.; Teng, Y. 5-Acetamido-1-(methoxybenzyl) isatin inhibits tumor cell proliferation, migration, and angiogenesis. RSC Adv. 2019, 9, 36690–36698. [Google Scholar] [CrossRef] [Green Version]
- Mekkawy, A.H.; Pourgholami, M.H.; Morris, D.L. Involvement of urokinase-type plasminogen activator system in cancer: An overview. Med. Res. Rev. 2014, 34, 918–956. [Google Scholar] [CrossRef]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Diagnostic, prognostic, and therapeutic applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vine, K.L.; Indira Chandran, V.; Locke, J.M.; Matesic, L.; Lee, J.; Skropeta, D.; Bremner, J.B.; Ranson, M. Targeting urokinase and the transferrin receptor with novel, anti-mitotic N-alkylisatin cytotoxin conjugates causes selective cancer cell death and reduces tumor growth. Curr. Cancer Drug Targets 2012, 12, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Maxhimer, J.B.; Quiros, R.M.; Stewart, R.; Dowlatshahi, K.; Gattuso, P.; Fan, M.; Prinz, R.A.; Xu, X. Heparanase-1 expression is associated with the metastatic potential of breast cancer. Surgery 2002, 132, 326–333. [Google Scholar] [CrossRef]
- El-Assal, O.N.; Yamanoi, A.; Ono, T.; Kohno, H.; Nagasue, N. The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin. Cancer Res. 2001, 7, 1299–1305. [Google Scholar] [PubMed]
- Coombe, D.R.; Gandhi, N.S. Heparanase: A challenging cancer drug target. Front. Oncol. 2019, 1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayatilleke, K.M.; Hulett, M.D. Heparanase and the hallmarks of cancer. J. Transl. Med. 2020, 18, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Knelson, E.H.; Nee, J.C.; Blobe, G.C. Heparan sulfate signaling in cancer. Trends Biochem. Sci. 2014, 39, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, L.; Jin, J.; Zhu, W.; Xu, Y.; Wu, Y.; Wang, Y.; Chen, H.; Webster, K.A.; Chen, H. Heparanase released from mesenchymal stem cells activates integrin beta1/HIF-2alpha/Flk-1 signaling and promotes endothelial cell migration and angiogenesis. Stem Cells 2015, 33, 1850–1862. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | HepG2 | Huh7 | RPE1 | |||
|---|---|---|---|---|---|---|
| Compound no. | IC50 (µM) | % Death Rate at 100 p.p.m (μg/mL) | IC50 (µM) | % Death Rate at 100 p.p.m (μg/mL) | CC50 (µM) | % Cell Viability at 100 p.p.m (μg/mL) |
| 2a | 54.60 ± 2.00 | 81.50 * | 40.00 ± 3.80 | 100 * | 40.30 ± 1.61 | 4.40 |
| 2b | >100 | 10.20 | >100 | N.A | - | - |
| 3a | 16.80 ± 1.44 | 70.10 * | 40.00 ± 2.20 | 100 * | >100 | 74 # |
| 3b | >100 | 1.20 | >100 | N.A | - | - |
| 3c | >100 | 11.50 | >100 | N.A | - | - |
| 3d | >100 | 19.50 | >100 | N.A | - | - |
| 4a | 12.00 ± 0.40 | 100 * | >100 | N.A | 21.90 ± 1.15 | 0 |
| 4b | 44.70 ± 1.55 | 94.20 * | 53.00 ± 3.00 | 76.00 * | >100 | 66.40 # |
| 4c | 39.70 ± 1.90 | 95.60 * | 35.00 ± 1.90 | 75.60 * | >100 | 80.70 # |
| 4d | 13.30 ± 0.75 | 100 * | 18.76 ± 0.8 | 0 | 11.90 ± 0.70 | 0 |
| DMSO | >100 | 1 | >100 | 5 | >100 | 95 |
| Doxorubicin | 21.60 ± 0.81 | 100 | 11.60 ± 0.90 | 100 | - | - |
| Tests | liver function | kidney function | |||||
|---|---|---|---|---|---|---|---|
| Parameters | S.ALT | S.AST | S.T.Bilirubin | S.D.Bilirubin | S.Creatinine | S.Urea | |
| Groups | /Units | (U/L) | (U/L) | (mg/dL) | (mg/dL) | (mg/dL) | (mg/dL) |
| Control | Normal | 28.92 ± 0.51 | 36.24 ± 0.94 | 0.69 ± 0.016 | 0.18 ± 0.005 | 0.67 ± 0.03 | 53.50 ± 1.01 |
| Negative | 30.06 ± 0.72 | 34.40 ± 0.89 | 0.74 ± 0.016 | 0.19 ± 0.002 | 0.79 ± 0.03 | 52.38 ± 0.69 | |
| Positive | 52.97 ± 0.92 *,# | 46.93 ± 0.30 *,# | 1.45 ± 0.072 *,# | 0.36 ± 0.026 *,# | 1.57 ± 0.02 *,# | 83.27 ± 0.95 *,# | |
| Treated | 2a | 28.42 ± 0.27 ^ | 37.11 ± 0.35 ^ | 0.65 ± 0.018 ^ | 0.16 ± 0.003 ^ | 0.59 ± 0.04 ^ | 52.24 ± 0.65 ^ |
| 3a | 27.05 ± 0.81 ^ | 36.78 ± 0.33 ^ | 0.65 ± 0.026 ^ | 0.16 ± 0.002 ^ | 0.82 ± 0.03 ^ | 51.25 ± 0.36 ^ | |
| 4a | 28.14 ± 0.56 ^ | 36.51 ± 0.15 ^ | 0.72 ± 0.019 ^ | 0.17 ± 0.004 ^ | 0.70 ± 0.02 ^ | 55.10 ± 2.96 ^ | |
| 4b | 27.95 ± 0.34 ^ | 35.18 ± 0.47 ^ | 0.70 ± 0.013 ^ | 0.17 ± 0.001 ^ | 0.88 ± 0.06 ^ | 62.60 ± 3.64 ^ | |
| 4c | 28.41 ± 0.28 ^ | 37.35 ± 1.47 ^ | 0.73 ± 0.014 ^ | 0.16 ± 0.007 ^ | 0.79 ± 0.04 ^ | 55.76 ± 5.64 ^ | |
| 4d | 30.14 ± 0.93 ^ | 38.72 ± 1.37 #,^ | 0.69 ± 0.016 ^ | 0.18 ± 0.004 ^ | 0.85 ± 0.03 ^ | 48.18 ± 0.53 ^ | |
| Compound no. | %Cell Cycle Arrest/Phase | %Cell Death | ||||||
|---|---|---|---|---|---|---|---|---|
| %G0-G1 | %S | %G2-M | %pre-G1 | Total | Early | Late | Necrosis | |
| Apoptosis | ||||||||
| 3a | 31.57 | 23.58 | 44.85 | 46.29 | 46.29 | 3.56 | 29.58 | 13.15 |
| 4b | 41.25 | 53.26 | 5.49 | 28.14 | 28.14 | 6.12 | 12.61 | 9.41 |
| 4c | 36.44 | 25.94 | 37.62 | 32.02 | 32.02 | 2.88 | 18.08 | 11.06 |
| Positive control | 29.74 | 31.26 | 39 | 42.38 | 42.38 | 1.85 | 26.27 | 14.26 |
| Negative control | 55.29 | 37.61 | 7.1 | 1.47 | 1.47 | 0.36 | 0.12 | 0.99 |
| HepG2 Cell Line | Control | Treated | |||
|---|---|---|---|---|---|
| Parameter/Groups | Positive | Negative | 3a | 4b | 4c |
| EGFR (pmol/mg protein) | 27.5 ± 1.7 a* | 306 ± 20 | 42 ± 2.3 a*,b* | 87.49 ± 3.4 a*,b* | 54 ± 2.4 a*,b* |
| uPA (nmol/mg protein) | 1244 ± 18 a* | 3149 ± 111 | 1258 ± 15 a* | 1916 ± 40 a*,b* | 1729 ± 38 a*,b* |
| Bcl-2 (nmol/mg protein) | 1.7 ± 0.10 a* | 7.8 ± 0.07 | 4.2 ± 0.10 a*,b* | 2.5 ± 0.04 a,b* | 3.6 ± 0.11 a*,b* |
| Heparanase (pmol/mg protein) | 902.1 ± 21 a* | 3097 ± 160 | 1262 ± 35 a*,b* | 1827 ± 30 a*, b* | 1449 ± 12.5 a*,b* |
| GSH (µmol/mg protein) | 0.8 ± 0.1 a* | 1.7 ± 0.2 | 1.6 ± 0.7 | 1.1 ± 0.7 | 2.1 ± 0.3 b* |
| MDA level (µmol/mg protein) | 18.5 ± 1.5 a* | 13.8 ± 1.0 | 2.0 ± 0.5 a*,b* | 3.7 ± 0.4 a*,b* | 14.5 ± 1.4 |
| Compound no. | S (Kcal/mol) | Amino Acids Residues | Ligand Atoms | Distance (Å A) | Strength (%) |
| 3a | −21.74 | Met769 | C=O of isatin | 2.86 | 15 |
| 4b | −19.21 | Met769 | C=O of isatin | 2.71 | 23 |
| 4c | −20.80 | Met769 | C=O of isatin | 2.85 | 33 |
| Asp831 | NH of acetanilide | 2.44 | 54 | ||
| Doxorubicin | −22.82 | Met769 | C=O pf anthraquinone | 2.74 | 32 |
| Lys721 | C=O of ethenone | 2.61 | 19 | ||
| Asp831 | Hydroxy group | 2.76 | 42 | ||
| Asn818 | NH2 group | 2.56 | 37 | ||
| Arg817 | NH2 group | 2.60 | 28 | ||
| Erlotinib | −25.65 | Met769 | C=O of quinazoline | 3.04 | 27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eldeeb, M.; Sanad, E.F.; Ragab, A.; Ammar, Y.A.; Mahmoud, K.; Ali, M.M.; Hamdy, N.M. Anticancer Effects with Molecular Docking Confirmation of Newly Synthesized Isatin Sulfonamide Molecular Hybrid Derivatives against Hepatic Cancer Cell Lines. Biomedicines 2022, 10, 722. https://doi.org/10.3390/biomedicines10030722
Eldeeb M, Sanad EF, Ragab A, Ammar YA, Mahmoud K, Ali MM, Hamdy NM. Anticancer Effects with Molecular Docking Confirmation of Newly Synthesized Isatin Sulfonamide Molecular Hybrid Derivatives against Hepatic Cancer Cell Lines. Biomedicines. 2022; 10(3):722. https://doi.org/10.3390/biomedicines10030722
Chicago/Turabian StyleEldeeb, Mahmoud, Eman F. Sanad, Ahmed Ragab, Yousry A. Ammar, Khaled Mahmoud, Mamdouh M. Ali, and Nadia M. Hamdy. 2022. "Anticancer Effects with Molecular Docking Confirmation of Newly Synthesized Isatin Sulfonamide Molecular Hybrid Derivatives against Hepatic Cancer Cell Lines" Biomedicines 10, no. 3: 722. https://doi.org/10.3390/biomedicines10030722
APA StyleEldeeb, M., Sanad, E. F., Ragab, A., Ammar, Y. A., Mahmoud, K., Ali, M. M., & Hamdy, N. M. (2022). Anticancer Effects with Molecular Docking Confirmation of Newly Synthesized Isatin Sulfonamide Molecular Hybrid Derivatives against Hepatic Cancer Cell Lines. Biomedicines, 10(3), 722. https://doi.org/10.3390/biomedicines10030722