
Whole-Exome Sequencing of Germline Variants in Non-BRCA Families with Hereditary Breast Cancer

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Family Selection
2.2. Family Characteristics
2.3. Whole-Exome Sequencing and Bioinformatics
2.4. Cell Culture, Transfection, and Plasmid Construction
2.5. Western Blot
2.6. Mass Spectrometry and Pathway Analysis
3. Results
3.1. Hereditary-Breast-Cancer-Associated Genes and Breast-Cancer-Associated High-Risk SNPs
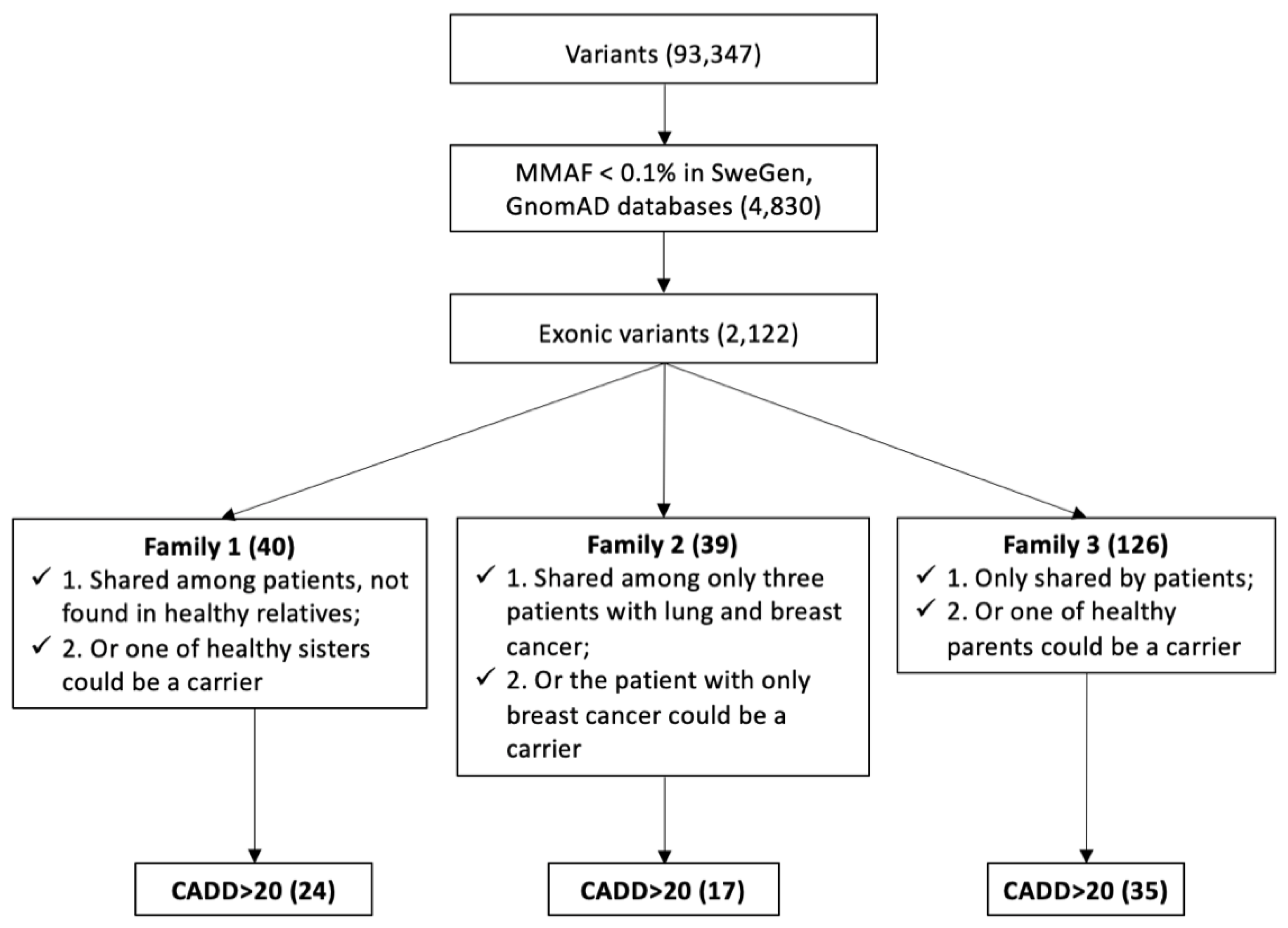
3.2. Identification of Candidate Genes by WES Analysis
3.2.1. Candidate Genes in Family 1
3.2.2. Candidate Genes in Family 2
3.2.3. Candidate Genes in Family 3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- King-Spohn, K.; Pilarski, R. Beyond BRCA1 and BRCA2. Curr. Probl. Cancer 2014, 38, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Terui-Kohbata, H.; Yoshida, M. Current Condition of Genetic Medicine for Hereditary Breast Cancer. Mol. Clin. Oncol. 2017, 7, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Pilarski, R. PTEN Hamartoma Tumor Syndrome: A Clinical Overview. Cancers 2019, 11, 844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.; Evans, D.G. Guidelines for the Li–Fraumeni and Heritable TP53-Related Cancer Syndromes. Eur. J. Hum. Genet. 2020, 28, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N. Realizing the Promise of Cancer Predisposition Genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, K.; Rhiem, K.; Wappenschmidt, B.; Hahnen, E.; Hauke, J.; Bluemcke, B.; Zarghooni, V.; Herold, N.; Ditsch, N.; Kiechle, M.; et al. Prevalence of BRCA1/2 Germline Mutations in 21 401 Families with Breast and Ovarian Cancer. J. Med. Genet. 2016, 53, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Levy-Lahad, E.; Friedman, E. Cancer Risks among BRCA1 and BRCA2 Mutation Carriers. Br. J. Cancer 2007, 96, 11–15. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [Green Version]
- McClellan, J.; King, M.-C. Genetic Heterogeneity in Human Disease. Cell 2010, 141, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Tung, N.M.; Boughey, J.C.; Pierce, L.J.; Robson, M.E.; Bedrosian, I.; Dietz, J.R.; Dragun, A.; Gelpi, J.B.; Hofstatter, E.W.; Isaacs, C.J.; et al. Management of Hereditary Breast Cancer: American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2080–2106. [Google Scholar] [CrossRef] [PubMed]
- Girdea, M.; Dumitriu, S.; Fiume, M.; Bowdin, S.; Boycott, K.M.; Chénier, S.; Chitayat, D.; Faghfoury, H.; Meyn, M.S.; Ray, P.N.; et al. PhenoTips: Patient Phenotyping Software for Clinical and Research Use. Hum. Mutat. 2013, 34, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Ameur, A.; Dahlberg, J.; Olason, P.; Vezzi, F.; Karlsson, R.; Martin, M.; Viklund, J.; Kähäri, A.K.; Lundin, P.; Che, H.; et al. SweGen: A Whole-Genome Data Resource of Genetic Variability in a Cross-Section of the Swedish Population. Eur. J. Hum. Genet. 2017, 25, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A More Comprehensive, Powerful, Flexible and Interactive Gene Set Enrichment Analysis Toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef]
- Li, H.; Feng, B.; Miron, A.; Chen, X.; Beesley, J.; Bimeh, E.; Barrowdale, D.; John, E.M.; Daly, M.B.; Andrulis, I.L.; et al. Breast Cancer Risk Prediction Using a Polygenic Risk Score in the Familial Setting: A Prospective Study from the Breast Cancer Family Registry and KConFab. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D. Localization of a Breast Cancer Susceptibility Gene, BRCA2, to Chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The Human Mutator Gene Homolog MSH2 and Its Association with Hereditary Nonpolyposis Colon Cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Leach, F.S.; Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Jen, J.; Parsons, R.; Peltomäki, P.; Sistonen, P.; Aaltonen, L.A.; Nyström-Lahti, M. Mutations of a MutS Homolog in Hereditary Nonpolyposis Colorectal Cancer. Cell 1993, 75, 1215–1225. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Nicolaides, N.C.; Wei, Y.F.; Ruben, S.M.; Carter, K.C.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M.; Adams, M.D. Mutation of a MutL Homolog in Hereditary Colon Cancer. Science 1994, 263, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Stadler, Z.K.; Schrader, K.A.; Vijai, J.; Robson, M.E.; Offit, K. Cancer Genomics and Inherited Risk. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 687–698. [Google Scholar] [CrossRef]
- Vahteristo, P.; Bartkova, J.; Eerola, H.; Syrjäkoski, K.; Ojala, S.; Kilpivaara, O.; Tamminen, A.; Kononen, J.; Aittomäki, K.; Heikkilä, P.; et al. A CHEK2 Genetic Variant Contributing to a Substantial Fraction of Familial Breast Cancer. Am. J. Hum. Genet. 2002, 71, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Bogdanova, N.; Cybulski, C.; Bermisheva, M.; Datsyuk, I.; Yamini, P.; Hillemanns, P.; Antonenkova, N.N.; Khusnutdinova, E.; Lubinski, J.; Dörk, T. A Nonsense Mutation (E1978X) in the ATM Gene Is Associated with Breast Cancer. Breast Cancer Res. Treat. 2009, 118, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Stankiewicz, E.; Mao, X.; Mangham, D.C.; Xu, L.; Yeste-Velasco, M.; Fisher, G.; North, B.; Chaplin, T.; Young, B.; Wang, Y.; et al. Identification of FBXL4 as a Metastasis Associated Gene in Prostate Cancer. Sci. Rep. 2017, 7, 5124. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Huo, D.; Ogundiran, T.O.; Ojengbede, O.; Zheng, W.; Nathanson, K.L.; Nemesure, B.; Ambs, S.; Olopade, O.I.; Zheng, Y. Association of Breast Cancer Risk and the MTOR Pathway in Women of African Ancestry in “The Root” Consortium. Carcinogenesis 2017, 38, 789–796. [Google Scholar] [CrossRef]
- Babu, N.; Pinto, S.M.; Biswas, M.; Subbannayya, T.; Rajappa, M.; Mohan, S.V.; Advani, J.; Rajagopalan, P.; Sathe, G.; Syed, N.; et al. Phosphoproteomic Analysis Identifies CLK1 as a Novel Therapeutic Target in Gastric Cancer. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2020, 23, 796–810. [Google Scholar] [CrossRef]
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H.; et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015, 75, 1516–1526. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.R.; An, C.H.; Yoo, N.J.; Lee, S.H. Frameshift Mutations of TAF1C Gene, a Core Component for Transcription by RNA Polymerase I, and Its Regional Heterogeneity in Gastric and Colorectal Cancers. Pathology 2015, 47, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-H.; Zhang, Y.-J.; Yue, J.-X.; Zhou, T. Comprehensive Analysis of the Expression and Prognosis for SFRPs in Breast Carcinoma. Cell Transplant. 2020, 29, 963689720962479. [Google Scholar] [CrossRef] [PubMed]
- Schlensog, M.; Magnus, L.; Heide, T.; Eschenbruch, J.; Steib, F.; Tator, M.; Kloten, V.; Rose, M.; Noetzel, E.; Gaisa, N.T.; et al. Epigenetic Loss of Putative Tumor Suppressor SFRP3 Correlates with Poor Prognosis of Lung Adenocarcinoma Patients. Epigenetics 2018, 13, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Storr, S.J.; Lee, K.W.; Woolston, C.M.; Safuan, S.; Green, A.R.; Macmillan, R.D.; Benhasouna, A.; Parr, T.; Ellis, I.O.; Martin, S.G. Calpain System Protein Expression in Basal-like and Triple-Negative Invasive Breast Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 2289–2296. [Google Scholar] [CrossRef]
- Cortesio, C.L.; Chan, K.T.; Perrin, B.J.; Burton, N.O.; Zhang, S.; Zhang, Z.-Y.; Huttenlocher, A. Calpain 2 and PTP1B Function in a Novel Pathway with Src to Regulate Invadopodia Dynamics and Breast Cancer Cell Invasion. J. Cell Biol. 2008, 180, 957–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reverter, D.; Strobl, S.; Fernandez-Catalan, C.; Sorimachi, H.; Suzuki, K.; Bode, W. Structural Basis for Possible Calcium-Induced Activation Mechanisms of Calpains. Biol. Chem. 2001, 382, 753–766. [Google Scholar] [CrossRef]
- Yodsurang, V.; Tanikawa, C.; Miyamoto, T.; Lo, P.H.Y.; Hirata, M.; Matsuda, K. Identification of a Novel P53 Target, COL17A1, That Inhibits Breast Cancer Cell Migration and Invasion. Oncotarget 2017, 8, 55790–55803. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Fu, Q.; Cao, Q.; Chen, X.; Wang, M.; Yu, J.; Long, J.; Yao, J.; Liu, H.; et al. AKR1B1 Promotes Basal-like Breast Cancer Progression by a Positive Feedback Loop That Activates the EMT Program. J. Exp. Med. 2017, 214, 1065–1079. [Google Scholar] [CrossRef] [Green Version]
- Logarajah, S.; Hunter, P.; Kraman, M.; Steele, D.; Lakhani, S.; Bobrow, L.; Venkitaraman, A.; Wagner, S. BCL-6 Is Expressed in Breast Cancer and Prevents Mammary Epithelial Differentiation. Oncogene 2003, 22, 5572–5578. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Rajaram, M.; Zhou, X.; Liu, Q.; Marchica, J.; Li, J.; Powers, R.S. Activation of Multiple Cancer Pathways and Tumor Maintenance Function of the 3q Amplified Oncogene FNDC3B. Cell Cycle Georget. Tex 2012, 11, 1773–1781. [Google Scholar] [CrossRef] [Green Version]
- Gucalp, A.; Traina, T.A.; Eisner, J.R.; Parker, J.S.; Selitsky, S.R.; Park, B.H.; Elias, A.D.; Baskin-Bey, E.S.; Cardoso, F. Male Breast Cancer: A Disease Distinct from Female Breast Cancer. Breast Cancer Res. Treat. 2019, 173, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Riabov, V.; Yin, S.; Song, B.; Avdic, A.; Schledzewski, K.; Ovsiy, I.; Gratchev, A.; Verdiell, M.L.; Sticht, C.; Schmuttermaier, C.; et al. Stabilin-1 Is Expressed in Human Breast Cancer and Supports Tumor Growth in Mammary Adenocarcinoma Mouse Model. Oncotarget 2016, 7, 31097–31110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, W.; Choi, S.-K.; Kim, D.; Kim, H.-G.; Park, J.-K.; Han, J.E.; Cho, G.-J.; Yun, S.; Yu, W.; Han, S.-H.; et al. ZNF507 Affects TGF-β Signaling via TGFBR1 and MAP3K8 Activation in the Progression of Prostate Cancer to an Aggressive State. J. Exp. Clin. Cancer Res. 2021, 40, 291. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.C.; Jorcyk, C.L.; Oxford, J.T. DICER1 Syndrome: DICER1 Mutations in Rare Cancers. Cancers 2018, 10, 143. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Filter | Gene | Position | Ref/Alt | SNP | Type | Change | MMAF | CADD |
|---|---|---|---|---|---|---|---|---|
| 1 | FBXL4 | chr6:993222281 | A/C | rs757154231 | missense | NM_012160:c.T1739G:p.L580R | 0.00001 | 32 |
| UBASH3A | chr21:43857670 | C/T | rs201756769 | missense | NM_001001895:c.C1352T:p.T451M | 0.0002 | 30 | |
| MYH13 | chr17:10206712 | T/C | rs767313943 | missense | NM_003802:c.A5570G:p.Q1857R | 0.00002 | 27.1 | |
| FRZB | chr2:183699592 | C/T | rs150679557 | missense | NM_001463:c.G962A:p.R321Q | 0.00083 | 24.3 | |
| TAF1C | chr16:84213125 | G/A | rs61730960 | missense | NM_001243158:c.C1036T:p.R346W | 0.00041 | 24.3 | |
| STRADB | chr2:202337785 | C/G | rs369992593 | missense | NM_001206864:c.C301G:p.L101V | 0.00008 | 23.6 | |
| CLK1 | chr2:201726049 | C/A | rs141755850 | missense | NM_001162407:c.G428T:p.S143I | 0.00041 | 23 | |
| 2 | SPTLC3 | chr20:13074186 | G/A | rs372930777 | missense | NM_018327:c.G788A:p.R263Q | 0.00008 | 35 |
| CAPN2 | chr1:223947063 | A/G | . | missense | NM_001146068:c.A1175G:p.E392G | 0 | 34 | |
| COL17A1 | chr10:105807514 | C/T | rs757388768 | missense | NM_000494:c.G2318A:p.G773E | 0.00041 | 34 | |
| HCLS1 | chr3:121363691 | C/T | rs757006680 | missense | NM_001292041:c.G373A:p.G125R | 0.00001 | 34 | |
| STARD9 | chr15:42930971 | C/T | rs369566419 | missense | NM_020759:c.C520T:p.R174W | 0.0001 | 33 | |
| CNST | chr1:246829203 | C/T | rs766380272 | missense | NM_152609:c.C2174T:p.S725F | 0.00001 | 28.4 | |
| STXBP5L | chr3:120976023 | C/T | rs184420053 | missense | NM_001308330:c.C1675T:p.L559F | 0.00083 | 26.4 | |
| DNAH3 | chr16:20975280 | G/A | rs376279103 | missense | NM_017539:c.C9926T:p.S3309L | 0.00008 | 26 | |
| TENM4 | chr11:78383268 | G/A | rs747100917 | missense | NM_001098816:c.C5603T:p.A1868V | 0.00001 | 25.6 | |
| MYOM3 | chr1:24421405 | G/C | rs200854393 | missense | NM_152372:c.C866G:p.S289C | 0.0004 | 25.1 | |
| ASIC2 | chr17:31351024 | C/T | rs199589382 | missense | NM_001094:c.G1051A:p.A351T | 0.0002 | 23.8 | |
| NMRK2 | chr19:3933697 | T/G | . | splicing | . | 0 | 23.8 | |
| APOB | chr2:21252574 | C/A | rs778274241 | missense | NM_000384:c.G1554T:p.K518N | 0.00002 | 23.4 | |
| DERL2 | chr17:5384651 | C/T | rs202210923 | missense | NM_001304777:c.G289A:p.V97I | 0.00083 | 22.9 | |
| RIC1 | chr9:5762545 | G/A | rs771929691 | missense | NM_001206557:c.G1886A:p.R629H | 0.00041 | 22.8 | |
| DLL3 | chr19:39996056 | G/A | . | missense | NM_016941:c.G1058A:p.R353K | 0 | 22.4 | |
| TMEM143 | chr19:48863405 | G/A | rs544787964 | missense | NM_001303539:c.C293T:p.A98V | 0.00083 | 20.2 |
| Filter | Gene | Position | Ref/Alt | SNP | Type | Change | MMAF | CADD |
|---|---|---|---|---|---|---|---|---|
| 1 | AKR1B1 | chr7:134136457 | C/T | rs201718247 | missense | NM_001628:c.G115A:p.G39R | 0.00083 | 34 |
| SLC25A25 | chr9:130864666 | C/T | rs748220703 | missense | NM_001006641:c.C494T:p.T165M | 0.0004 | 34 | |
| RYR3 | chr15:33893707 | C/T | rs760906719 | missense | NM_001036:c.C1876T:p.R626W | 0.00041 | 34 | |
| RUFY1 | chr5:178987155 | A/T | rs754852607 | missense | NM_001040451:c.A116T:p.Q39L | 0.00008 | 28.6 | |
| TIPIN | chr15:66641436 | T/C | rs200514985 | missense | NM_001289986:c.A134G:p.D45G | 0.00041 | 27.9 | |
| C1orf228 | chr17:45166747 | C/T | rs575641425 | missense | NM_001145636:c.C595T:p.P199S | 0.00041 | 26.3 | |
| HECTD4 | chr12:112677734 | T/C | rs779868916 | missense | NM_001109662:c.A4654G:p.I1552V | 0.00002 | 25.2 | |
| BCL6 | chr3:187444624 | T/C | rs747910667 | missense | NM_001130845:c.A1603G:p.R535G | 0.0001 | 23.4 | |
| CA12 | chr15:63618533 | C/T | rs149256486 | missense | NM_001293642:c.G803A:p.G268E | 0.0002 | 23.3 | |
| 2 | UTP11L | chr1:38489295 | C/T | rs771377582 | missense | NM_016037:c.C757T:p.R253C | 0.0002 | 35 |
| PAX7 | chr1:18961015 | G/A | rs369607271 | missense | NM_001135254:c.G304A:p.G102S | 0.00008 | 34 | |
| FNDC3B | chr3:171969145 | C/T | rs190147254 | missense | NM_001135095:c.C604T:p.R202C | 0.00083 | 31 | |
| ZMYM4 | chr1:35836075 | G/A | . | missense | NM_005095:c.G1028A:p.G343D | 0 | 24.6 | |
| WDFY4 | chr10:50013303 | A/G | rs748753983 | splicing | . | 0.00005 | 23.7 | |
| NSD2 | chr4:1957024 | C/G | rs748922675 | missense | NM_001042424:c.C2475G:p.H825Q | 0.00041 | 23.6 | |
| MED14 | chrX:40552004 | G/A | rs763899660 | missense | NM_004229:c.C1801T:p.R601C | 0.00026 | 22.7 | |
| C17orf53 | chr17:42225596 | G/A | rs377372267 | missense | NM_001171251:c.G425A:p.S142N | 0.0004 | 21.8 |
| Filter | Gene | Position | Ref/Alt | SNP | Type | Change | MMAF | CADD |
|---|---|---|---|---|---|---|---|---|
| 1 | PRKD1 | chr14:30108088 | A/C | . | missense | NM_001330069.2:c.743T > G:p.F248C | 0 | 22.3 |
| 2 | TNRC6C | chr17:76094616 | C/T | rs367710467 | missense | NM_001142640.1:c.4607C > T:p.S1536L | 0 | 33 |
| STAB1 | chr3:52554136 | G/C | . | missense | NM_015136.3:c.5412G > C:p.E1804D | 0 | 33 | |
| THOP1 | chr19:2799789 | G/A | . | missense | NM_003249.5:c.589G > A:p.G197R | 0 | 33 | |
| SPDYA | chr2:29072798 | TG/T | . | splicing | NM_001142634.2:c.934del:p.E312KfsTer20 | 0 | 32 | |
| TMX2-CTNND1 | chr11:57505465 | C/T | rs375390370 | missense | NM_001347890.1:c.331C > T:p.R111C | 0 | 32 | |
| ZNF793 | chr19:38023257 | G/A | rs200503532 | splicing | NM_001013659.3:c.16-1G > A | 0 | 32 | |
| NOMO3 | chr16:16363998 | C/T | . | missense | NM_001004067.4:c.1915C > T:p.R639C | 0.00017 | 32 | |
| USP17L10 | chr4:9213003 | T/A | . | nonsense | NM_001256852.1:c.621T > A:p.C207Ter | 0 | 32 | |
| CNR2 | chr1:24201377 | G/A | rs201829495 | missense | NM_001841.3:c.731C > T:p.A244V | 0 | 28.2 | |
| ZNF507 | chr19:32845840 | T/C | . | missense | NM_001136156.2:c.2104T > C:p.C702R | 0 | 27.2 | |
| USP40 | chr2:234399901 | G/GA | . | splicing | NM_001365479.1:c.2923dup:p.S975FfsTer65 | 0 | 27 | |
| TUBGCP6 | chr22:50656236 | G/A | rs138609686 | missense | NM_020461.4:c.5389C > T:p.R1797C | 0.0000649 | 26.6 | |
| AGAP3 | chr7:150839000 | T/G | . | missense | NM_001281300.2:c.827T > G:p.F276C | 0 | 25.9 | |
| ITPR1 | chr3:4706906 | G/A | . | missense | NM_001099952.3:c.1639G > A:p.A547T | 0 | 25.8 | |
| SPDL1 | chr5:169028384 | AG/A | . | splicing | NM_001329639.2:c.1426del:p.E476KfsTer19 | 0 | 25.7 | |
| RUNX1 | chr21:36164605 | A/C | . | missense | NM_001001890.3:c.1189T > G:p.S397A | 0 | 25.5 | |
| TCF3 | chr19:1646413 | G/C | . | missense | NM_001136139.4:c.86C > G:p.P29R | 0 | 25.2 | |
| NT5C1B-RDH14 | chr2:18745234 | C/T | rs147855687 | missense | NM_001002006.3:c.1661G > A:p.R554H | 0.000454 | 25 | |
| ABHD6 | chr3:58279442 | G/A | rs144907290 | missense | NM_001320126.2:c.964G > A:p.D322N | 0 | 24.9 | |
| EPHA2 | chr1:16456073 | C/T | . | missense | NM_001329090.2:c.2519G > A:p.R840Q | 0 | 24.8 | |
| HSPA6 | chr1:161494581 | G/C | . | missense | NM_002155.5:c.133G > C:p.A45P | 0 | 24.4 | |
| MYO16 | chr13:109507831 | C/T | . | missense | NM_001198950.3:c.1289C > T:p.T430M | 0 | 23.8 | |
| SMC4 | chr3:160135704 | T/C | rs41272953 | missense | NM_001002800.3:c.1631T > C:p.I544T | 0.000115 | 23.5 | |
| PRKAR2A | chr3:48884770 | T/G | . | missense | NM_001321982.2:c.260A > C:p.E87A | 0 | 23.5 | |
| USP17L22 | chr4:9270417 | G/GA | . | splicing | NM_001256863.1:c.1073_1074insA:p.S358RfsTer20 | 0 | 23.3 | |
| ZYX | chr7:143079991 | G/T | rs150223874 | missense | NM_001010972.2:c.599G > T:p.W200L | 0 | 23.1 | |
| ITFG1 | chr16:47494745 | T/G | rs376408976 | splicing | NM_001305002.1:c.−132 + 281A > C | 0 | 23.1 | |
| NYNRIN | chr14:24883816 | T/C | . | missense | NM_025081.3:c.2861T > C:p.I954T | 0 | 22.6 | |
| NIM1K | chr5:43280642 | C/G | . | missense | NM_153361.4:c.1122C > G:p.N374K | 0 | 22.3 | |
| DICER1 | chr14:95570153 | T/C | . | missense | NM_001271282.3:c.3580A > G:p.R1194G | 0 | 21.8 | |
| NXNL1 | chr19:17571431 | G/A | rs377352923 | missense | NM_138454.2:c.248C > T:p.T83M | 0 | 21.5 | |
| TRPV6 | chr7:142574988 | G/T | rs139115329 | missense | NM_018646.6:c.514C > A:p.L172M | 0 | 21.2 | |
| ZNF320 | chr19:53384145 | G/C | . | missense | NM_001351773.1:c.1234C > G:p.L412V | 0 | 21 | |
| ZNF320 | chr19:53384768 | A/G | rs144964547 | missense | NM_001351773.1:c.611T > C:p.L204P | 0.000907 | 20.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Helgadottir, H.T.; Kharaziha, P.; Choi, J.; López-Giráldez, F.; Mane, S.M.; Höiom, V.; Juhlin, C.C.; Larsson, C.; Bajalica-Lagercrantz, S. Whole-Exome Sequencing of Germline Variants in Non-BRCA Families with Hereditary Breast Cancer. Biomedicines 2022, 10, 1004. https://doi.org/10.3390/biomedicines10051004
Liu Y, Helgadottir HT, Kharaziha P, Choi J, López-Giráldez F, Mane SM, Höiom V, Juhlin CC, Larsson C, Bajalica-Lagercrantz S. Whole-Exome Sequencing of Germline Variants in Non-BRCA Families with Hereditary Breast Cancer. Biomedicines. 2022; 10(5):1004. https://doi.org/10.3390/biomedicines10051004
Chicago/Turabian StyleLiu, Yaxuan, Hafdis T. Helgadottir, Pedram Kharaziha, Jungmin Choi, Francesc López-Giráldez, Shrikant M. Mane, Veronica Höiom, Carl Christofer Juhlin, Catharina Larsson, and Svetlana Bajalica-Lagercrantz. 2022. "Whole-Exome Sequencing of Germline Variants in Non-BRCA Families with Hereditary Breast Cancer" Biomedicines 10, no. 5: 1004. https://doi.org/10.3390/biomedicines10051004
APA StyleLiu, Y., Helgadottir, H. T., Kharaziha, P., Choi, J., López-Giráldez, F., Mane, S. M., Höiom, V., Juhlin, C. C., Larsson, C., & Bajalica-Lagercrantz, S. (2022). Whole-Exome Sequencing of Germline Variants in Non-BRCA Families with Hereditary Breast Cancer. Biomedicines, 10(5), 1004. https://doi.org/10.3390/biomedicines10051004