
The Trinity: Interplay among Cancer Cells, Fibroblasts, and Immune Cells in Pancreatic Cancer and Implication of CD8+ T Cell-Orientated Therapy

Abstract
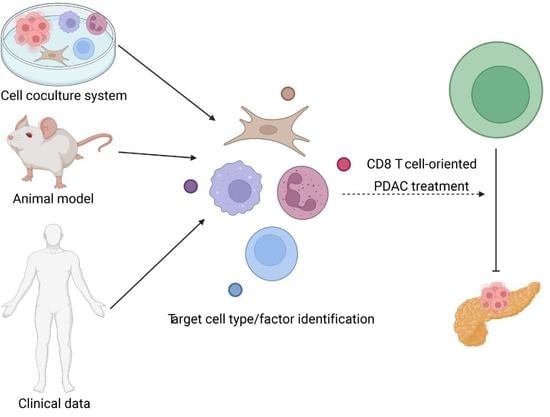
:
1. Introduction
2. Fibroblasts and Immune Cells in Pancreatic Cancer
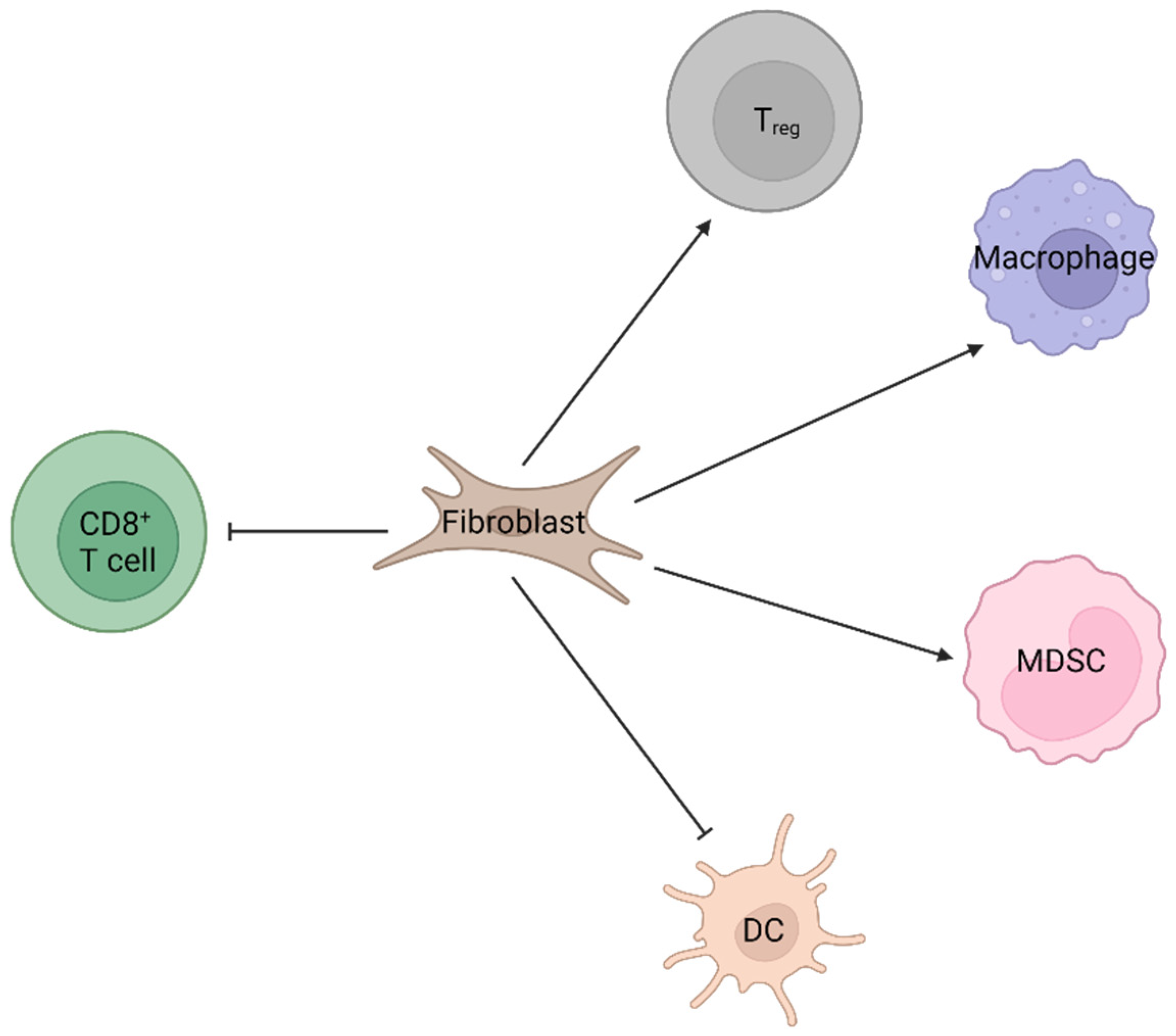
2.1. Fibroblast
2.2. Immune Cell
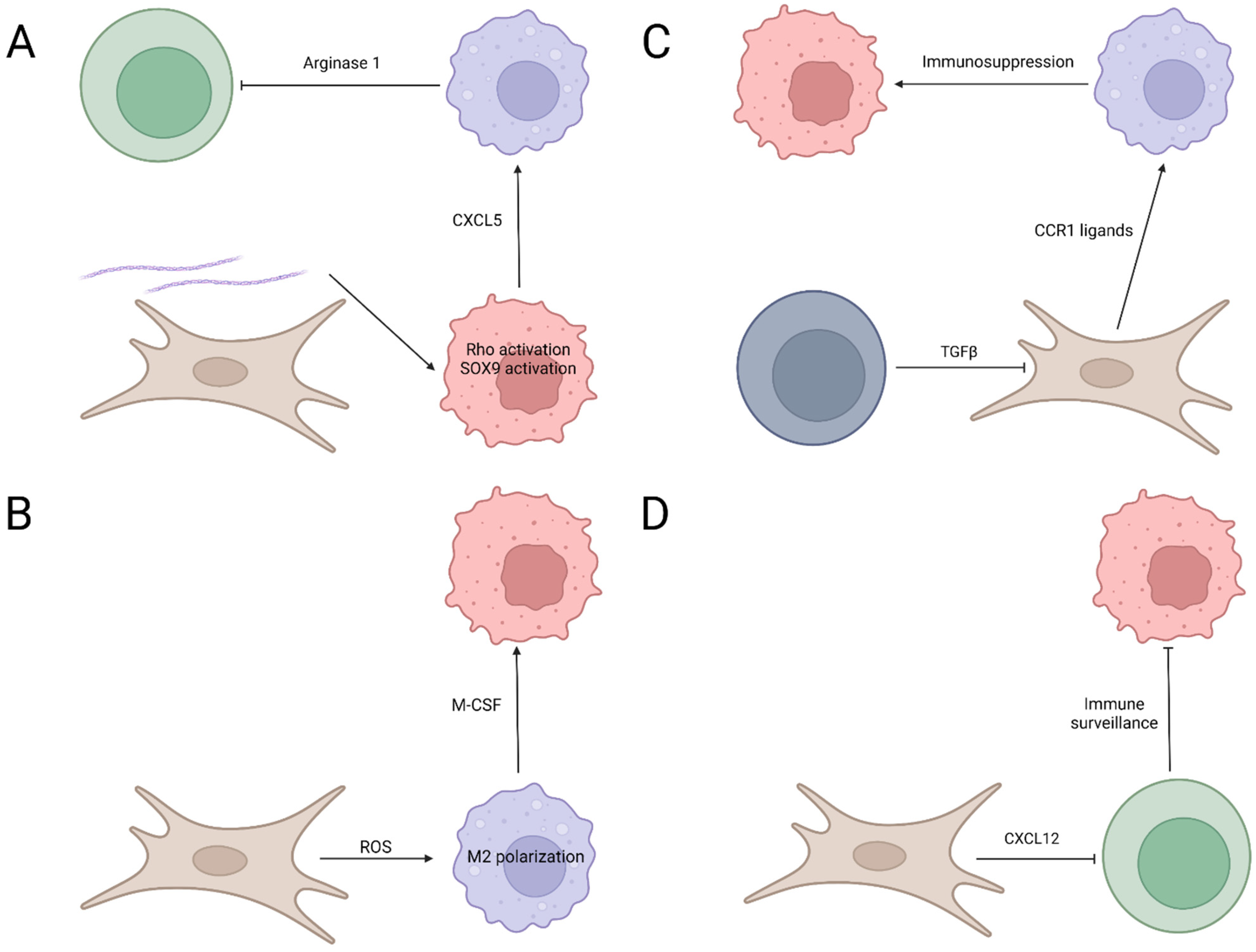
3. Mechanisms Underlying the Interplay among Pancreatic Cancer Cells, Fibroblasts, and Immune Cells
3.1. Cytokine and Chemokine
3.2. Extracellular Signal
3.3. Pathway Modulation
4. Correlation of Alterations in the “Trinity” Population in Preclinical Model and Clinical Setting
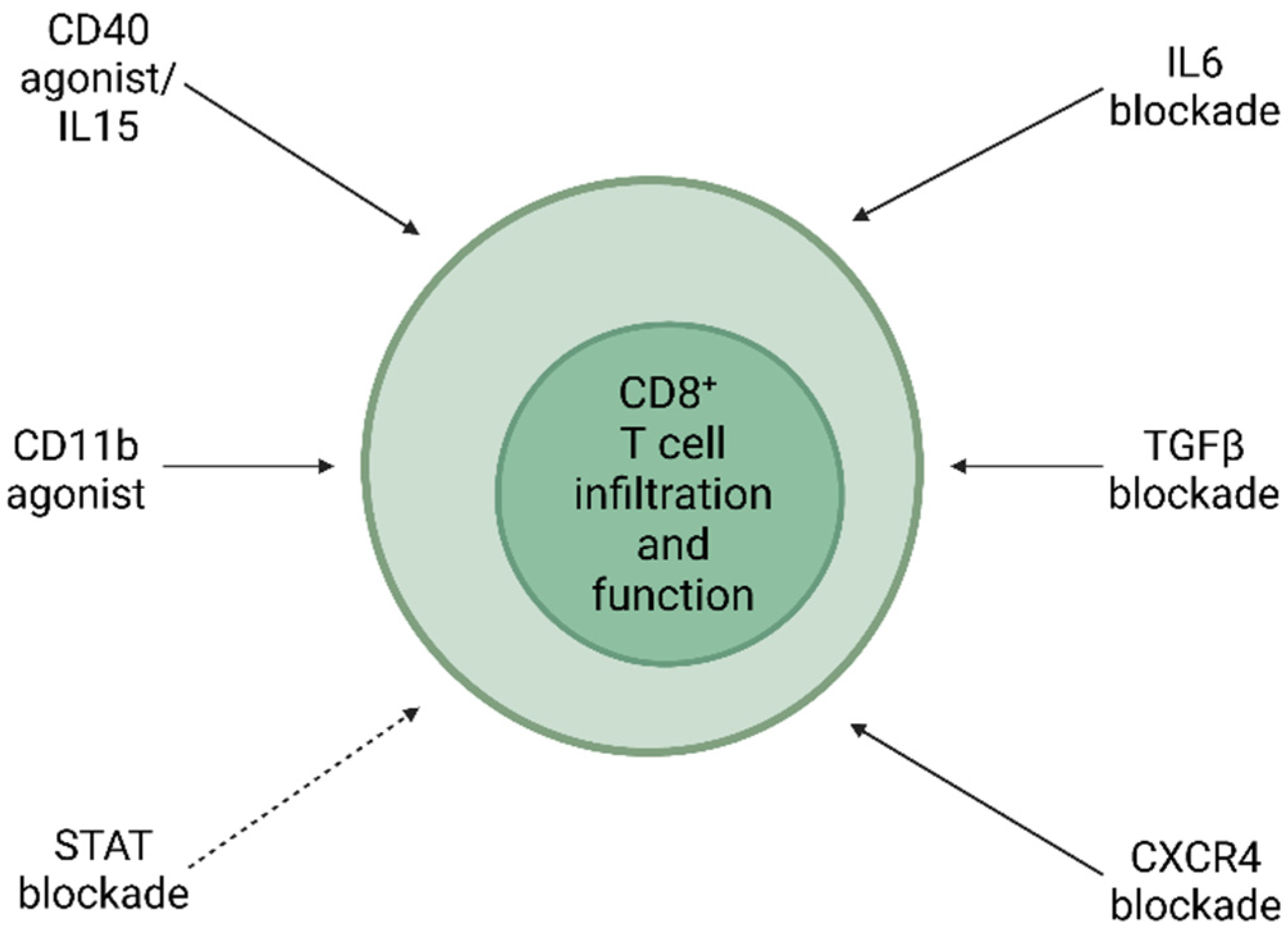
5. Treating Pancreatic Cancer with CD8+ T Cell-Orientated Approach
5.1. Preclinical Study
5.2. Clinical Trial
5.3. Clinical Correlation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Traub, B.; Link, K.H.; Kornmann, M. Curing pancreatic cancer. Semin. Cancer Biol. 2021, 76, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Yazal, T.; Bailleul, J.; Ruan, Y.; Sung, D.; Chu, F.I.; Palomera, D.; Dao, A.; Sehgal, A.; Gurunathan, V.; Aryan, L.; et al. Radiosensitizing Pancreatic Cancer via Effective Autophagy Inhibition. Mol. Cancer Ther. 2022, 21, 79–88. [Google Scholar] [CrossRef]
- Macedo, F.I.; Ryon, E.; Maithel, S.K.; Lee, R.M.; Kooby, D.A.; Fields, R.C.; Hawkins, W.G.; Williams, G.; Maduekwe, U.; Kim, H.J.; et al. Survival Outcomes Associated with Clinical and Pathological Response Following Neoadjuvant FOLFIRINOX or Gemcitabine/Nab-Paclitaxel Chemotherapy in Resected Pancreatic Cancer. Ann. Surg. 2019, 270, 400–413. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, C.; Xie, K.P. Therapeutic resistance of pancreatic cancer: Roadmap to its reversal. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188461. [Google Scholar] [CrossRef]
- Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers 2021, 13, 4138. [Google Scholar] [CrossRef]
- Principe, D.R.; Korc, M.; Kamath, S.D.; Munshi, H.G.; Rana, A. Trials and tribulations of pancreatic cancer immunotherapy. Cancer Lett. 2021, 504, 1–14. [Google Scholar] [CrossRef]
- Kang, J.S.; Choi, Y.J.; Byun, Y.; Han, Y.; Kim, J.H.; Lee, J.M.; Sohn, H.J.; Kim, H.; Kwon, W.; Jang, J.Y. Radiological tumour invasion of splenic artery or vein in patients with pancreatic body or tail adenocarcinoma and effect on recurrence and survival. Br. J. Surg. 2021, 109, 105–113. [Google Scholar] [CrossRef]
- Zhu, X.; Cao, Y.; Liu, W.; Ju, X.; Zhao, X.; Jiang, L.; Ye, Y.; Jin, G.; Zhang, H. Stereotactic body radiotherapy plus pembrolizumab and trametinib versus stereotactic body radiotherapy plus gemcitabine for locally recurrent pancreatic cancer after surgical resection: An open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2021, 22, 1093–1102. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Jiang, Y.; Song, L.; Liu, Y.; Cheng, F.; Fan, X.; Cao, X.; Gong, A.; Wang, D.; et al. Radiotherapy-induced cell death activates paracrine HMGB1-TLR2 signaling and accelerates pancreatic carcinoma metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shi, H.; Chen, H.; Gong, A.; Liu, Y.; Song, L.; Xu, X.; You, T.; Fan, X.; Wang, D.; et al. Dedifferentiation process driven by radiotherapy-induced HMGB1/TLR2/YAP/HIF-1α signaling enhances pancreatic cancer stemness. Cell Death Dis. 2019, 10, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, K.; Takano, S.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Ohtsuka, M. The effect of the low stromal ratio induced by neoadjuvant chemotherapy on recurrence patterns in borderline resectable pancreatic ductal adenocarcinoma. Clin. Exp. Metastasis 2022, 39, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Hellmann, M.D.; Wolchok, J.D.; Vyas, M.; Shia, J.; Stadler, Z.K.; Diaz, L.A., Jr.; O’Reilly, E.M. Acquired resistance to immunotherapy in MMR-D pancreatic cancer. J. Immunother. Cancer 2018, 6, 127. [Google Scholar] [CrossRef]
- Jiang, J.; Zhou, H.; Ni, C.; Hu, X.; Mou, Y.; Huang, D.; Yang, L. Immunotherapy in pancreatic cancer: New hope or mission impossible? Cancer Lett. 2019, 445, 57–64. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Upstill-Goddard, R.; Legrini, A.; Biankin, A.V.; Jamieson, N.B.; Chang, D.K.; Jamieson, N.B.; Chang, D.K. Genomic and Molecular Analyses Identify Molecular Subtypes of Pancreatic Cancer Recurrence. Gastroenterology 2022, 162, 320–324.e324. [Google Scholar] [CrossRef]
- Asimgil, H.; Ertetik, U.; Çevik, N.C.; Ekizce, M.; Doğruöz, A.; Gökalp, M.; Arık-Sever, E.; Istvanffy, R.; Friess, H.; Ceyhan, G.O.; et al. Targeting the undruggable oncogenic KRAS: The dawn of hope. JCI Insight 2022, 7, e153688. [Google Scholar] [CrossRef]
- Saiki, Y.; Jiang, C.; Ohmuraya, M.; Furukawa, T. Genetic Mutations of Pancreatic Cancer and Genetically Engineered Mouse Models. Cancers 2021, 14, 71. [Google Scholar] [CrossRef]
- Ala, M. Target c-Myc to treat pancreatic cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Ou, A.; Zhao, X.; Lu, Z. The potential roles of p53 signaling reactivation in pancreatic cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188662. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.S.; Kanani, R.; Roylance, R.; Mittnacht, S. Systematic Review of Molecular Biomarkers Predictive of Resistance to CDK4/6 Inhibition in Metastatic Breast Cancer. JCO Precis. Oncol. 2022, 6, e2100002. [Google Scholar] [CrossRef] [PubMed]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 kinases: From basic science to cancer therapy. Science 2022, 375, eabc1495. [Google Scholar] [CrossRef]
- Killock, D. Abemaciclib crowned in monarchE. Nat. Rev. Clin. Oncol. 2022, 19, 5. [Google Scholar] [CrossRef]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef]
- Prados-Carvajal, R.; Irving, E.; Lukashchuk, N.; Forment, J.V. Preventing and Overcoming Resistance to PARP Inhibitors: A Focus on the Clinical Landscape. Cancers 2021, 14, 44. [Google Scholar] [CrossRef]
- Ji, Y.; Liu, X.; Li, J.; Xie, X.; Huang, M.; Jiang, J.; Liao, Y.P.; Donahue, T.; Meng, H. Use of ratiometrically designed nanocarrier targeting CDK4/6 and autophagy pathways for effective pancreatic cancer treatment. Nat. Commun. 2020, 11, 4249. [Google Scholar] [CrossRef]
- Wang, S.P.; Li, Y.; Huang, S.H.; Wu, S.Q.; Gao, L.L.; Sun, Q.; Lin, Q.W.; Huang, L.; Meng, L.Q.; Zou, Y.; et al. Discovery of Potent and Novel Dual PARP/BRD4 Inhibitors for Efficient Treatment of Pancreatic Cancer. J. Med. Chem. 2021, 64, 17413–17435. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. The efficacy of PD-1/PD-L1 blockade in cold cancers and future perspectives. Clin. Immunol. 2021, 226, 108707. [Google Scholar] [CrossRef]
- Torphy, R.J.; Schulick, R.D.; Zhu, Y. Understanding the immune landscape and tumor microenvironment of pancreatic cancer to improve immunotherapy. Mol. Carcinog. 2020, 59, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Wandmacher, A.M.; Letsch, A.; Sebens, S. Challenges and Future Perspectives of Immunotherapy in Pancreatic Cancer. Cancers 2021, 13, 4235. [Google Scholar] [CrossRef] [PubMed]
- Saka, D.; Gökalp, M.; Piyade, B.; Cevik, N.C.; Arik Sever, E.; Unutmaz, D.; Ceyhan, G.O.; Demir, I.E.; Asimgil, H. Mechanisms of T-Cell Exhaustion in Pancreatic Cancer. Cancers 2020, 12, 2274. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.B.; Pasca di Magliano, M.; Crawford, H.C. Myeloid Cell Mediated Immune Suppression in Pancreatic Cancer. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1531–1542. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Shi, Y.; Qian, F. Opportunities and delusions regarding drug delivery targeting pancreatic cancer-associated fibroblasts. Adv. Drug Deliv. Rev. 2021, 172, 37–51. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Sunami, Y.; Häußler, J.; Kleeff, J. Cellular Heterogeneity of Pancreatic Stellate Cells, Mesenchymal Stem Cells, and Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers 2020, 12, 3770. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Bauer, C.A.; Öhlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.T.; Abuwarwar, M.H.; Poly, L.; Wilkins, S.; Fletcher, A.L. Cancer-Associated Fibroblasts and T Cells: From Mechanisms to Outcomes. J. Immunol. 2021, 206, 310–320. [Google Scholar] [CrossRef]
- Huang, H.; Brekken, R.A. Recent advances in understanding cancer-associated fibroblasts in pancreatic cancer. Am. J. Physiol. Cell Physiol. 2020, 319, C233–C243. [Google Scholar] [CrossRef]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Polani, F.; Grierson, P.M.; Lim, K.H. Stroma-targeting strategies in pancreatic cancer: Past lessons, challenges and prospects. World J. Gastroenterol. 2021, 27, 2105–2121. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Affo, S.; Nair, A.; Brundu, F.; Ravichandra, A.; Bhattacharjee, S.; Matsuda, M.; Chin, L.; Filliol, A.; Wen, W.; Song, X.; et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021, 39, 883. [Google Scholar] [CrossRef] [PubMed]
- Strating, E.; Wassenaar, E.; Verhagen, M.; Rauwerdink, P.; van Schelven, S.; de Hingh, I.; Rinkes, I.B.; Boerma, D.; Witkamp, A.; Lacle, M.; et al. Fibroblast activation protein identifies Consensus Molecular Subtype 4 in colorectal cancer and allows its detection by (68)Ga-FAPI-PET imaging. Br. J. Cancer 2022. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, L.; Liu, L.; Hou, Y.; Xiong, M.; Yang, Y.; Hu, J.; Chen, K. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 2020, 11, 5077. [Google Scholar] [CrossRef]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef]
- Mhaidly, R.; Mechta-Grigoriou, F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol. Rev. 2021, 302, 259–272. [Google Scholar] [CrossRef]
- Ganguly, K.; Shah, A.; Atri, P.; Rauth, S.; Ponnusamy, M.P.; Kumar, S.; Batra, S.K. Chemokine-mucinome interplay in shaping the heterogeneous tumor microenvironment of pancreatic cancer. Semin. Cancer Biol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Bernard, V.; Semaan, A.; Monberg, M.E.; Huang, J.; Stephens, B.M.; Lin, D.; Rajapakshe, K.I.; Weston, B.R.; Bhutani, M.S.; et al. Elucidation of Tumor-Stromal Heterogeneity and the Ligand-Receptor Interactome by Single-Cell Transcriptomics in Real-world Pancreatic Cancer Biopsies. Clin. Cancer Res. 2021, 27, 5912–5921. [Google Scholar] [CrossRef] [PubMed]
- Domen, A.; Quatannens, D.; Zanivan, S.; Deben, C.; Van Audenaerde, J.; Smits, E.; Wouters, A.; Lardon, F.; Roeyen, G.; Verhoeven, Y.; et al. Cancer-Associated Fibroblasts as a Common Orchestrator of Therapy Resistance in Lung and Pancreatic Cancer. Cancers 2021, 13, 987. [Google Scholar] [CrossRef]
- Gorchs, L.; Kaipe, H. Interactions between Cancer-Associated Fibroblasts and T Cells in the Pancreatic Tumor Microenvironment and the Role of Chemokines. Cancers 2021, 13, 2995. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Boyd, L.N.C.; Andini, K.D.; Peters, G.J.; Kazemier, G.; Giovannetti, E. Heterogeneity and plasticity of cancer-associated fibroblasts in the pancreatic tumor microenvironment. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef]
- Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 13408. [Google Scholar] [CrossRef]
- Van Audenaerde, J.R.M.; Roeyen, G.; Darcy, P.K.; Kershaw, M.H.; Peeters, M.; Smits, E.L.J. Natural killer cells and their therapeutic role in pancreatic cancer: A systematic review. Pharmacol. Ther. 2018, 189, 31–44. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Sun, Z.; Zhan, H. Macrophages in pancreatic cancer: An immunometabolic perspective. Cancer Lett. 2021, 498, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.L.; Hii, L.W.; Wong, S.F.; Leong, C.O.; Mai, C.W. Molecular Mechanisms and Potential Therapeutic Reversal of Pancreatic Cancer-Induced Immune Evasion. Cancers 2020, 12, 1872. [Google Scholar] [CrossRef] [PubMed]
- Pergamo, M.; Miller, G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017, 24, 100–105. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Bear, A.S. Tumor-Derived Myeloid Cell Chemoattractants and T Cell Exclusion in Pancreatic Cancer. Front. Immunol. 2020, 11, 605619. [Google Scholar] [CrossRef] [PubMed]
- Ware, M.B.; El-Rayes, B.F.; Lesinski, G.B. Mirage or long-awaited oasis: Reinvigorating T-cell responses in pancreatic cancer. J. Immunother. Cancer 2020, 8, e001100. [Google Scholar] [CrossRef] [PubMed]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing pancreatic cancer: Combat with a double edged sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fincham, R.E.A.; Delvecchio, F.R.; Goulart, M.R.; Yeong, J.P.S.; Kocher, H.M. Natural killer cells in pancreatic cancer stroma. World J. Gastroenterol. 2021, 27, 3483–3501. [Google Scholar] [CrossRef] [PubMed]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; Gögenur, I. The prognostic value of tumour-infiltrating lymphocytes in pancreatic cancer: A systematic review and meta-analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef]
- Xu, C.; Sui, S.; Shang, Y.; Yu, Z.; Han, J.; Zhang, G.; Ntim, M.; Hu, M.; Gong, P.; Chen, H.; et al. The landscape of immune cell infiltration and its clinical implications of pancreatic ductal adenocarcinoma. J. Adv. Res. 2020, 24, 139–148. [Google Scholar] [CrossRef]
- Theoharides, T.C. Mast cells and pancreatic cancer. N. Engl. J. Med. 2008, 358, 1860–1861. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Ullrich, S.E. Intratumoral mast cells promote the growth of pancreatic cancer. Oncoimmunology 2013, 2, e25964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, V.; Tamma, R.; Brunetti, O.; Pisconti, S.; Argentiero, A.; Silvestris, N.; Ribatti, D. Mast cells and angiogenesis in pancreatic ductal adenocarcinoma. Clin. Exp. Med. 2018, 18, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, N.G.; Griffith, D.; Hammoda, M.; Shingler, G.; Kambal, A.; Al-Sarireh, B. A meta-analysis of the utility of the neutrophil-to-lymphocyte ratio in predicting survival after pancreatic cancer resection. HPB 2018, 20, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wei, Q.; Fan, J.; Cheng, S.; Ding, W.; Hua, Z. Prognostic role of the neutrophil-to-lymphocyte ratio in pancreatic cancer: A meta-analysis containing 8252 patients. Clin. Chim. Acta 2018, 479, 181–189. [Google Scholar] [CrossRef]
- Yu, M.; Guan, R.; Hong, W.; Zhou, Y.; Lin, Y.; Jin, H.; Hou, B.; Jian, Z. Prognostic value of tumor-associated macrophages in pancreatic cancer: A meta-analysis. Cancer Manag. Res. 2019, 11, 4041–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallal, R.M.; Christakos, P.; Lee, K.; Egawa, S.; Son, Y.I.; Lotze, M.T. Paucity of dendritic cells in pancreatic cancer. Surgery 2002, 131, 135–138. [Google Scholar] [CrossRef]
- Bauer, C.; Dauer, M.; Saraj, S.; Schnurr, M.; Bauernfeind, F.; Sterzik, A.; Junkmann, J.; Jakl, V.; Kiefl, R.; Oduncu, F.; et al. Dendritic cell-based vaccination of patients with advanced pancreatic carcinoma: Results of a pilot study. Cancer Immunol. Immunother. 2011, 60, 1097–1107. [Google Scholar] [CrossRef]
- Ochi, A.; Nguyen, A.H.; Bedrosian, A.S.; Mushlin, H.M.; Zarbakhsh, S.; Barilla, R.; Zambirinis, C.P.; Fallon, N.C.; Rehman, A.; Pylayeva-Gupta, Y.; et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 2012, 209, 1671–1687. [Google Scholar] [CrossRef]
- Kobayashi, M.; Shimodaira, S.; Nagai, K.; Ogasawara, M.; Takahashi, H.; Abe, H.; Tanii, M.; Okamoto, M.; Tsujitani, S.; Yusa, S.; et al. Prognostic factors related to add-on dendritic cell vaccines on patients with inoperable pancreatic cancer receiving chemotherapy: A multicenter analysis. Cancer Immunol. Immunother. 2014, 63, 797–806. [Google Scholar] [CrossRef]
- Mayanagi, S.; Kitago, M.; Sakurai, T.; Matsuda, T.; Fujita, T.; Higuchi, H.; Taguchi, J.; Takeuchi, H.; Itano, O.; Aiura, K.; et al. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci. 2015, 106, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, M.; Kobayashi, M.; Yonemitsu, Y.; Koido, S.; Homma, S. Dendritic cell-based vaccine for pancreatic cancer in Japan. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307.e289. [Google Scholar] [CrossRef]
- Shangguan, A.; Shang, N.; Figini, M.; Pan, L.; Yang, J.; Ma, Q.; Hu, S.; Eresen, A.; Sun, C.; Wang, B.; et al. Prophylactic dendritic cell vaccination controls pancreatic cancer growth in a mouse model. Cytotherapy 2020, 22, 6–15. [Google Scholar] [CrossRef]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef]
- Cui, C.; Lan, P.; Fu, L. The role of myeloid-derived suppressor cells in gastrointestinal cancer. Cancer Commun. 2021, 41, 442–471. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Trovato, R.; Fiore, A.; Sartori, S.; Canè, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Zhang, Q.; Liu, Y.; Guo, X.; Ju, J.; Xu, L.; Gao, Y.; Chen, D.; Mu, D.; Zhang, R. Apolipoprotein A-I Mimetic Peptide L-4F Suppresses Granulocytic-Myeloid-Derived Suppressor Cells in Mouse Pancreatic Cancer. Front. Pharmacol. 2020, 11, 576. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.D.; Abrams, S.I. Granulocytic Myeloid-Derived Suppressor Cells as Negative Regulators of Anticancer Immunity. Front. Immunol. 2020, 11, 1963. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Lenehan, J.G.; Burton, J.P.; Maleki Vareki, S. Immunomodulation in Pancreatic Cancer. Cancers 2020, 12, 3340. [Google Scholar] [CrossRef]
- Mucciolo, G.; Curcio, C.; Roux, C.; Li, W.Y.; Capello, M.; Curto, R.; Chiarle, R.; Giordano, D.; Satolli, M.A.; Lawlor, R.; et al. IL17A critically shapes the transcriptional program of fibroblasts in pancreatic cancer and switches on their protumorigenic functions. Proc. Natl. Acad. Sci. USA 2021, 118, e2020395118. [Google Scholar] [CrossRef]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Ghoreschi, K.; Steward-Tharp, S.M.; Rodriguez-Canales, J.; Zhu, J.; Grainger, J.R.; Hirahara, K.; Sun, H.W.; Wei, L.; Vahedi, G.; et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011, 12, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.J.; Moon, S.J.; Lee, E.J.; Jung, K.A.; Kim, E.K.; Kim, D.S.; Lee, J.H.; Kwok, S.K.; Min, J.K.; Park, S.H.; et al. IL-1-IL-17 Signaling Axis Contributes to Fibrosis and Inflammation in Two Different Murine Models of Systemic Sclerosis. Front. Immunol. 2018, 9, 1611. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e20190354. [Google Scholar] [CrossRef]
- Chen, J.; Ye, X.; Pitmon, E.; Lu, M.; Wan, J.; Jellison, E.R.; Adler, A.J.; Vella, A.T.; Wang, K. IL-17 inhibits CXCL9/10-mediated recruitment of CD8(+) cytotoxic T cells and regulatory T cells to colorectal tumors. J. Immunother. Cancer 2019, 7, 324. [Google Scholar] [CrossRef]
- Dadaglio, G.; Fayolle, C.; Oberkampf, M.; Tang, A.; Rudilla, F.; Couillin, I.; Torheim, E.A.; Rosenbaum, P.; Leclerc, C. IL-17 suppresses the therapeutic activity of cancer vaccines through the inhibition of CD8(+) T-cell responses. Oncoimmunology 2020, 9, 1758606. [Google Scholar] [CrossRef]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.L.; Cooke, M.E.; Alliston, T. ECM stiffness primes the TGFβ pathway to promote chondrocyte differentiation. Mol. Biol. Cell 2012, 23, 3731–3742. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhou, W. The Emerging Regulation of VEGFR-2 in Triple-Negative Breast Cancer. Front. Endocrinol. 2015, 6, 159. [Google Scholar] [CrossRef] [Green Version]
- Tirpe, A.; Gulei, D.; Tirpe, G.R.; Nutu, A.; Irimie, A.; Campomenosi, P.; Pop, L.A.; Berindan-Neagoe, I. Beyond Conventional: The New Horizon of Anti-Angiogenic microRNAs in Non-Small Cell Lung Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 8002. [Google Scholar] [CrossRef]
- Zhao, J.; Xiao, Z.; Li, T.; Chen, H.; Yuan, Y.; Wang, Y.A.; Hsiao, C.H.; Chow, D.S.; Overwijk, W.W.; Li, C. Stromal Modulation Reverses Primary Resistance to Immune Checkpoint Blockade in Pancreatic Cancer. ACS Nano 2018, 12, 9881–9893. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Y.; Hu, M.; Wang, M.; Liu, X.; Huang, L. Relaxin gene delivery modulates macrophages to resolve cancer fibrosis and synergizes with immune checkpoint blockade therapy. Sci. Adv. 2021, 7, eabb6596. [Google Scholar] [CrossRef]
- Kanai, A.J.; Konieczko, E.M.; Bennett, R.G.; Samuel, C.S.; Royce, S.G. Relaxin and fibrosis: Emerging targets, challenges, and future directions. Mol. Cell. Endocrinol. 2019, 487, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Gorchs, L.; Fernández Moro, C.; Bankhead, P.; Kern, K.P.; Sadeak, I.; Meng, Q.; Rangelova, E.; Kaipe, H. Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-inhibitory Markers on CD4(+) and CD8(+) T-Cells. Front. Immunol. 2019, 10, 847. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.; Luckett, T.; Schmid, M.C.; Mielgo, A. Blockade of Stromal Gas6 Alters Cancer Cell Plasticity, Activates NK Cells, and Inhibits Pancreatic Cancer Metastasis. Front. Immunol. 2020, 11, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef]
- Zdżalik-Bielecka, D.; Poświata, A.; Kozik, K.; Jastrzębski, K.; Schink, K.O.; Brewińska-Olchowik, M.; Piwocka, K.; Stenmark, H.; Miączyńska, M. The GAS6-AXL signaling pathway triggers actin remodeling that drives membrane ruffling, macropinocytosis, and cancer-cell invasion. Proc. Natl. Acad. Sci. USA 2021, 118, e2024596118. [Google Scholar] [CrossRef]
- Kamionka, E.M.; Qian, B.; Gross, W.; Bergmann, F.; Hackert, T.; Beretta, C.A.; Dross, N.; Ryschich, E. Collagen Organization Does Not Influence T-Cell Distribution in Stroma of Human Pancreatic Cancer. Cancers 2021, 13, 3648. [Google Scholar] [CrossRef]
- Lawal, B.; Lin, L.C.; Lee, J.C.; Chen, J.H.; Bekaii-Saab, T.S.; Wu, A.T.H.; Ho, C.L. Multi-Omics Data Analysis of Gene Expressions and Alterations, Cancer-Associated Fibroblast and Immune Infiltrations, Reveals the Onco-Immune Prognostic Relevance of STAT3/CDK2/4/6 in Human Malignancies. Cancers 2021, 13, 954. [Google Scholar] [CrossRef]
- MacNeil, T.; Vathiotis, I.A.; Shafi, S.; Aung, T.N.; Zugazagoitia, J.; Gruver, A.M.; Driscoll, K.; Rimm, D.L. Multiplex Quantitative Analysis of Tumor-Infiltrating Lymphocytes, Cancer-Associated Fibroblasts, and CD200 in Pancreatic Cancer. Cancers 2021, 13, 5501. [Google Scholar] [CrossRef]
- Tahkola, K.; Ahtiainen, M.; Mecklin, J.P.; Kellokumpu, I.; Laukkarinen, J.; Tammi, M.; Tammi, R.; Väyrynen, J.P.; Böhm, J. Stromal hyaluronan accumulation is associated with low immune response and poor prognosis in pancreatic cancer. Sci. Rep. 2021, 11, 12216. [Google Scholar] [CrossRef]
- Nizri, E.; Bar-David, S.; Aizic, A.; Sternbach, N.; Lahat, G.; Wolf, I.; Klausner, J. Desmoplasia in Lymph Node Metastasis of Pancreatic Adenocarcinoma Reveals Activation of Cancer-Associated Fibroblasts Pattern and T-helper 2 Immune Cell Infiltration. Pancreas 2019, 48, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Sadozai, H.; Acharjee, A.; Eppenberger-Castori, S.; Gloor, B.; Gruber, T.; Schenk, M.; Karamitopoulou, E. Distinct Stromal and Immune Features Collectively Contribute to Long-Term Survival in Pancreatic Cancer. Front. Immunol. 2021, 12, 643529. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8(+) T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef] [Green Version]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Van Audenaerde, J.R.; Marcq, E.; von Scheidt, B.; Davey, A.S.; Oliver, A.J.; De Waele, J.; Quatannens, D.; Van Loenhout, J.; Pauwels, P.; Roeyen, G.; et al. Novel combination immunotherapy for pancreatic cancer: Potent anti-tumor effects with CD40 agonist and interleukin-15 treatment. Clin. Transl. Immunol. 2020, 9, e1165. [Google Scholar] [CrossRef]
- Panni, R.Z.; Herndon, J.M.; Zuo, C.; Hegde, S.; Hogg, G.D.; Knolhoff, B.L.; Breden, M.A.; Li, X.; Krisnawan, V.E.; Khan, S.Q.; et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci. Transl. Med. 2019, 11, eaau9240. [Google Scholar] [CrossRef]
- Michaelis, K.A.; Norgard, M.A.; Zhu, X.; Levasseur, P.R.; Sivagnanam, S.; Liudahl, S.M.; Burfeind, K.G.; Olson, B.; Pelz, K.R.; Angeles Ramos, D.M.; et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat. Commun. 2019, 10, 4682. [Google Scholar] [CrossRef] [Green Version]
- Ajina, R.; Malchiodi, Z.X.; Fitzgerald, A.A.; Zuo, A.; Wang, S.; Moussa, M.; Cooper, C.J.; Shen, Y.; Johnson, Q.R.; Parks, J.M.; et al. Antitumor T-cell Immunity Contributes to Pancreatic Cancer Immune Resistance. Cancer Immunol. Res. 2021, 9, 386–400. [Google Scholar] [CrossRef]
- Herting, C.J.; Karpovsky, I.; Lesinski, G.B. The tumor microenvironment in pancreatic ductal adenocarcinoma: Current perspectives and future directions. Cancer Metastasis Rev. 2021, 40, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Sonbol, M.B.; Ahn, D.H.; Goldstein, D.; Okusaka, T.; Tabernero, J.; Macarulla, T.; Reni, M.; Li, C.P.; O’Neil, B.; Van Cutsem, E.; et al. CanStem111P trial: A Phase III study of napabucasin plus nab-paclitaxel with gemcitabine. Future Oncol. 2019, 15, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; Nott, L.; Yoshino, T.; Gill, S.; Shapiro, J.; Ohtsu, A.; Zalcberg, J.; Vickers, M.M.; Wei, A.C.; Gao, Y.; et al. Napabucasin versus placebo in refractory advanced colorectal cancer: A randomised phase 3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 263–270. [Google Scholar] [CrossRef]
- Taniguchi, H.; Masuishi, T.; Kawazoe, A.; Muro, K.; Kadowaki, S.; Bando, H.; Iino, S.; Kageyama, R.; Yoshino, T. Phase I study of napabucasin in combination with FOLFIRI + bevacizumab in Japanese patients with metastatic colorectal cancer. Int. J. Clin. Oncol. 2021, 26, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.; Bladé, J.; Shpilberg, O.; Grosicki, S.; Maloisel, F.; Min, C.K.; Polo Zarzuela, M.; Robak, T.; Prasad, S.V.; Tee Goh, Y.; et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti–IL-6) in multiple myeloma. Blood 2014, 123, 4136–4142. [Google Scholar] [CrossRef] [Green Version]
- Brighton, T.A.; Khot, A.; Harrison, S.J.; Ghez, D.; Weiss, B.M.; Kirsch, A.; Magen, H.; Gironella, M.; Oriol, A.; Streetly, M.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Study of Siltuximab in High-Risk Smoldering Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3772–3775. [Google Scholar] [CrossRef] [Green Version]
- Uy, G.L.; Rettig, M.P.; Stone, R.M.; Konopleva, M.Y.; Andreeff, M.; McFarland, K.; Shannon, W.; Fletcher, T.R.; Reineck, T.; Eades, W.; et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J. 2017, 7, e542. [Google Scholar] [CrossRef]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef]
- Dessureault, S.; Noyes, D.; Lee, D.; Dunn, M.; Janssen, W.; Cantor, A.; Sotomayor, E.; Messina, J.; Antonia, S.J. A phase-I trial using a universal GM-CSF-producing and CD40L-expressing bystander cell line (GM.CD40L) in the formulation of autologous tumor cell-based vaccines for cancer patients with stage IV disease. Ann. Surg. Oncol. 2007, 14, 869–884. [Google Scholar] [CrossRef]
- Gray, J.E.; Chiappori, A.; Williams, C.C.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.C.; Kim, J.; Boyle, T.A.; Pinder-Schenck, M.; Khalil, F.; et al. A phase I/randomized phase II study of GM.CD40L vaccine in combination with CCL21 in patients with advanced lung adenocarcinoma. Cancer Immunol. Immunother. 2018, 67, 1853–1862. [Google Scholar] [CrossRef] [Green Version]
- Plesca, I.; Benešová, I.; Beer, C.; Sommer, U.; Müller, L.; Wehner, R.; Heiduk, M.; Aust, D.; Baretton, G.; Bachmann, M.P.; et al. Clinical Significance of Tumor-Infiltrating Conventional and Plasmacytoid Dendritic Cells in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 1216. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Benedetti, R.; Piconese, S.; Pulcinelli, F.M.; Timperio, A.M.; Romeo, M.A.; Masuelli, L.; Mattei, M.; Bei, R.; D’Orazi, G.; et al. PGE2 Released by Pancreatic Cancer Cells Undergoing ER Stress Transfers the Stress to DCs Impairing Their Immune Function. Mol. Cancer Ther. 2021, 20, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liao, M.; Hu, X.; Wang, J. Tumour-Derived Reg3A Educates Dendritic Cells to Promote Pancreatic Cancer Progression. Mol. Cells 2021, 44, 647–657. [Google Scholar] [CrossRef]
- Giri, B.; Sharma, P.; Jain, T.; Ferrantella, A.; Vaish, U.; Mehra, S.; Garg, B.; Iyer, S.; Sethi, V.; Malchiodi, Z.; et al. Hsp70 modulates immune response in pancreatic cancer through dendritic cells. Oncoimmunology 2021, 10, 1976952. [Google Scholar] [CrossRef] [PubMed]
- Sadeghlar, F.; Vogt, A.; Mohr, R.U.; Mahn, R.; van Beekum, K.; Kornek, M.; Weismüller, T.J.; Branchi, V.; Matthaei, H.; Toma, M.; et al. Induction of cytotoxic effector cells towards cholangiocellular, pancreatic, and colorectal tumor cells by activation of the immune checkpoint CD40/CD40L on dendritic cells. Cancer Immunol. Immunother. 2021, 70, 1451–1464. [Google Scholar] [CrossRef]
- Kim, D.; Lee, S.; Na, K. Immune Stimulating Antibody-Photosensitizer Conjugates via Fc-Mediated Dendritic Cell Phagocytosis and Phototriggered Immunogenic Cell Death for KRAS-Mutated Pancreatic Cancer Treatment. Small 2021, 17, 2006650. [Google Scholar] [CrossRef]
- Salah, A.; Wang, H.; Li, Y.; Ji, M.; Ou, W.B.; Qi, N.; Wu, Y. Insights into Dendritic Cells in Cancer Immunotherapy: From Bench to Clinical Applications. Front. Cell Dev. Biol. 2021, 9, 686544. [Google Scholar] [CrossRef]
- Kvedaraite, E.; Ginhoux, F. Human dendritic cells in cancer. Sci. Immunol. 2022, 7, eabm9409. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, L.; Song, X.; Li, C.; Zhang, J.; Wang, L. A TP53-associated immune prognostic signature for the prediction of the overall survival and therapeutic responses in pancreatic cancer. Math. Biosci. Eng. 2022, 19, 191–208. [Google Scholar] [CrossRef]
- Kandimalla, R.; Tomihara, H.; Banwait, J.K.; Yamamura, K.; Singh, G.; Baba, H.; Goel, A. A 15-Gene Immune, Stromal, and Proliferation Gene Signature that Significantly Associates with Poor Survival in Patients with Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2020, 26, 3641–3648. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yao, P.; Zhao, K.; Ye, Z.; Zhang, H.; Cao, J.; Zhang, S.; Xing, C. Individualized prognostic signature for pancreatic carcinoma validated by integrating immune-related gene pairs (IRGPs). Bioengineered 2021, 12, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Ren, D.; Zhang, K.; Zhao, J.; Jin, X.; Wu, H. Using ESTIMATE algorithm to establish an 8-mRNA signature prognosis prediction system and identify immunocyte infiltration-related genes in Pancreatic adenocarcinoma. Aging 2020, 12, 5048–5070. [Google Scholar] [CrossRef]
- Zhang, C.; Ding, J.; Xu, X.; Liu, Y.; Huang, W.; Da, L.; Ma, Q.; Chen, S. Tumor Microenvironment Characteristics of Pancreatic Cancer to Determine Prognosis and Immune-Related Gene Signatures. Front. Mol. Biosci. 2021, 8, 645024. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Q.; Zhang, X.; Cui, M.; Li, T.; Zhang, Y.; Liao, Q. Immune subtyping for pancreatic cancer with implication in clinical outcomes and improving immunotherapy. Cancer Cell Int. 2021, 21, 137. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune Cell and Stromal Signature Associated with Progression-Free Survival of Patients with Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology 2018, 155, 1625–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santiago, I.; Yau, C.; Heij, L.; Middleton, M.R.; Markowetz, F.; Grabsch, H.I.; Dustin, M.L.; Sivakumar, S. Immunophenotypes of pancreatic ductal adenocarcinoma: Meta-analysis of transcriptional subtypes. Int. J. Cancer 2019, 145, 1125–1137. [Google Scholar] [CrossRef]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Cell Type | Mechanism | Reference |
|---|---|---|---|
| TGFβ | Treg | TGFβ from Treg activates SMA+ myofibroblasts and their expression of CCR1 ligands to recruit MDSC | [109] |
| CXCL12 | CAF | CXCL12 from FAP+ CAF increases T cell exclusion and cancer growth | [110] |
| Type I collagen | Myofibroblast | Type I collagen from myofibroblast decreases SOX9 expression and subsequently increases CXCL5 in PDAC to recruit MDSC | [113] |
| ROS | CAF | ROS from CAF increases monocyte differentiation into M2 macrophages and their production of M-CSF to increase the invasiveness of PDAC | [114] |
| Molecule | Cell Type | Association | Reference | |
|---|---|---|---|---|
| NA | PDAC | Collagen/fibroblast | ↓ post FAKi treatment | [122] |
| Macrophage | ↓ post FAKi treatment | |||
| G-MDSC | ↓ post FAKi treatment | |||
| NA | PDAC | CAF | PD-L1/PD-L2↑ | [123] |
| CD4/CD8 proliferation ↓ | ||||
| CD4/CD8 co-inhibitory marker↑ | ||||
| CD8 function ↓ | ||||
| NA | PDAC | Vimentin | ↓ post α-Gas6 treatment | [124] |
| NK | ↓ post α-Gas6 treatment | |||
| NA | PDAC | Collagen | Density not altered | [127] |
| T cell | Infiltration not altered | |||
| CDK2/4/6 | PDAC | CAF | Co-occurrence ↑ | [128] |
| STAT3 | CAF | Co-occurrence ↑ | ||
| Immunity | Onco-immune signature ↑ | |||
| NA | PDAC | CD4 | Disease progression ↓ | [129] |
| CD8 | Disease progression ↓ | |||
| Thy-1+ CAF | Disease progression ↓ | |||
| FAP+ CAF | Disease progression ↑ | |||
| Stromal hyaluronan accumulation | NA | CD8/CD3-based immune cell score | ↓ | [130] |
| NA | PDAC | Desmoplasia | COL11A1/COL11A2/COL1A1/TGF-β mRNA ↑ | [131] |
| Th2 immunity | GATA3 ↑ | |||
| NA | PDAC | α-SMA/fibrosis | ↑ in STS | [132] |
| NA | PDAC | CD68/CD163 | ↑ in STS | |
| CD4 | ↓ in STS | |||
| iNOS | ↓ in STS | |||
| Foxp3 | ↑ in STS | |||
| B cell/DC | ↓ in STS | |||
| NA | PDAC | Metabolic active CAF (meCAF) | ↑ in dense (high desmoplasia) group | [55] |
| CD8 | ↑ in loose (low desmoplasia) group | |||
| Response to α-PD-1 | ↑ in loose group | |||
| Clinical Trial ID | Treatment | Cancer Type | Reference |
|---|---|---|---|
| NCT02993731 | Napabucasin + nab-paclitaxel(+gemcitabine) | Pancreas | [142] |
| NCT00911859 | Siltuximab(+velcade-melphalan-prednisone) | Multiple myeloma | [145] |
| NCT01484275 | Siltuximab | Smoldering multiple myeloma | [146] |
| NCT00906945 | G-CSF + plerixafor + mitoxantrone + etoposide + cytarabine | Acute myeloid leukemia | [147] |
| NCT00512252 | plerixafor + mitoxantrone + etoposide + cytarabine | Acute myeloid leukemia | [148] |
| NCT00101166 | GM.CD40L vaccination | Melanoma | [149] |
| NCT01433172 | GM.CD40L vaccination (+CCL21) | Lung | [150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, Y.-H.; Chen, L.-T.; Hung, W.-C. The Trinity: Interplay among Cancer Cells, Fibroblasts, and Immune Cells in Pancreatic Cancer and Implication of CD8+ T Cell-Orientated Therapy. Biomedicines 2022, 10, 926. https://doi.org/10.3390/biomedicines10040926
Hung Y-H, Chen L-T, Hung W-C. The Trinity: Interplay among Cancer Cells, Fibroblasts, and Immune Cells in Pancreatic Cancer and Implication of CD8+ T Cell-Orientated Therapy. Biomedicines. 2022; 10(4):926. https://doi.org/10.3390/biomedicines10040926
Chicago/Turabian StyleHung, Yu-Hsuan, Li-Tzong Chen, and Wen-Chun Hung. 2022. "The Trinity: Interplay among Cancer Cells, Fibroblasts, and Immune Cells in Pancreatic Cancer and Implication of CD8+ T Cell-Orientated Therapy" Biomedicines 10, no. 4: 926. https://doi.org/10.3390/biomedicines10040926
APA StyleHung, Y. -H., Chen, L. -T., & Hung, W. -C. (2022). The Trinity: Interplay among Cancer Cells, Fibroblasts, and Immune Cells in Pancreatic Cancer and Implication of CD8+ T Cell-Orientated Therapy. Biomedicines, 10(4), 926. https://doi.org/10.3390/biomedicines10040926