
Hormonal and Genetic Regulatory Events in Breast Cancer and Its Therapeutics: Importance of the Steroidogenic Acute Regulatory Protein

 ,
, 

Abstract
:1. Introduction
2. Overview of BC Subtypes
3. Molecular Pathogenesis of BCs and Frequently Altered Pathways
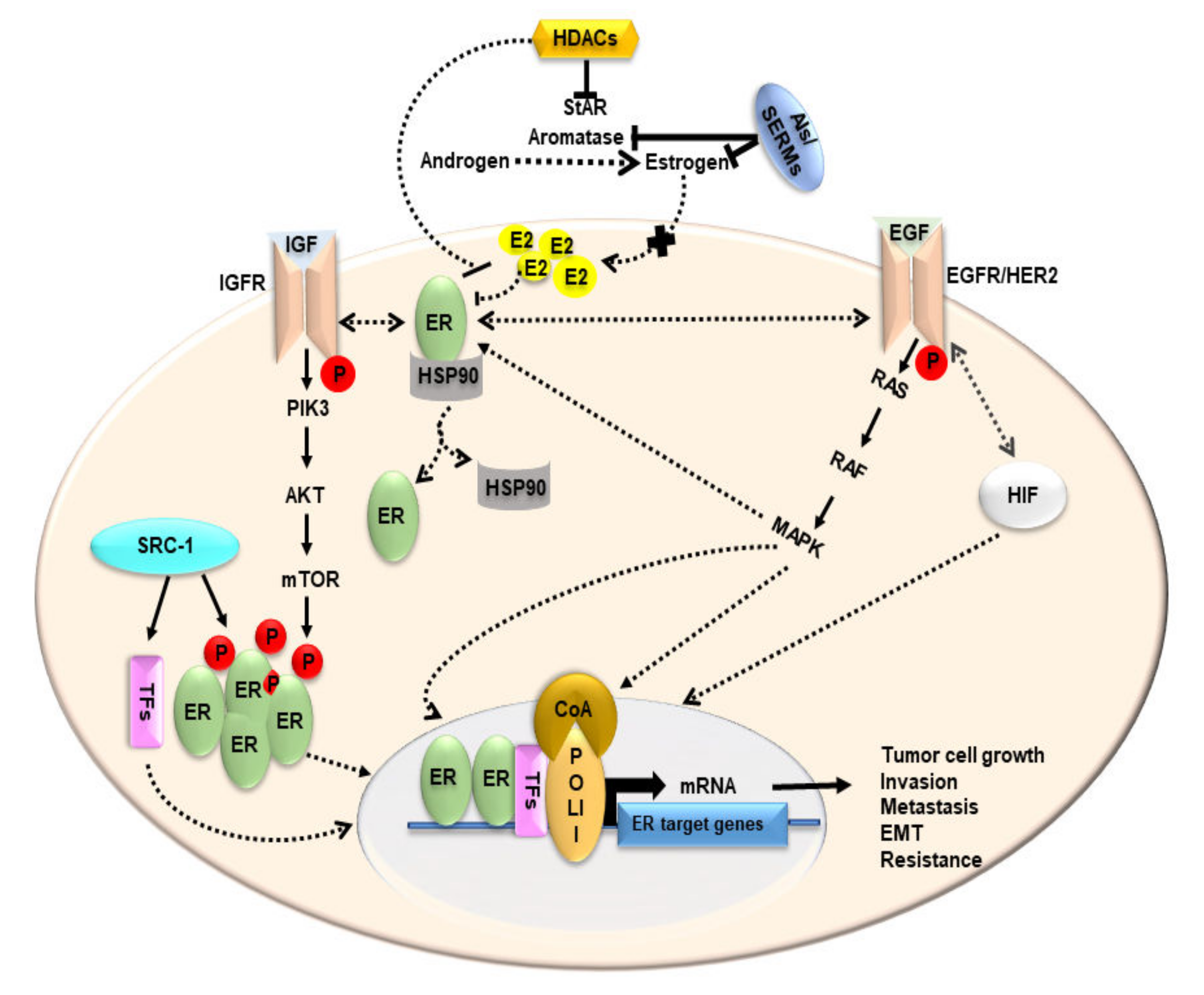
4. Estrogen and Its Receptors in BCs
5. Functional Relevance of StAR and Steroidogenic Machinery in BCs
6. Steroidogenic Enzyme and HR Genes, and Their Correlation to BC Survival
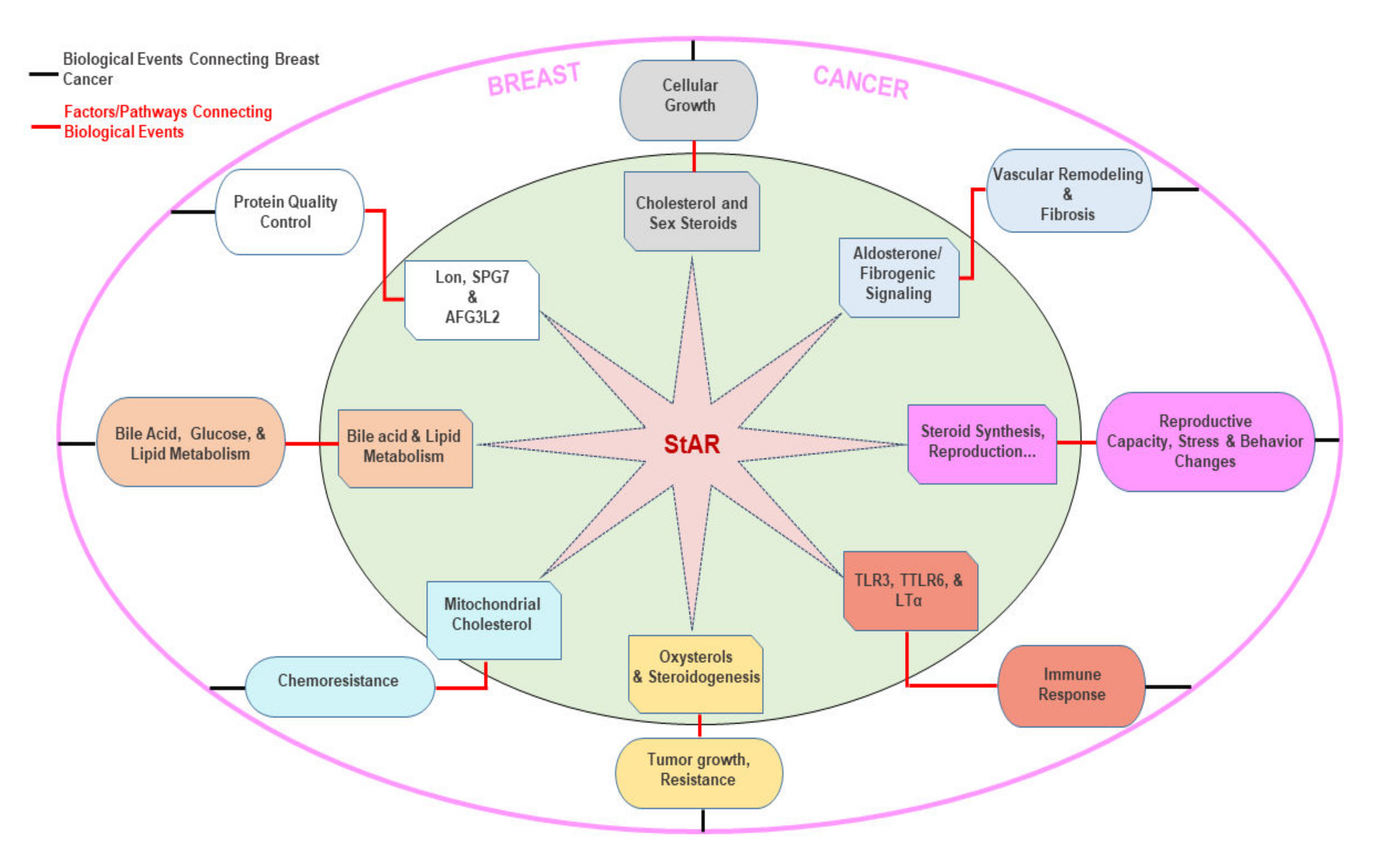
7. Putative Roles of Multiple Factors/Signaling in StAR Linked BCs
7.1. Cholesterol and Sex Steroids
7.2. Aldosterone and Fibrogenic Signaling
7.3. Reproductive Hormone, Behavior, and Stress Changes
7.4. Toll-Like Receptors and Immuno-Inflammation
7.5. Oxysterols
7.6. Mitochondrial Cholesterol
7.7. Bile Acid and Lipid Metabolism
7.8. LON, SPG7, and AFG3L2
8. Therapeutic Strategies for the Treatment and Management of BCs
9. Impact of HDACIs in BCs: Possible Alternatives to Endocrine Therapies
10. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Yue, W.; Yager, J.D.; Wang, J.P.; Jupe, E.R.; Santen, R.J. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids 2013, 78, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.R.; Molehin, D.; Ahmed, A.U. Dysregulation of aromatase in rreast, endometrial, and ovarian cancers: An Overview of Therapeutic Strategies. Prog. Mol. Biol. Transl. Sci. 2016, 144, 487–537. [Google Scholar] [PubMed]
- Renoir, J.M.; Marsaud, V.; Lazennec, G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem. Pharmacol. 2013, 85, 449–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulun, S.E.; Lin, Z.; Imir, G.; Amin, S.; Demura, M.; Yilmaz, B.; Martin, R.; Utsunomiya, H.; Thung, S.; Gurates, B.; et al. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: From bench to treatment. Pharmacol. Rev. 2005, 57, 359–383. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, L.; Shangguan, A.J.; Bulun, S.E. Aromatase expression and regulation in breast and endometrial cancer. J. Mol. Endocrinol. 2016, 57, R19–R33. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Li, H.; Yuan, Y.C.; Chen, S. Molecular characterization of aromatase. Ann. N. Y. Acad. Sci. 2009, 1155, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Sjoquist, K.M.; Martyn, J.; Edmondson, R.J.; Friedlander, M.L. The role of hormonal therapy in gynecological cancers-current status and future directions. Int. J. Gynecol. Cancer 2011, 21, 1328–1333. [Google Scholar] [CrossRef] [Green Version]
- Jahan, N.; Jones, C.; Rahman, R.L. Endocrine prevention of breast cancer. Mol. Cell. Endocrinol. 2021, 530, 111284. [Google Scholar] [CrossRef]
- Bulun, S.E.; Lin, Z.; Zhao, H.; Lu, M.; Amin, S.; Reierstad, S.; Chen, D. Regulation of aromatase expression in breast cancer tissue. Ann. N. Y. Acad. Sci. 2009, 1155, 121–131. [Google Scholar] [CrossRef]
- Simpson, E.; Santen, R.J. Celebrating 75 years of oestradiol. J. Mol. Endocrinol. 2015, 55, T1–T20. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.J.; Petrossian, K.; Chen, S. Structural and functional characterization of aromatase, estrogen receptor, and their genes in endocrine-responsive and -resistant breast cancer cells. J. Steroid Biochem. Mol. Biol. 2016, 161, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Caldon, C.E. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front. Oncol. 2014, 4, 106. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network Research. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network Research. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network Research. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Stocco, D.M.; Wang, X.; Jo, Y.; Manna, P.R. Multiple signaling pathways regulating steroidogenesis and steroidogenic acute regulatory protein expression: More complicated than we thought. Mol. Endocrinol. 2005, 19, 2647–2659. [Google Scholar] [CrossRef] [Green Version]
- Manna, P.R.; Dyson, M.T.; Stocco, D.M. Regulation of the steroidogenic acute regulatory protein gene expression: Present and future perspectives. Mol. Hum. Reprod. 2009, 15, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.L. Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 2013, 379, 62–73. [Google Scholar] [CrossRef]
- Manna, P.R.; Stetson, C.L.; Slominski, A.T.; Pruitt, K. Role of the steroidogenic acute regulatory protein in health and disease. Endocrine 2016, 51, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Molehin, D.; Castro-Piedras, I.; Sharma, M.; Sennoune, S.R.; Arena, D.; Manna, P.R.; Pruitt, K. Aromatase Acetylation Patterns and Altered Activity in Response to Sirtuin Inhibition. Mol. Cancer Res. 2018, 16, 1530–1542. [Google Scholar] [CrossRef] [Green Version]
- Castro-Piedras, I.; Sharma, M.; Brelsfoard, J.; Vartak, D.; Martinez, E.G.; Rivera, C.; Molehin, D.; Bright, R.K.; Fokar, M.; Guindon, J.; et al. Nuclear Dishevelled targets gene regulatory regions and promotes tumor growth. EMBO Rep. 2021, 22, e50600. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.R.; Ahmed, A.U.; Vartak, D.; Molehin, D.; Pruitt, K. Overexpression of the steroidogenic acute regulatory protein in breast cancer: Regulation by histone deacetylase inhibition. Biochem. Biophys. Res. Commun. 2019, 509, 476–482. [Google Scholar] [CrossRef]
- Manna, P.R.; Ahmed, A.U.; Yang, S.; Narasimhan, M.; Cohen-Tannoudji, J.; Slominski, A.T.; Pruitt, K. Genomic profiling of the steroidogenic acute regulatory protein in breast cancer: In silico sssessments and a mechanistic perspective. Cancers 2019, 11, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Piedras, I.; Sharma, M.; den Bakker, M.; Molehin, D.; Martinez, E.G.; Vartak, D.; Pruitt, W.M.; Deitrick, J.; Almodovar, S.; Pruitt, K. DVL1 and DVL3 differentially localize to CYP19A1 promoters and regulate aromatase mRNA in breast cancer cells. Oncotarget 2018, 9, 35639–35654. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, D.; Lockman, P.R.; Thomas, F.C.; Hua, E.; Herring, J.; Hargrave, E.; Johnson, M.; Flores, N.; Qian, Y.; Vega-Valle, E.; et al. Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clin. Cancer Res. 2009, 15, 6148–6157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Chen, S.; Zhang, W.; Zheng, X.; Hu, S.; Pang, C.; Lu, J.; Xing, J.; Dong, Y. Salvianolic acid A reverses paclitaxel resistance in human breast cancer MCF-7 cells via targeting the expression of transgelin 2 and attenuating PI3 K/Akt pathway. Phytomedicine 2014, 21, 1725–1732. [Google Scholar] [CrossRef]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of histone deacetylases and inhibitors in anticancer therapy. Cancers 2020, 12, 1664. [Google Scholar] [CrossRef]
- Schnitt, S.J. Classification and prognosis of invasive breast cancer: From morphology to molecular taxonomy. Mod. Pathol. 2010, 23, S60–S64. [Google Scholar] [CrossRef] [Green Version]
- Amjad, E.; Asnaashari, S.; Sokouti, B.; Dastmalchi, S. Systems biology comprehensive analysis on breast cancer for identification of key gene modules and genes associated with TNM-based clinical stages. Sci. Rep. 2020, 10, 10816. [Google Scholar] [CrossRef] [PubMed]
- Dunbier, A.K.; Anderson, H.; Ghazoui, Z.; Folkerd, E.J.; A’Hern, R.; Crowder, R.J.; Hoog, J.; Smith, I.E.; Osin, P.; Nerurkar, A.; et al. Relationship between plasma estradiol levels and estrogen-responsive gene expression in estrogen receptor-positive breast cancer in postmenopausal women. J. Clin. Oncol. 2010, 28, 1161–1167. [Google Scholar] [CrossRef]
- Lonning, P.E.; Haynes, B.P.; Straume, A.H.; Dunbier, A.; Helle, H.; Knappskog, S.; Dowsett, M. Exploring breast cancer estrogen disposition: The basis for endocrine manipulation. Clin. Cancer Res. 2011, 17, 4948–4958. [Google Scholar] [CrossRef] [Green Version]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.Y.; Askarian-Amiri, M.E.; Sarkar, D.; Ferraro-Peyret, C.; Joseph, W.R.; Finlay, G.J.; Baguley, B.C. Endocrine therapy of estrogen receptor-positive breast cancer cells: Early differential effects on stem cell markers. Front. Oncol. 2017, 7, 184. [Google Scholar] [CrossRef] [Green Version]
- Bulun, S.E.; Chen, D.; Lu, M.; Zhao, H.; Cheng, Y.; Demura, M.; Yilmaz, B.; Martin, R.; Utsunomiya, H.; Thung, S.; et al. Aromatase excess in cancers of breast, endometrium and ovary. J. Steroid Biochem. Mol. Biol. 2007, 106, 81–96. [Google Scholar] [CrossRef] [Green Version]
- Maximov, P.Y.; Abderrahman, B.; Hawsawi, Y.M.; Chen, Y.; Foulds, C.E.; Jain, A.; Malovannaya, A.; Fan, P.; Curpan, R.F.; Han, R.; et al. The structure-function relationship of angular sstrogens and estrogen receptor alpha to initiate estrogen-induced apoptosis in breast cancer cells. Mol. Pharmacol. 2020, 98, 24–37. [Google Scholar] [CrossRef]
- Abderrahman, B.; Maximov, P.Y.; Curpan, R.F.; Hanspal, J.S.; Fan, P.; Xiong, R.; Tonetti, D.A.; Thatcher, G.R.J.; Jordan, V.C. Pharmacology and molecular mechanisms of clinically relevant estrogen estetrol and estrogen mimic BMI-135 for the treatment of endocrine-resistant breast cancer. Mol. Pharmacol. 2020, 98, 364–381. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Cheskis, B.J.; Greger, J.; Cooch, N.; McNally, C.; McLarney, S.; Lam, H.S.; Rutledge, S.; Mekonnen, B.; Hauze, D.; Nagpal, S.; et al. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids 2008, 73, 901–905. [Google Scholar] [CrossRef]
- Aksamitiene, E.; Achanta, S.; Kolch, W.; Kholodenko, B.N.; Hoek, J.B.; Kiyatkin, A. Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell Signal. 2011, 23, 1794–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkoyluoglu-Cotul, E.; Arca, A.; Madak-Erdogan, Z. Crosstalk between estrogen signaling and breast cancer Metabolism. Trends Endocrinol. Metab. 2019, 30, 25–38. [Google Scholar] [CrossRef]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.W.; Balko, J.M.; Arteaga, C.L. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J. Clin. Oncol. 2011, 29, 4452–4461. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Liu, L.; Sun, S.; Zhang, D.; Sheng, J.; Li, B.; Yang, W. The impact of PI3K inhibitors on breast cancer cell and its tumor microenvironment. PeerJ 2018, 6, e5092. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, T.C.; Paull, E.O.; Ellis, M.J.; Stuart, J.M. Molecular pathways: Extracting medical knowledge from high-throughput genomic data. Clin. Cancer Res. 2013, 19, 3114–3120. [Google Scholar] [CrossRef] [Green Version]
- Ellis, M.J.; Perou, C.M. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013, 3, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 2006, 38, 1289–1297. [Google Scholar] [CrossRef]
- Chen, G.G.; Zeng, Q.; Tse, G.M. Estrogen and its receptors in cancer. Med. Res. Rev. 2008, 28, 954–974. [Google Scholar] [CrossRef] [PubMed]
- Lonard, D.M.; O’Malley, B.W. Nuclear receptor coregulators: Judges, juries, and executioners of cellular regulation. Mol. Cell 2007, 27, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Samanthapudi, V.S.; Gajulapalli, V.N. Estrogen receptor coregulators and pioneer factors: The orchestrators of mammary gland cell fate and development. Front. Cell Dev. Biol. 2014, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartella, V.; Rizza, P.; Barone, I.; Zito, D.; Giordano, F.; Catalano, S.; Mauro, L.; Sisci, D.; Panno, M.L.; Fuqua, S.A.W.; et al. Estrogen receptor beta binds Sp1 and recruits a corepress.sor complex to the estrogen receptor alpha gene promoter. Breast Cancer Res. Treat. 2012, 134, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Kurebayashi, J.; Otsuki, T.; Kunisue, H.; Tanaka, K.; Yamamoto, S.; Sonoo, H. Expression levels of estrogen receptor-alpha, estrogen receptor-beta, coactivators, and corepressors in breast cancer. Clin. Cancer Res. 2000, 6, 512–518. [Google Scholar]
- Brisken, C.; O’Malley, B. Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2010, 2, a003178. [Google Scholar] [CrossRef]
- Jordan, V.C. SERMs: Meeting the promise of multifunctional medicines. J. Natl. Cancer Inst. 2007, 99, 350–356. [Google Scholar] [CrossRef]
- Jordan, V.C.; O’Malley, B.W. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J. Clin. Oncol. 2007, 25, 5815–5824. [Google Scholar] [CrossRef] [Green Version]
- Baumann, C.K.; Castiglione-Gertsch, M. Estrogen receptor modulators and down regulators: Optimal use in postmenopausal women with breast cancer. Drugs 2007, 67, 2335–2353. [Google Scholar] [CrossRef]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef] [Green Version]
- Silvente-Poirot, S.; Poirot, M. Cancer. Cholesterol and cancer, in the balance. Science 2014, 343, 1445–1446. [Google Scholar] [CrossRef]
- Poirot, M.; Soules, R.; Mallinger, A.; Dalenc, F.; Silvente-Poirot, S. Chemistry, biochemistry, metabolic fate and mechanism of action of 6-oxo-cholestan-3beta,5alpha-diol (OCDO), a tumor promoter and cholesterol metabolite. Biochimie 2018, 153, 139–149. [Google Scholar] [CrossRef]
- Wu, Q.; Ishikawa, T.; Sirianni, R.; Tang, H.; McDonald, J.G.; Yuhanna, I.S.; Thompson, B.; Girard, L.; Mineo, C.; Brekken, R.A.; et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. 2013, 5, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Voisin, M.; de Medina, P.; Mallinger, A.; Dalenc, F.; Huc-Claustre, E.; Leignadier, J.; Serhan, N.; Soules, R.; Segala, G.; Mougel, A.; et al. Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2017, 114, E9346–E9355. [Google Scholar] [CrossRef] [Green Version]
- De Medina, P.; Diallo, K.; Huc-Claustre, E.; Attia, M.; Soules, R.; Silvente-Poirot, S.; Poirot, M. The 5,6-epoxycholesterol metabolic pathway in breast cancer: Emergence of new pharmacological targets. Br. J. Pharmacol. 2021, 178, 3248–3260. [Google Scholar] [CrossRef]
- Leignadier, J.; Dalenc, F.; Poirot, M.; Silvente-Poirot, S. Improving the efficacy of hormone therapy in breast cancer: The role of cholesterol metabolism in SERM-mediated autophagy, cell differentiation and death. Biochem. Pharmacol. 2017, 144, 18–28. [Google Scholar] [CrossRef]
- Gomez-Coronado, D.; Lasuncion, M.A.; Martinez-Botas, J.; Fernandez-Suarez, M.E. Role of cholesterol metabolism in the anticancer pharmacology of selective estrogen receptor modulators. Semin. Cancer Biol. 2021, 73, 101–115. [Google Scholar] [CrossRef]
- Fernandez-Suarez, M.E.; Daimiel, L.; Villa-Turegano, G.; Pavon, M.V.; Busto, R.; Escola-Gil, J.C.; Platt, F.M.; Lasuncion, M.A.; Martinez-Botas, J.; Gomez-Coronado, D. Selective estrogen receptor modulators (SERMs) affect cholesterol homeostasis through the master regulators SREBP and LXR. Biomed. Pharmacother. 2021, 141, 111871. [Google Scholar] [CrossRef]
- Grober, O.M.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef]
- Powles, T.J. Anti-oestrogenic prevention of breast cancer—The make or break point. Nat. Rev. Cancer 2002, 2, 787–794. [Google Scholar] [CrossRef]
- Bulun, S.E.; Simpson, E.R. Aromatase expression in women’s cancers. Adv. Exp. Med. Biol. 2008, 630, 112–132. [Google Scholar]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [Green Version]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Deb, M.; Sengupta, D.; Shilpi, A.; Bhutia, S.K.; Patra, S.K. Intricacies of hedgehog signaling pathways: A perspective in tumorigenesis. Exp. Cell Res. 2012, 318, 1959–1972. [Google Scholar] [CrossRef]
- Diao, Y.; Azatyan, A.; Rahman, M.F.; Zhao, C.; Zhu, J.; Dahlman-Wright, K.; Zaphiropoulos, P.G. Blockade of the Hedgehog pathway downregulates estrogen receptor alpha signaling in breast cancer cells. Oncotarget 2016, 7, 71580–71593. [Google Scholar] [CrossRef] [Green Version]
- Molehin, D.; Rasha, F.; Rahman, R.L.; Pruitt, K. Regulation of aromatase in cancer. Mol. Cell. Biochem. 2021, 476, 2449–2464. [Google Scholar] [CrossRef]
- Molehin, D.; Filleur, S.; Pruitt, K. Regulation of aromatase expression: Potential therapeutic insight into breast cancer treatment. Mol. Cell. Endocrinol. 2021, 531, 111321. [Google Scholar] [CrossRef]
- Bulun, S.E.; Chen, D.; Moy, I.; Brooks, D.C.; Zhao, H. Aromatase, breast cancer and obesity: A complex interaction. Trends Endocrinol. Metab. 2012, 23, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev, B.; Sihto, H.; Li, S.; Holtta-Vuori, M.; Ilola, J.; Lundin, J.; Isola, J.; Kellokumpu-Lehtinen, P.L.; Joensuu, H.; Ikonen, E. Elevated levels of StAR-related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: Potential mechanisms contributing to progression of HER2-positive breast cancers. Am. J. Pathol. 2015, 185, 987–1000. [Google Scholar] [CrossRef]
- Manna, P.R.; Cohen-Tannoudji, J.; Counis, R.; Garner, C.W.; Huhtaniemi, I.; Kraemer, F.B.; Stocco, D.M. Mechanisms of action of hormone-sensitive lipase in mouse Leydig cells: Its role in the regulation of the steroidogenic acute regulatory protein. J. Biol. Chem. 2013, 288, 8505–8518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Zhang, X.; Dhakal, I.B.; Beggs, M.; Kadlubar, S.; Luo, D. Induction of cell proliferation and survival genes by estradiol-repressed microRNAs in breast cancer cells. BMC Cancer 2012, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chlebowski, R.T.; Anderson, G.L.; Prentice, R.L.; Rossouw, J.E.; Aragaki, A.K.; Manson, J.E. Reliable evidence from placebo-controlled, randomized, clinical trials for menopausal hormone therapy’s influence on incidence and deaths from breast cancer. Climacteric 2015, 18, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Emons, G.; Grundker, C. Estrogen signaling in ERalpha-negative breast cancer: ERbeta and GPER. Front. Endocrinol. 2018, 9, 781. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.A.; Oldham, W.M.; Chan, S.Y.; Vargas, S.O.; Arons, E.; Zhang, Y.Y.; Loscalzo, J.; Leopold, J.A. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation 2014, 130, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Boscaro, M.; Giacchetti, G.; Ronconi, V. Visceral adipose tissue: Emerging role of gluco- and mineralocorticoid hormones in the setting of cardiometabolic alterations. Ann. N. Y. Acad. Sci. 2012, 1264, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Phoenix, K.N.; Vumbaca, F.; Fox, M.M.; Evans, R.; Claffey, K.P. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res. Treat. 2010, 123, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Mollica, H.; Coclite, A.; Miali, M.E.; Pereira, R.C.; Paleari, L.; Manneschi, C.; DeCensi, A.; Decuzzi, P. Deciphering the relative contribution of vascular inflammation and blood rheology in metastatic spreading. Biomicrofluidics 2018, 12, 042205. [Google Scholar] [CrossRef]
- Suraj, J.; Kurpinska, A.; Zakrzewska, A.; Sternak, M.; Stojak, M.; Jasztal, A.; Walczak, M.; Chlopicki, S. Early and late endothelial response in breast cancer metastasis in mice: Simultaneous quantification of endothelial biomarkers using a mass spectrometry-based method. Dis. Model. Mech. 2019, 12, dmm036269. [Google Scholar] [CrossRef] [Green Version]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; Avino, S.; De Marco, P.; Bussolati, B.; Maggiolini, M.; De Francesco, E.M. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget 2016, 7, 94–111. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.L.; Bose, H.S. Early steps in steroidogenesis: Intracellular cholesterol trafficking. J. Lipid Res. 2011, 52, 2111–2135. [Google Scholar] [CrossRef] [Green Version]
- Manna, P.R.; Stetson, C.L.; Daugherty, C.; Shimizu, I.; Syapin, P.J.; Garrel, G.; Cohen-Tannoudji, J.; Huhtaniemi, I.; Slominski, A.T.; Pruitt, K.; et al. Up-regulation of steroid biosynthesis by retinoid signaling: Implications for aging. Mech. Ageing Dev. 2015, 150, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, J.P.; Alliende, M.I.; Molina, N.; Serrano, F.G.; Molina, S.; Vigil, P. Steroid hormones and their action in women’s brains: The importance of hormonal balance. Front. Public Health 2018, 6, 141. [Google Scholar] [CrossRef] [Green Version]
- Jensen, A.; Sharif, H.; Svare, E.I.; Frederiksen, K.; Kjaer, S.K. Risk of breast cancer after exposure to fertility drugs: Results from a large Danish cohort study. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Lerner-Geva, L.; Rabinovici, J.; Olmer, L.; Blumstein, T.; Mashiach, S.; Lunenfeld, B. Are infertility treatments a potential risk factor for cancer development? Perspective of 30 years of follow-up. Gynecol. Endocrinol. 2012, 28, 809–814. [Google Scholar]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 2012, 37, 137–162. [Google Scholar] [CrossRef]
- Drutskaya, M.S.; Efimov, G.A.; Kruglov, A.A.; Kuprash, D.V.; Nedospasov, S.A. Tumor necrosis factor, lymphotoxin and cancer. IUBMB Life 2010, 62, 283–289. [Google Scholar] [CrossRef]
- Green, T.L.; Santos, M.F.; Ejaeidi, A.A.; Craft, B.S.; Lewis, R.E.; Cruse, J.M. Toll-like receptor (TLR) expression of immune system cells from metastatic breast cancer patients with circulating tumor cells. Exp. Mol. Pathol. 2014, 97, 44–48. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, S.; Marin, L.; Gonzalez, L.; Gonza.alez, L.O.; del Casar, J.M.; Lamelas, M.L.; Gonzalez-Quintana, J.M.; Vizoso, F.J. Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer 2010, 10, 665. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Ud Din, N.; Browning, D.D.; Abrams, S.I.; Liu, K. Targeting lymphotoxin beta receptor with tumor-specific T lymphocytes for tumor regression. Clin. Cancer Res. 2007, 13, 5202–5210. [Google Scholar] [CrossRef] [Green Version]
- Fakheri, R.J.; Javitt, N.B. Autoregulation of cholesterol synthesis: Physiologic and pathophysiologic consequences. Steroids 2011, 76, 211–215. [Google Scholar] [CrossRef]
- Simigdala, N.; Gao, Q.; Pancholi, S.; Roberg-Larsen, H.; Zvelebil, M.; Ribas, R.; Folkerd, E.; Thompson, A.; Bhamra, A.; Dowsett, M.; et al. Cholesterol biosynthesis pathway as a novel mechanism of resistance to estrogen deprivation in estrogen receptor-positive breast cancer. Breast Cancer Res. 2016, 18, 58. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.R.; Ishikawa, T.; Umetani, M. The interaction between metabolism, cancer and cardiovascular disease, connected by 27-hydroxycholesterol. Clin. Lipidol. 2014, 9, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Basaria, S. Male hypogonadism. Lancet 2014, 383, 1250–1263. [Google Scholar] [CrossRef]
- Hall, E.A.; Ren, S.; Hylemon, P.B.; Redford, K.; del Castillo, A.; Gil, G.; Pandak, W.M. Mitochondrial cholesterol transport: A possible target in the management of hyperlipidemia. Lipids 2005, 40, 1237–1244. [Google Scholar] [CrossRef]
- Baek, A.E.; Yu, Y.A.; He, S.; Wardell, S.E.; Chang, C.Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 2017, 8, 864. [Google Scholar] [CrossRef]
- Liu, B.; Fan, Y.; Song, Z.; Han, B.; Meng, Y.; Cao, P.; Tan, K. Identification of DRP1 as a prognostic factor correlated with immune infiltration in breast cancer. Int. Immunopharmacol. 2020, 89, 107078. [Google Scholar] [CrossRef]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basanez, G.; Antonsson, B.; Prieto, J.; Garcia-Ruiz, C.; Colell, A.; et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronco, A.L.; De Stefani, E.; Stoll, M. Hormonal and metabolic modulation through nutrition: Towards a primary prevention of breast cancer. Breast 2010, 19, 322–332. [Google Scholar] [CrossRef]
- Warner, M.; Gustafsson, J.A. On estrogen, cholesterol metabolism, and breast cancer. N. Engl. J. Med. 2014, 370, 572–573. [Google Scholar] [CrossRef]
- Zimber, A.; Gespach, C. Bile acids and derivatives, their nuclear receptors FXR, PXR and ligands: Role in health and disease and their therapeutic potential. Anticancer Agents Med. Chem. 2008, 8, 540–563. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, H.; Duparc, C.; Cartier, D.; Contesse, V.; Perraudin, V.; Bertherat, J.; Plouin, P.F.; Reznik, Y.; Kuhn, J.M.; Louiset, E. Autocrine/paracrine regulations of steroidogenesis in adrenocortical hyperplasias and tumors. Ann. Endocrinol. 2009, 70, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Costarelli, V.; Sanders, T.A. Plasma deoxycholic acid concentration is elevated in postmenopausal women with newly diagnosed breast cancer. Eur. J. Clin. Nutr. 2002, 56, 925–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granot, Z.; Melamed-Book, N.; Bahat, A.; Orly, J. Turnover of StAR protein: Roles for the proteasome and mitochondrial proteases. Mol. Cell Endocrinol. 2007, 265–266, 51–58. [Google Scholar] [CrossRef]
- Bahat, A.; Perlberg, S.; Melamed-Book, N.; Lauria, I.; Langer, T.; Orly, J. StAR enhances transcription of genes encoding the mitochondrial proteases involved in its own degradation. Mol. Endocrinol. 2014, 28, 208–224. [Google Scholar] [CrossRef] [Green Version]
- Glynn, S.E. Multifunctional Mitochondrial AAA Proteases. Front. Mol. Biosci 2017, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef]
- Sannino, S.; Brodsky, J.L. Targeting protein quality control pathways in breast cancer. BMC Biol. 2017, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013, 4, e681. [Google Scholar] [CrossRef]
- Bultel, S.; Helin, L.; Clavey, V.; Chinetti-Gbaguidi, G.; Rigamonti, E.; Colin, M.; Fruchart, J.C.; Staels, B.; Lestavel, S. Liver X receptor activation induces the uptake of cholesteryl esters from high density lipoproteins in primary human macrophages. Arter. Thromb. Vasc. Biol. 2008, 28, 2288–2295. [Google Scholar] [CrossRef] [Green Version]
- Toss, A.; Palazzo, J.; Berger, A.; Guiles, F.; Sendecki, J.A.; Simone, N.; Anne, R.; Avery, T.; Jaslow, R.; Lazar, M.; et al. Clinical-pathological features and treatment modalities associated with recurrence in DCIS and micro-invasive carcinoma: Who to treat more and who to treat less. Breast 2016, 29, 223–230. [Google Scholar] [CrossRef]
- Harano, K.; Lei, X.; Gonzalez-Angulo, A.M.; Murthy, R.K.; Valero, V.; Mittendorf, E.A.; Ueno, N.T.; Hortobagyi, G.N.; Chavez-MacGregor, M. Clinicopathological and surgical factors associated with long-term survival in patients with HER2-positive metastatic breast cancer. Breast Cancer Res. Treat. 2016, 159, 367–374. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef]
- Byrski, T.; Dent, R.; Blecharz, P.; Foszczynska-Kloda, M.; Gronwald, J.; Huzarski, T.; Cybulski, C.; Marczyk, E.; Chrzan, R.; Eisen, A.; et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res. 2012, 14, 110. [Google Scholar] [CrossRef] [Green Version]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer 2016, 8, 93–107. [Google Scholar]
- Jahan, N.; Rehman, S.; Khan, R.; Jones, C. Relative risk of peripheral neuropathy with ado-trastuzumab emtansine (T-DM1) compared to taxane-based regimens in human epidermal growth factor receptor 2 (HER2)-positive cancers: A systematic review and meta-analysis. Cureus 2021, 13, e15282. [Google Scholar] [CrossRef]
- Kriege, M.; Jager, A.; Hooning, M.J.; Huijskens, E.; Blom, J.; van Deurzen, C.H.; Bontenbal, M.; Collee, J.M.; Menke-Pluijmers, M.B.; Martens, J.W.; et al. The efficacy of taxane chemotherapy for metastatic breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer 2012, 118, 899–907. [Google Scholar] [CrossRef]
- Santa-Maria, C.A.; Gradishar, W.J. Changing treatment paradigms in metastatic breast cancer: Lessons learned. JAMA Oncol. 2015, 1, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Mittendorf, E.A.; Peoples, G.E. Injecting hope-A review of breast cancer vaccines. Oncology 2016, 30, 475–481, 485. [Google Scholar]
- Wojtowicz, M.E.; Dunn, B.K.; Umar, A. Immunologic approaches to cancer prevention-current status, challenges, and future perspectives. Semin Oncol. 2016, 43, 161–172. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, L.; Zhang, Y.; Yu, X.; Wang, C.; Wang, H.; Yang, Y.; Chong, X.; Xia, T.; Meng, Y.; et al. An anti-ErbB2 fully human antibody circumvents trastuzumab resistance. Oncotarget 2016, 7, 67129–67141. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, G.R.; Franchina, T.; Russo, A.; Schifano, S.; Ferraro, G.; Adamo, V. Nab-paclitaxel and trastuzumab combination: A promising approach for neoadjuvant treatment in HER2-positive breast cancer. OncoTargets Ther. 2016, 9, 4351–4355. [Google Scholar]
- Li, H.; Shao, B.; Yan, Y.; Song, G.; Liu, X.; Wang, J.; Liang, X. Efficacy and safety of trastuzumab combined with chemotherapy for first-line treatment and beyond progression of HER2-overexpressing advanced breast cancer. Chin. J. Cancer Res. 2016, 28, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Mukai, H.; Saeki, T.; Aogi, K.; Naito, Y.; Matsubara, N.; Shigekawa, T.; Ueda, S.; Takashima, S.; Hara, F.; Yamashita, T.; et al. Patritumab plus trastuzumab and paclitaxel in human epidermal growth factor receptor 2-overexpressing metastatic breast cancer. Cancer Sci. 2016, 107, 1465–1470. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Clifton, G.T.; Holmes, J.P.; Clive, K.S.; Patil, R.; Benavides, L.C.; Gates, J.D.; Sears, A.K.; Stojadinovic, A.; Ponniah, S.; et al. Clinical trial results of the HER-2/neu (E75) vaccine to prevent breast cancer recurrence in high-risk patients: From US military cancer institute clinical trials group study I-01 and I-02. Cancer 2012, 118, 2594–2602. [Google Scholar] [CrossRef] [Green Version]
- Joshi, T.; Elias, D.; Stenvang, J.; Alves, C.L.; Teng, F.; Lyng, M.B.; Lykkesfeldt, A.E.; Brunner, N.; Wang, J.; Gupta, R.; et al. Integrative analysis of miRNA and gene expression reveals regulatory networks in tamoxifen-resistant breast cancer. Oncotarget 2016, 7, 57239–57253. [Google Scholar] [CrossRef] [Green Version]
- Cummings, S.R.; Ensrud, K.; Delmas, P.D.; LaCroix, A.Z.; Vukicevic, S.; Reid, D.M.; Goldstein, S.; Sriram, U.; Lee, A.; Thompson, J.; et al. Lasofoxifene in postmenopausal women with osteoporosis. N. Engl. J. Med. 2010, 362, 686–696. [Google Scholar] [CrossRef] [Green Version]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90, 2–6. [Google Scholar] [CrossRef]
- Mishra, A.K.; Abrahamsson, A.; Dabrosin, C. Fulvestrant inhibits growth of triple negative breast cancer and synergizes with tamoxifen in ERa positive breast cancer by up-regulation of ERbeta. Oncotarget 2016, 7, 56876–56888. [Google Scholar] [CrossRef]
- Mitwally, M.F.; Casper, R.F.; Diamond, M.P. The role of aromatase inhibitors in ameliorating deleterious effects of ovarian stimulation on outcome of infertility treatment. Reprod. Biol. Endocrinol. 2005, 3, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasha, F.; Sharma, M.; Pruitt, K. Mechanisms of endocrine therapy resistance in breast cancer. Mol. Cell Endocrinol. 2021, 532, 111322. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.; Schiavone, P.; Fedele, P.; Calvani, N.; Nacci, A.; Rizzo, P.; Marino, A.; D’Amico, M.; Sponziello, F.; Mazzoni, E.; et al. Molecularly targeted endocrine therapies for breast cancer. Cancer Treat. Rev. 2010, 36, S67–S71. [Google Scholar] [CrossRef]
- Fan, P.; Maximov, P.Y.; Curpan, R.F.; Abderrahman, B.; Jordan, V.C. The molecular, cellular and clinical consequences of targeting the estrogen receptor following estrogen deprivation therapy. Mol. Cell. Endocrinol. 2015, 418, 245–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruitt, K. Molecular and cellular changes during cancer progression resulting from genetic and epigenetic alterations. Prog. Mol. Biol. Transl. Sci. 2016, 144, 3–47. [Google Scholar]
- Kaypee, S.; Sudarshan, D.; Shanmugam, M.K.; Mukherjee, D.; Sethi, G.; Kundu, T.K. Aberrant lysine acetylation in tumorigenesis: Implications in the development of therapeutics. Pharmacol. Ther. 2016, 162, 98–119. [Google Scholar] [CrossRef]
- Holloway, K.R.; Barbieri, A.; Malyarchuk, S.; Saxena, M.; Nedeljkovic-Kurepa, A.; Cameron Mehl, M.; Wang, A.; Gu, X.; Pruitt, K. SIRT1 positively regulates breast cancer associated human aromatase (CYP19A1) expression. Mol. Endocrinol. 2013, 27, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Damaskos, C.; Garmpis, N.; Karatzas, T.; Nikolidakis, L.; Kostakis, I.D.; Garmpi, A.; Karamaroudis, S.; Boutsikos, G.; Damaskou, Z.; Kostakis, A.; et al. Histone deacetylase (HDAC) inhibitors: Current evidence for therapeutic activities in pancreatic cancer. Anticancer. Res. 2015, 35, 3129–3135. [Google Scholar]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Legare, S.; Basik, M. Minireview: The link between ERalpha corepressors and histone deacetylases in tamoxifen resistance in breast cancer. Mol. Endocrinol. 2016, 30, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.W.; Allred, J.B.; Moreno-Aspitia, A.; Northfelt, D.W.; Ingle, J.N.; Goetz, M.P.; Perez, E.A. Phase I study of panobinostat (LBH589) and letrozole in postmenopausal metastatic breast cancer patients. Clin. Breast Cancer 2016, 16, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Chen, Y.A.; Chiou, Y.S.; Ho, S.Y.; Lin, P.; Wang, Y.J. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo. PLoS ONE 2013, 8, e76340. [Google Scholar] [CrossRef] [Green Version]
- Tate, C.R.; Rhodes, L.V.; Segar, H.C.; Driver, J.L.; Pounder, F.N.; Burow, M.E.; Collins-Burow, B.M. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012, 14, 79. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Ye, J.; Kijima, I.; Evans, D. The HDAC inhibitor LBH589 (panobinostat) is an inhibitory modulator of aromatase gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 11032–11037. [Google Scholar] [CrossRef] [Green Version]
- Kubo, M.; Kanaya, N.; Petrossian, K.; Ye, J.; Warden, C.; Liu, Z.; Nishimura, R.; Osako, T.; Okido, M.; Shimada, K.; et al. Inhibition of the proliferation of acquired aromatase inhibitor-resistant breast cancer cells by histone deacetylase inhibitor LBH589 (panobinostat). Breast Cancer Res. Treat. 2013, 137, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.; Li, J.; Atadja, P.; Bhalla, K.; Haura, E.B. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor-dependent human lung cancer cells. Mol. Cancer Ther. 2007, 6, 2515–2524. [Google Scholar] [CrossRef] [Green Version]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Agoston, A.T.; Atadja, P.; Nelson, W.G.; Davidson, N.E. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol. Cancer Res. 2008, 6, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Shaw, P.G.; Davidson, N.E. Inhibition of histone deacetylase suppresses EGF signaling pathways by destabilizing EGFR mRNA in ER-negative human breast cancer cells. Breast Cancer Res. Treat. 2009, 117, 443–451. [Google Scholar] [CrossRef]
- Robertson, F.M.; Chu, K.; Boley, K.M.; Ye, Z.; Liu, H.; Wright, M.C.; Moraes, R.; Zhang, X.; Green, T.L.; Barsky, S.H.; et al. The class I HDAC inhibitor Romidepsin targets inflammatory breast cancer tumor emboli and synergizes with paclitaxel to inhibit metastasis. J. Exp. Ther. Oncol. 2013, 10, 219–233. [Google Scholar]
- Urbinati, G.; Marsaud, V.; Plassat, V.; Fattal, E.; Lesieur, S.; Renoir, J.M. Liposomes loaded with histone deacetylase inhibitors for breast cancer therapy. Int. J. Pharm. 2010, 397, 184–193. [Google Scholar] [CrossRef]
- Schech, A.; Kazi, A.; Yu, S.; Shah, P.; Sabnis, G. Histone deacetylase inhibitor entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol. Cancer Ther. 2015, 14, 1848–1857. [Google Scholar] [CrossRef] [Green Version]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar]
- Tomita, Y.; Lee, M.-J.; Lee, S.; Tomita, S.; Chumsri, S.; Cruickshank, S.; Ordentlich, P.; Trepel, J.B. The interplay of epigenetic therapy and immunity in locally recurrent or metastatic estrogen receptor-positive breast cancer: Correlative analysis of ENCORE 301, a randomized, placebo-controlled phase II trial of exemestane with or without entinostat. OncoImmunology 2016, 5, e1219008. [Google Scholar] [CrossRef] [Green Version]
- Mawatari, T.; Ninomiya, I.; Inokuchi, M.; Harada, S.; Hayashi, H.; Oyama, K.; Makino, I.; Nakagawara, H.; Miyashita, T.; Tajima, H.; et al. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int. J. Oncol. 2015, 47, 2073–2081. [Google Scholar] [CrossRef] [Green Version]
- Fortunati, N.; Bertino, S.; Costantino, L.; Bosco, O.; Vercellinatto, I.; Catalano, M.G.; Boccuzzi, G. Valproic acid is a selective antiproliferative agent in estrogen-sensitive breast cancer cells. Cancer Lett. 2008, 259, 156–164. [Google Scholar] [CrossRef]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar]
- Ajabnoor, G.M.; Crook, T.; Coley, H.M. Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells. Cell Death Dis. 2012, 3, e260. [Google Scholar] [CrossRef] [Green Version]
- Cheang, M.C.; Martin, M.; Nielsen, T.O.; Prat, A.; Voduc, D.; Rodriguez-Lescure, A.; Ruiz, A.; Chia, S.; Shepherd, L.; Ruiz-Borrego, M.; et al. Defining breast cancer intrinsic subtypes by quantitative receptor expression. Oncologist 2015, 20, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Tavakoli-Yaraki, M.; Karami-Tehrani, F.; Salimi, V.; Sirati-Sabet, M. Induction of apoptosis by Trichostatin A in human breast cancer cell lines: Involvement of 15-Lox-1. Tumor Biol. 2013, 34, 241–249. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, H.J.; Na, H.; Lee, M.O. Trichostatin A enhances acetylation as well as protein stability of ERalpha through induction of p300 protein. Breast Cancer Res 2010, 12, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, A.; Berdeja, J.G.; Patel, M.R.; Flinn, I.; Gerecitano, J.F.; Neelapu, S.S.; Kelly, K.R.; Copeland, A.R.; Akins, A.; Clancy, M.S.; et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: An open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016, 17, 622–631. [Google Scholar] [CrossRef] [Green Version]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Raha, P.; Thomas, S.; Thurn, K.T.; Park, J.; Munster, P.N. Combined histone deacetylase inhibition and tamoxifen induces apoptosis in tamoxifen-resistant breast cancer models, by reversing Bcl-2 overexpression. Breast Cancer Res. 2015, 17, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hait, N.C.; Avni, D.; Yamada, A.; Nagahashi, M.A.; Aoyagi, T.; Aoki, H.; Dumur, C.I.; Zelenko, Z.; Gallagher, E.J.; Leroith, D.; et al. The phosphorylated prodrug FTY720 is a histone deacetylase inhibitor that reactivates ERα expression and enhances hormonal therapy for breast cancer. Oncogenesis 2015, 4, e156. [Google Scholar] [CrossRef]
- Swaby, R.; Wang, M.; Sparano, J.; Bhalla, K.; Meropol, N.; Falkson, C.; Pellegrino, C.; Klein, P.; Goldstein, L.; Sledge, J.G. A phase II study of the histone deacetylase inhibitor, vorinostat, in combination with trastuzumab in patients with advanced metastatic and/or local chest wall recurrent HER-2 amplified breast cancer resistant to transtuzumab-containing therapy: (E1104) a trial of the eastern cooperative oncology group. Cancer Res. 2009, 69, 5084. [Google Scholar]
- Ramaswamy, B.; Fiskus, W.; Cohen, B.; Pellegrino, C.; Hershman, D.L.; Chuang, E.; Luu, T.; Somlo, G.; Goetz, M.; Swaby, R.; et al. Phase I-II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: Evidence for vorinostat-induced tubulin acetylation and Hsp90 inhibition in vivo. Breast Cancer Res. Treat. 2012, 132, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Connolly, R.M.; Jeter, S.; Zorzi, J.; Zhang, Z.; Armstrong, D.K.; Fetting, J.H.; Wolff, A.C.; Goetz, M.P.; Storniolo, A.M.; Stearns, V. A multi-institutional double-blind phase II study evaluating response and surrogate biomarkers to carboplatin and nab-paclitaxel (CP) with or without vorinostat as preoperative systemic therapy (PST) in HER2-negative primary operable breast cancer (TBCRC008). J. Clin. Oncol. 2010, 28, TPS111. [Google Scholar]
- Tu, Y.; Hershman, D.L.; Bhalla, K.; Fiskus, W.; Pellegrino, C.M.; Andreopoulou, E.; Makower, D.; Kalinsky, K.; Fehn, K.; Fineberg, S.; et al. A phase I-II study of the histone deacetylase inhibitor vorinostat plus sequential weekly paclitaxel and doxorubicin-cyclophosphamide in locally advanced breast cancer. Breast Cancer Res. Treat. 2014, 146, 145–152. [Google Scholar] [CrossRef]
- Conte, P.; Campone, M.; Pronzato, P.; Amadori, D.; Frank, R.; Schuetz, F.; Rea, D.; Wardley, A.; Britten, C.; Elias, A. Phase I trial of panobinostat (LBH589) in combination with trastuzumab in pretreated HER2-positive metastatic breast cancer (mBC): Preliminary safety and tolerability results. J. Clin. Oncol. 2009, 27, 1081. [Google Scholar] [CrossRef]
- Peacock, N.; Jones, S.; Yardley, D.; Bendell, J.; Infante, J.; Murphy, P.; Burris, H. Abstract P5-06-06: The Safety and tolerability of panobinostat (LBH589) in combination with capecitabine +/− lapatinib: A phase I study in HER2+ breast cancer. Cancer Res. 2010, 70, P5-06-06. [Google Scholar]
- Ryan, Q.C.; Headlee, D.; Acharya, M.; Sparreboom, A.; Trepel, J.B.; Ye, J.; Figg, W.D.; Hwang, K.; Chung, E.J.; Murgo, A.; et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J. Clin. Oncol. 2005, 23, 3912–3922. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). Exemestane with or without entinostat in treating patients with recurrent hormone receptor-positive breast cancer that is locally advanced or metastatic. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA, 2016. Available online: http://clinicaltrials.gov/show/NCT02115282 (accessed on 6 March 2018).
- Connolly, R.; Jankowitz, R.; Andreopoulou, E.; Allred, J.; Jeter, S.; Zorzi, J.; Adam, B.; Espinoza-Delgado, I.; Baylin, S.; Zahnow, C.; et al. OT3-01-06: A Phase 2 Study Investigating the Safety, Efficacy and Surrogate Biomarkers of Response of 5-Azacitidine (5-AZA) and Entinostat (MS-275) in Patients with Advanced Breast Cancer. Cancer Res 2011, 71, OT3-01-06. [Google Scholar]
- National Cancer Institute (NCI). Entinostat, nivolumab, and ipilimumab in treating patients with solid tumors that are metastatic or cannot be removed by surgery or locally advanced or metastatic HER2-negative breast ccancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA, 2016. Available online: http://clinicaltrials.gov/show/NCT02453620 (accessed on 12 April 2018).
- Lim, B.; Jackson, S.; Alvarez, R.; Ibrahim, N.; Willey, J.; Murthy, R.; Booser, D.; Giordano, S.; Barcenas, C.; Brewster, A.; et al. Abstract P4-14-22: A single-center, open-label phase 1b study of entinostat, and lapatinib alone, and in combination with and trastuzumab in patients with HER2+ metastatic breast cancer after progression on trastuzumab. Cancer Res. 2016, 76, P4-14-22. [Google Scholar]
- Sharma, P. Celgene Corporation: Romidepsin plus cisplatin in locally recurrent or metastatic triple negative breast cancer (TNBC). In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA, 2016. Available online: http://clinicaltrials.gov/show/NCT02393794 (accessed on 9 September 2018).
- Kalinsky, K. Acetylon Pharmaceuticals Incorporated: ACY-1215+Nab-paclitaxel in metastatic breast cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA, 2016. Available online: http://clinicaltrials.gov/show/NCT02632071 (accessed on 27 January 2018).
- Curis Inc. Open label, multi-center study to assess the safety, tolerability and pharmacokinetics of CUDC-907 in subjects with advanced/relapsed solid tumors. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA, 2016. Available online: http://clinicaltrials.gov/show/NCT02307240NLM (accessed on 16 August 2018).
- McGrogan, B.T.; Gilmartin, B.; Carney, D.N.; McCann, A. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys. Acta 2008, 1785, 96–132. [Google Scholar] [CrossRef]
- Chen, S.Y.; Zheng, X.W.; Cai, J.X.; Zhang, W.P.; You, H.S.; Xing, J.F.; Dong, Y.L. Histone deacetylase inhibitor reverses multidrug resistance by attenuating the nucleophosmin level through PI3K/Akt pathway in breast cancer. Int. J. Oncol. 2016, 49, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Steward, L.T.; Gao, F.; Taylor, M.A.; Margenthaler, J.A. Impact of radiation therapy on survival in patients with triple-negative breast cancer. Oncol. Lett. 2014, 7, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Baschnagel, A.; Russo, A.; Burgan, W.E.; Carter, D.; Beam, K.; Palmieri, D.; Steeg, P.S.; Tofilon, P.; Camphausen, K. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol. Cancer Ther. 2009, 8, 1589–1595. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Wang, H.; Zhao, X.; Dong, C.; Zhang, F.; Guo, G.; Guo, G.; Wang, X.; Powell, S.N.; Feng, Z.Y. Valproic acid causes radiosensitivity of breast cancer cells via disrupting.g the DNA repair pathway. Toxicol. Res. 2016, 5, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Chanda, N.; Kan, P.; Watkinson, L.D.; Shukla, R.; Zambre, A.; Carmack, T.L.; Engelbrecht, H.; Lever, J.R.; Katti, K.; Fent, G.M.; et al. Radioactive gold nanoparticles in cancer therapy: Therapeutic efficacy studies of GA-198AuNP nanoconstruct in prostate tumor-bearing mice. Nanomedicine 2010, 6, 201–209. [Google Scholar] [CrossRef]
- Sharma, M.; Molehin, D.; Castro-Piedras, I.; Martinez, E.G.; Pruitt, K. Acetylation of conserved DVL-1 lysines regulates its nuclear translocation and binding to gene promoters in triple-negative breast cancer. Sci. Rep. 2019, 9, 16257. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alterations | Luminal A | Luminal B | HER2E | Basal-Like |
|---|---|---|---|---|
| ER+/HER2− (%) | 87 | 82 | 20 | 10 |
| HER2+ (%) | 7 | 15 | 68 | 2 |
| TNBCs (%) | 2 | 1 | 9 | 80 |
| mRNAexpression | High ER, low proliferation | Lower ER, high proliferation | HER2 amplicon signature, high proliferation | Basal signature, high proliferation |
| TP53mutation | 12% | 32% | 75% | 84% |
| MAP3K1mutation | 14% | 5% | - | - |
| GATA3mutation | 14% | - | - | - |
| PIK3R1mutation | - | - | 8% | - |
| PIK3CAmutation | 49% | 32% | 42% | 7% |
| PTENmutation/loss | 13% | 24% | 19% | 35% |
| INPP4Bloss | 9% | 16% | 30% | 30% |
| RB1expression | high | - | - | low |
| RB1mutation/loss | - | - | - | 20% |
| Cyclin D1 amplification | 29% | 58% | 38% | - |
| Cyclin E1 amplification | - | - | - | 9% |
| CDK4gain | 14% | 25% | 24% | - |
| Grade | Low | Moderate | High | High |
| Prognosis | Good | Intermediate | Poor | Poor |
| Targeted Therapies | Endocrine | HER2 targeted therapy (e.g., Trastuzumab) | Chemotherapy, Investigational | |
| HDACIs | Targets | Phase(s) | Type of Therapy | BC Subtypes | References |
|---|---|---|---|---|---|
| Vorinostat (SAHA) | Class I, II, IV | II | Combination | ER+/PR+ | [172] |
| I/II | Combination | HER2 amplified | [175] | ||
| I/II | Combination | Metastatic or recurrent | [176] | ||
| II | Combination | HER2+ | [177] | ||
| I/II | Combination | HER2+ | [178] | ||
| Panobinostat (LBH589) | Class I, II, IV | I | Combination | TNBC | [179] |
| I/II | Combination | TNBC | [150] | ||
| I | Combination | Metastatic or recurrent | [180] | ||
| Entinostat (MS275) | Class I | I | Single | Metastatic/unresectable with no effective treatment | [181] |
| II | Combination | ER+, relapsed/progressed | [162,163] | ||
| III | Combination | ER+/PR+, metastatic/locally advanced | [182] | ||
| II | Combination | TNBC, metastatic/locally advanced | [183] | ||
| I | Combination | HER2-, metastatic/locally advanced | [184] | ||
| I | Combination | HER2+, metastatic or recurrent | [185] | ||
| Romidepsin | Class I | I/II | Combination | TNBC or BRCA1/BRCA2, metastatic/locally recurrent | [186] |
| Valproic Acid | Class I, II | I | Combination | Metastatic/locally advanced | [172] |
| Rocilinostat (ACY-1215) | Class II | I | Combination | Metastatic/unresectable with no effective treatment | [187] |
| CUDC-907 | Class I, II | I | Single | ER+/PR+, HER2-, advanced | [188] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manna, P.R.; Ahmed, A.U.; Molehin, D.; Narasimhan, M.; Pruitt, K.; Reddy, P.H. Hormonal and Genetic Regulatory Events in Breast Cancer and Its Therapeutics: Importance of the Steroidogenic Acute Regulatory Protein. Biomedicines 2022, 10, 1313. https://doi.org/10.3390/biomedicines10061313
Manna PR, Ahmed AU, Molehin D, Narasimhan M, Pruitt K, Reddy PH. Hormonal and Genetic Regulatory Events in Breast Cancer and Its Therapeutics: Importance of the Steroidogenic Acute Regulatory Protein. Biomedicines. 2022; 10(6):1313. https://doi.org/10.3390/biomedicines10061313
Chicago/Turabian StyleManna, Pulak R., Ahsen U. Ahmed, Deborah Molehin, Madhusudhanan Narasimhan, Kevin Pruitt, and P. Hemachandra Reddy. 2022. "Hormonal and Genetic Regulatory Events in Breast Cancer and Its Therapeutics: Importance of the Steroidogenic Acute Regulatory Protein" Biomedicines 10, no. 6: 1313. https://doi.org/10.3390/biomedicines10061313
APA StyleManna, P. R., Ahmed, A. U., Molehin, D., Narasimhan, M., Pruitt, K., & Reddy, P. H. (2022). Hormonal and Genetic Regulatory Events in Breast Cancer and Its Therapeutics: Importance of the Steroidogenic Acute Regulatory Protein. Biomedicines, 10(6), 1313. https://doi.org/10.3390/biomedicines10061313