
Epigenetic and Transcriptional Signaling in Ewing Sarcoma—Disease Etiology and Therapeutic Opportunities


Abstract
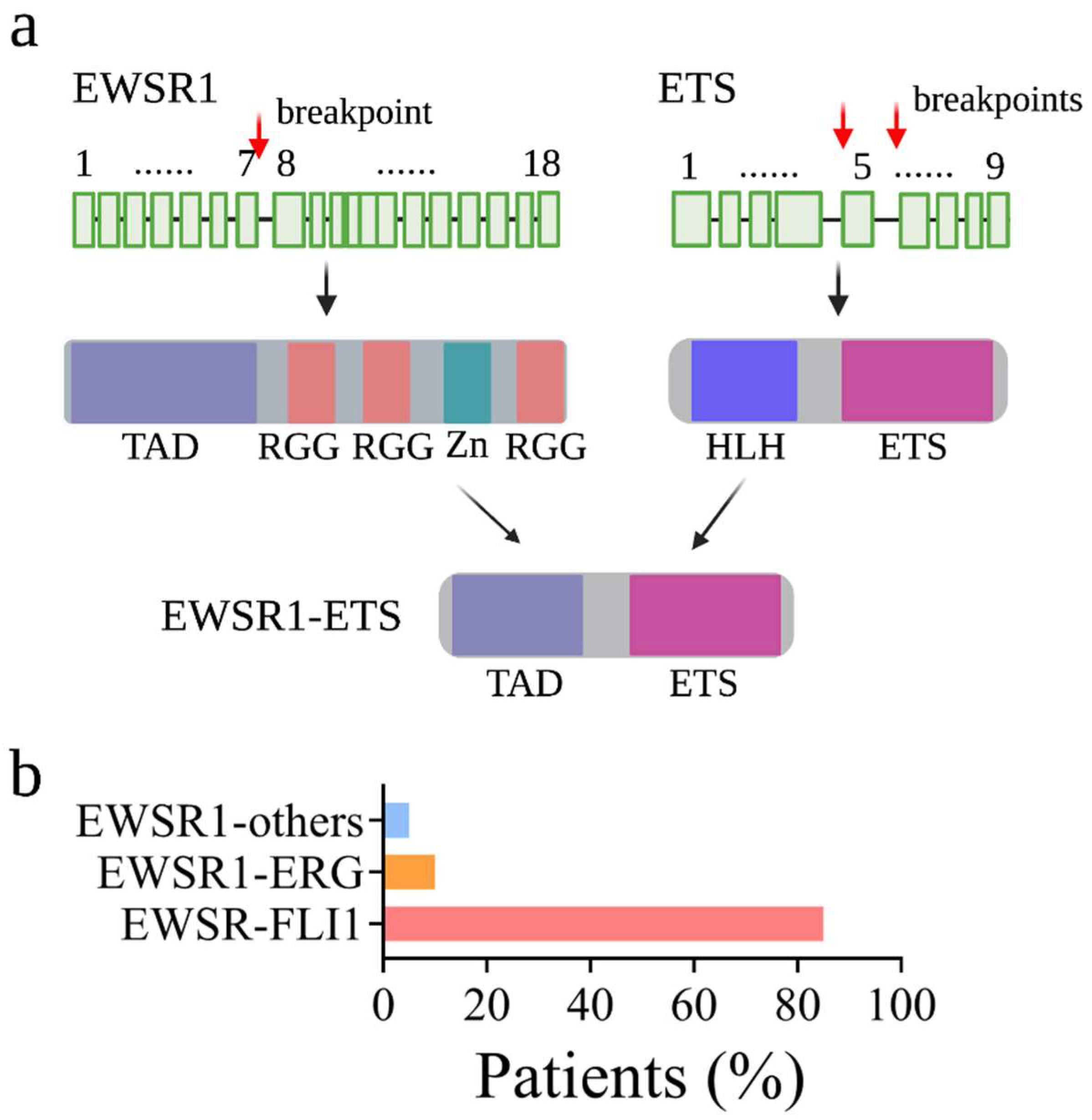
:1. Introduction
2. The Epigenetic Regulation Mediated by EWSR1-FLI1
2.1. Epigenetic Regulation
2.2. EWSR1-FLI1 Binding Sites on Chromatin
2.3. EWSR1-FLI1 Establishes Open Chromatin State at GGAA Microsatellites
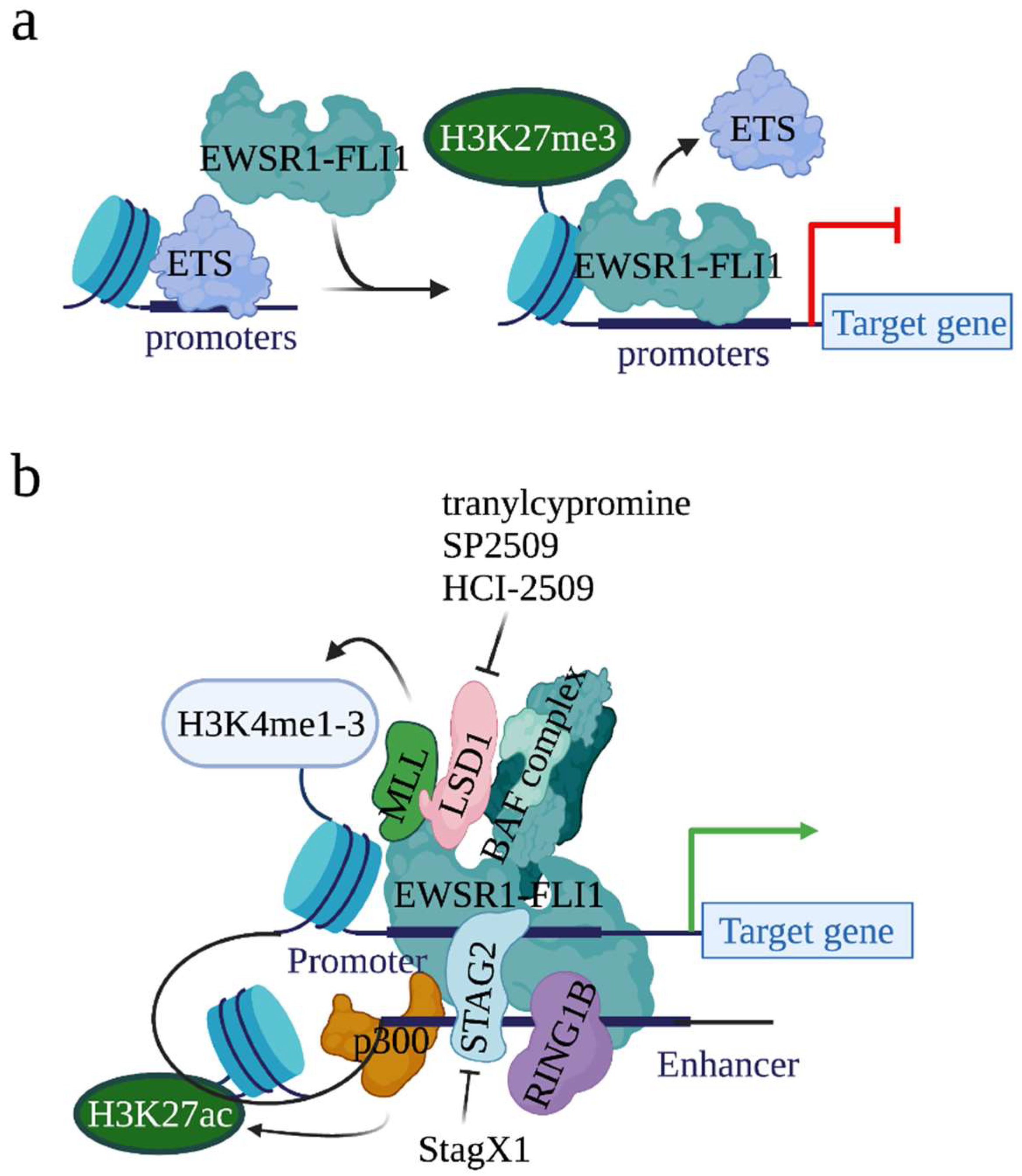
2.4. EWSR1-FLI1 Alters Epigenetic State by Recruiting Chromatin Modifiers to Its Binding Sites on Chromatin
2.4.1. Histone Acetyltransferase p300
2.4.2. BAF (BRG1/BRM-Associated Factor) Chromatin Remodeling Complex
2.4.3. LSD1 (Lysine-Specific Demethylase 1)
2.4.4. RING1B (Ring Finger Protein 2)
2.4.5. STAG2 (Stromal Antigen 2)
2.4.6. HDACs (Histone Deacetylases)
3. The Transcriptional Regulation Mediated by EWSR1-FLI1
3.1. EWSR1-FLI1 Regulates Gene Transcription by Interacting with Non-Chromatin Modifers on Chromatin
3.1.1. E2F Family Transcription Factors
3.1.2. NR0B1 (Nuclear Receptor Subfamily 0, Group B, Member 1)
3.1.3. RHA (RNA Helicase A)
3.2. Roles of EWSR1-FLI1 in Activiating Gene Expression
3.2.1. NR0B1
3.2.2. EZH2 (Enhancer of Zeste)
3.2.3. NKX2.2 (NK2 Homeobox 2)
3.2.4. KDM3A (Lysine Demethylase 3A)
3.2.5. GLI1 (Glioma-Associated Oncogene Homolog 1)
3.2.6. Homeobox Protein MEIS1
3.2.7. IL1RAP (Interleukin 1 Receptor Accessory Protein)
3.3. Roles of EWSR1-FLI1 in Repressing Gene Expression
3.3.1. TGFBR2 (Transforming Growth Factor Beta Receptor 2)
3.3.2. IGFBP3 (Insulin Like Growth Factor Binding Protein 3)
3.3.3. LOX (Lysyl Oxidase)
3.3.4. FOXO1 (Forkhead Box O1)
4. High Throughput Screens in EwS

{kind=link}
{kind=link}
{kind=link}
| Types | Aims | Inhibitors and Targets | Libraries | References |
|---|---|---|---|---|
| Chemical screens | Identify compounds to attenuate EWSR1-FLI1 activity | Cytosine arabinoside (ARA-C) | 1040 cpds (NCI) | Stegmaier et al., 2007 [129] |
| Mithramycin | 50,000 cpds (NCI) | Grohar et al., 2011 [130] | ||
| Midostaurin | 1280 cpds (Sigma) | Boro et al., 2012 [131] | ||
| Identify compounds to alter activity FAK, a highly activated kinase in EwS | AURKB inhibitor AZD-1152 | 1912 cpds (MIPE 4.0) | Wang et al., 2019 [132] | |
| Identify compounds to inhibit growth of mutant STAG2 EwS cells | StagX1 | 8000 cpds | Zhang et al., 2022 [62] | |
| RNAi screens | Identify kinase targets involved in EwS growth | STK10 and TNK2 | Kinase siRNA library (572 genes) | Arora et al., 2010 [133] |
| Identify regulators which modulate EWSR1-FLI1 activity | HNRNPH1 and SF3B1 | Genome-scale siRNA library | Grohar et al., 2016 [134] | |
| Identify regulators involved in EWSR1-FLI1 driven cell viability | LRWD1 | Druggable siRNA library (6781 genes) | He et al., 2016 [135] | |
| CRISPR-Cas9 screens | Identify genetic dependencies specific for growth of TP53 wild-type EwS cells | MDM2, MDM4, USP7, and PPM1D | Genome-scale CRISPR-Cas9 library | Stolte et al., 2018 [136] |
| Identify regulators which alter EWSR1-FLI1 stability | TRIM8 | Seong et al., 2021 [137] | ||
| Identify genes whose knockout conferred resistance to the LSD1 inhibition in EwS | Mitochondrial electron transport chain complexes III and IV | Tokarsky et al., 2022 [138] |
4.1. Chemical Screens Conducted in EwS
4.2. RNA Interference-Based Screens Conducted in EwS
4.3. CRISPR-Cas9-Based Screens Conducted in EwS
5. Discussion and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Harms, D.; Burdach, S. Malignant peripheral neuroectodermal tumours of childhood and adolescence. Virchows Archiv. A Pathol. Anat. Histopathol. 1985, 406, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Staege, M.S.; Hutter, C.; Neumann, I.; Foja, S.; Hattenhorst, U.E.; Hansen, G.; Afar, D.; Burdach, S.E. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res. 2004, 64, 8213–8221. [Google Scholar] [CrossRef] [Green Version]
- Ewing, J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA A Cancer J. Clin. 1972, 22, 95–98. [Google Scholar] [CrossRef]
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007, 11, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A first-generation pediatric cancer dependency map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Cervera, S.T.; Rodríguez-Martín, C.; Fernández-Tabanera, E.; de Mera, M.-F.R.M.; Morin, M.; Fernández-Peñalver, S.; Iranzo-Martínez, M.; Amhih-Cardenas, J.; García-García, L.; González-González, L.; et al. Therapeutic Potential of EWSR1-FLI1 Inactivation by CRISPR/Cas9 in Ewing Sarcoma. Cancers 2021, 13, 3783. [Google Scholar] [CrossRef]
- Riggi, N.; Suvà, M.L.; Suvà, D.; Cironi, L.; Provero, P.; Tercier, S.; Joseph, J.M.; Stehle, J.C.; Baumer, K.; Kindler, V.; et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008, 68, 2176–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Levetzow, C.; Jiang, X.; Gwye, Y.; von Levetzow, G.; Hung, L.; Cooper, A.; Hsu, J.H.; Lawlor, E.R. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS ONE 2011, 6, e19305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessnick, S.L.; Dacwag, C.S.; Golub, T.R. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell 2002, 1, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Cironi, L.; Provero, P.; Suvà, M.L.; Kaloulis, K.; Garcia-Echeverria, C.; Hoffmann, F.; Trumpp, A.; Stamenkovic, I. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. 2005, 65, 11459–11468. [Google Scholar] [CrossRef] [Green Version]
- Torchia, E.C.; Jaishankar, S.; Baker, S.J. Ewing tumor fusion proteins block the differentiation of pluripotent marrow stromal cells. Cancer Res. 2003, 63, 3464–3468. [Google Scholar]
- Eliazer, S.; Spencer, J.; Ye, D.; Olson, E.; Ilaria, R.L., Jr. Alteration of mesodermal cell differentiation by EWS/FLI-1, the oncogene implicated in Ewing’s sarcoma. Mol. Cell. Biol. 2003, 23, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Zhang, J.; Wu, L.; Shimada, H.; Schofield, D.E.; Triche, T.J. EWS-FLI1 fusion protein up-regulates critical genes in neural crest development and is responsible for the observed phenotype of Ewing’s family of tumors. Cancer Res. 2005, 65, 4633–4644. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Suvà, M.L.; De Vito, C.; Provero, P.; Stehle, J.C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suvà, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932. [Google Scholar] [CrossRef] [Green Version]
- Gangwal, K.; Sankar, S.; Hollenhorst, P.C.; Kinsey, M.; Haroldsen, S.C.; Shah, A.A.; Boucher, K.M.; Watkins, W.S.; Jorde, L.B.; Graves, B.J.; et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc. Natl. Acad. Sci. USA 2008, 105, 10149–10154. [Google Scholar] [CrossRef] [Green Version]
- Guillon, N.; Tirode, F.; Boeva, V.; Zynovyev, A.; Barillot, E.; Delattre, O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS ONE 2009, 4, e4932. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suvà, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schönegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deneen, B.; Welford, S.M.; Ho, T.; Hernandez, F.; Kurland, I.; Denny, C.T. PIM3 proto-oncogene kinase is a common transcriptional target of divergent EWS/ETS oncoproteins. Mol. Cell. Biol. 2003, 23, 3897–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunreiter, C.L.; Hancock, J.D.; Coffin, C.M.; Boucher, K.M.; Lessnick, S.L. Expression of EWS-ETS fusions in NIH3T3 cells reveals significant differences to Ewing’s sarcoma. Cell Cycle 2006, 5, 2753–2759. [Google Scholar] [CrossRef] [Green Version]
- Prieur, A.; Tirode, F.; Cohen, P.; Delattre, O. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol. Cell. Biol. 2004, 24, 7275–7283. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006, 9, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Seale, K.; Horvath, S.; Teschendorff, A.; Eynon, N.; Voisin, S. Making sense of the ageing methylome. Nat. Rev. Genet. 2022, 1–21. [Google Scholar] [CrossRef]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.; Zhao, K. The epigenetic basis of cellular heterogeneity. Nat. Rev. Genet. 2021, 22, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.; Bell, R.; Stephens, B.; Zhuo, R.; Sharma, S.; Bearss, D.J.; Lessnick, S.L. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene 2013, 32, 5089–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangwal, K.; Close, D.; Enriquez, C.A.; Hill, C.P.; Lessnick, S.L. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing’s Sarcoma. Genes Cancer 2010, 1, 177–187. [Google Scholar] [CrossRef]
- Monument, M.J.; Johnson, K.M.; Grossmann, A.H.; Schiffman, J.D.; Randall, R.L.; Lessnick, S.L. Microsatellites with macro-influence in ewing sarcoma. Genes 2012, 3, 444–460. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.M.; Mahler, N.R.; Saund, R.S.; Theisen, E.R.; Taslim, C.; Callender, N.W.; Crow, J.C.; Miller, K.R.; Lessnick, S.L. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proc. Natl. Acad. Sci. USA 2017, 114, 9870–9875. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.M.; Taslim, C.; Saund, R.S.; Lessnick, S.L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLoS ONE 2017, 12, e0186275. [Google Scholar] [CrossRef]
- Boulay, G.; Volorio, A.; Iyer, S.; Broye, L.C.; Stamenkovic, I.; Riggi, N.; Rivera, M.N. Epigenome editing of microsatellite repeats defines tumor-specific enhancer functions and dependencies. Genes Dev. 2018, 32, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178.e119. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [Green Version]
- Sankar, S.; Theisen, E.R.; Bearss, J.; Mulvihill, T.; Hoffman, L.M.; Sorna, V.; Beckerle, M.C.; Sharma, S.; Lessnick, S.L. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin. Cancer Res. 2014, 20, 4584–4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Gamero, C.; Malla, S.; Aguilo, F. LSD1: Expanding Functions in Stem Cells and Differentiation. Cells 2021, 10, 3252. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Schildhaus, H.U.; Riegel, R.; Hartmann, W.; Steiner, S.; Wardelmann, E.; Merkelbach-Bruse, S.; Tanaka, S.; Sonobe, H.; Schüle, R.; Buettner, R.; et al. Lysine-specific demethylase 1 is highly expressed in solitary fibrous tumors, synovial sarcomas, rhabdomyosarcomas, desmoplastic small round cell tumors, and malignant peripheral nerve sheath tumors. Hum. Pathol. 2011, 42, 1667–1675. [Google Scholar] [CrossRef]
- Kahl, P.; Gullotti, L.; Heukamp, L.C.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006, 66, 11341–11347. [Google Scholar] [CrossRef] [Green Version]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef]
- Theisen, E.R.; Pishas, K.I.; Saund, R.S.; Lessnick, S.L. Therapeutic opportunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget 2016, 7, 17616–17630. [Google Scholar] [CrossRef] [Green Version]
- Theisen, E.R.; Selich-Anderson, J.; Miller, K.R.; Tanner, J.M.; Taslim, C.; Pishas, K.I.; Sharma, S.; Lessnick, S.L. Chromatin profiling reveals relocalization of lysine-specific demethylase 1 by an oncogenic fusion protein. Epigenetics 2021, 16, 405–424. [Google Scholar] [CrossRef]
- García-Domínguez, D.J.; Hontecillas-Prieto, L.; Rodríguez-Núñez, P.; Pascual-Pasto, G.; Vila-Ubach, M.; García-Mejías, R.; Robles, M.J.; Tirado, O.M.; Mora, J.; Carcaboso, A.M.; et al. The combination of epigenetic drugs SAHA and HCI-2509 synergistically inhibits EWS-FLI1 and tumor growth in Ewing sarcoma. Oncotarget 2018, 9, 31397–31410. [Google Scholar] [CrossRef] [Green Version]
- Welch, D.; Kahen, E.; Fridley, B.; Brohl, A.S.; Cubitt, C.L.; Reed, D.R. Small molecule inhibition of lysine-specific demethylase 1 (LSD1) and histone deacetylase (HDAC) alone and in combination in Ewing sarcoma cell lines. PLoS ONE 2019, 14, e0222228. [Google Scholar] [CrossRef]
- Pishas, K.I.; Drenberg, C.D.; Taslim, C.; Theisen, E.R.; Johnson, K.M.; Saund, R.S.; Pop, I.L.; Crompton, B.D.; Lawlor, E.R.; Tirode, F.; et al. Therapeutic Targeting of KDM1A/LSD1 in Ewing Sarcoma with SP-2509 Engages the Endoplasmic Reticulum Stress Response. Mol. Cancer Ther. 2018, 17, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Muñoz, I.; Figuerola, E.; Sanchez-Molina, S.; Rodriguez, E.; Fernández-Mariño, A.I.; Pardo-Pastor, C.; Bahamonde, M.I.; Fernández-Fernández, J.M.; García-Domínguez, D.J.; Hontecillas-Prieto, L.; et al. RING1B contributes to Ewing sarcoma development by repressing the NaV1.6 sodium channel and the NF-κB pathway, independently of the fusion oncoprotein. Oncotarget 2016, 7, 46283–46300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Molina, S.; Figuerola-Bou, E.; Blanco, E.; Sánchez-Jiménez, M.; Táboas, P.; Gómez, S.; Ballaré, C.; García-Domínguez, D.J.; Prada, E.; Hontecillas-Prieto, L.; et al. RING1B recruits EWSR1-FLI1 and cooperates in the remodeling of chromatin necessary for Ewing sarcoma tumorigenesis. Sci. Adv. 2020, 6, eaba3058. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Lee, P.; Stafford, J.M.; von Schimmelmann, M.; Schaefer, A.; Reinberg, D. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 2014, 516, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, S.; Mas, G.; Di Croce, L. Regulation of gene transcription by Polycomb proteins. Sci. Adv. 2015, 1, e1500737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surdez, D.; Zaidi, S.; Grossetête, S.; Laud-Duval, K.; Ferre, A.S.; Mous, L.; Vourc’h, T.; Tirode, F.; Pierron, G.; Raynal, V.; et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell 2021, 39, 810–826.e819. [Google Scholar] [CrossRef]
- Adane, B.; Alexe, G.; Seong, B.K.A.; Lu, D.; Hwang, E.E.; Hnisz, D.; Lareau, C.A.; Ross, L.; Lin, S.; Dela Cruz, F.S.; et al. STAG2 loss rewires oncogenic and developmental programs to promote metastasis in Ewing sarcoma. Cancer Cell 2021, 39, 827–844.e810. [Google Scholar] [CrossRef]
- Bintu, B.; Mateo, L.J.; Su, J.H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef] [Green Version]
- Haering, C.H.; Farcas, A.-M.; Arumugam, P.; Metson, J.; Nasmyth, K. The cohesin ring concatenates sister DNA molecules. Nature 2008, 454, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Solomon, D.A.; Kim, J.-S.; Bondaruk, J.; Shariat, S.F.; Wang, Z.-F.; Elkahloun, A.G.; Ozawa, T.; Gerard, J.; Zhuang, D.; Zhang, S. Frequent truncating mutations of STAG2 in bladder cancer. Nat. Genet. 2013, 45, 1428–1430. [Google Scholar] [CrossRef]
- Balbás-Martínez, C.; Sagrera, A.; Carrillo-de-Santa-Pau, E.; Earl, J.; Márquez, M.; Vazquez, M.; Lapi, E.; Castro-Giner, F.; Beltran, S.; Bayés, M.; et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat. Genet. 2013, 45, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Lin, K.F.; Yang, C.; Peruski, S.; Pati, D.; Gilbertson, S.R. Synthesis and Evaluation of a Class of Compounds Inhibiting the Growth of Stromal Antigen 2 (STAG2)-Mutant Ewing Sarcoma Cells. ChemMedChem 2022, 17, e202100653. [Google Scholar] [CrossRef] [PubMed]
- Owen, L.A.; Kowalewski, A.A.; Lessnick, S.L. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing’s sarcoma. PLoS ONE 2008, 3, e1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzi, C.; Cassinelli, G. Combinatorial strategies to potentiate the efficacy of HDAC inhibitors in fusion-positive sarcomas. Biochem. Pharmacol. 2022, 198, 114944. [Google Scholar] [CrossRef] [PubMed]
- Jaboin, J.; Wild, J.; Hamidi, H.; Khanna, C.; Kim, C.J.; Robey, R.; Bates, S.E.; Thiele, C.J. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res. 2002, 62, 6108–6115. [Google Scholar]
- Sakimura, R.; Tanaka, K.; Nakatani, F.; Matsunobu, T.; Li, X.; Hanada, M.; Okada, T.; Nakamura, T.; Matsumoto, Y.; Iwamoto, Y. Antitumor effects of histone deacetylase inhibitor on Ewing’s family tumors. Int. J. Cancer 2005, 116, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, F.; Tanaka, K.; Sakimura, R.; Matsumoto, Y.; Matsunobu, T.; Li, X.; Hanada, M.; Okada, T.; Iwamoto, Y. Identification of p21WAF1/CIP1 as a direct target of EWS-Fli1 oncogenic fusion protein. J. Biol. Chem. 2003, 278, 15105–15115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, B.K.; da Costa Lopez, P.L.; Menegotto, P.R.; Vieira, I.A.; Kersting, N.; Abujamra, A.L.; Brunetto, A.T.; Brunetto, A.L.; Gregianin, L.; de Farias, C.B.; et al. Targeting Histone Deacetylase Activity to Arrest Cell Growth and Promote Neural Differentiation in Ewing Sarcoma. Mol. Neurobiol. 2018, 55, 7242–7258. [Google Scholar] [CrossRef]
- Bilke, S.; Schwentner, R.; Yang, F.; Kauer, M.; Jug, G.; Walker, R.L.; Davis, S.; Zhu, Y.J.; Pineda, M.; Meltzer, P.S.; et al. Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Res. 2013, 23, 1797–1809. [Google Scholar] [CrossRef] [Green Version]
- DeGregori, J.; Johnson, D.G. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr. Mol. Med. 2006, 6, 739–748. [Google Scholar] [CrossRef]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Kauer, M.; Ban, J.; Kofler, R.; Walker, B.; Davis, S.; Meltzer, P.; Kovar, H. A molecular function map of Ewing’s sarcoma. PLoS ONE 2009, 4, e5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwentner, R.; Papamarkou, T.; Kauer, M.O.; Stathopoulos, V.; Yang, F.; Bilke, S.; Meltzer, P.S.; Girolami, M.; Kovar, H. EWS-FLI1 employs an E2F switch to drive target gene expression. Nucleic Acids Res. 2015, 43, 2780–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, M.; Smith, R.; Lessnick, S.L. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol. Cancer Res. 2006, 4, 851–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendiola, M.; Carrillo, J.; García, E.; Lalli, E.; Hernández, T.; de Alava, E.; Tirode, F.; Delattre, O.; García-Miguel, P.; López-Barea, F.; et al. The orphan nuclear receptor DAX1 is up-regulated by the EWS/FLI1 oncoprotein and is highly expressed in Ewing tumors. Int. J. Cancer 2006, 118, 1381–1389. [Google Scholar] [CrossRef]
- Monument, M.J.; Johnson, K.M.; McIlvaine, E.; Abegglen, L.; Watkins, W.S.; Jorde, L.B.; Womer, R.B.; Beeler, N.; Monovich, L.; Lawlor, E.R.; et al. Clinical and biochemical function of polymorphic NR0B1 GGAA-microsatellites in Ewing sarcoma: A report from the Children’s Oncology Group. PLoS ONE 2014, 9, e104378. [Google Scholar] [CrossRef] [Green Version]
- Lalli, E.; Sassone-Corsi, P. DAX-1, an unusual orphan receptor at the crossroads of steroidogenic function and sexual differentiation. Mol. Endocrinol. 2003, 17, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, M.; Smith, R.; Iyer, A.K.; McCabe, E.R.; Lessnick, S.L. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res. 2009, 69, 9047–9055. [Google Scholar] [CrossRef] [Green Version]
- García-Aragoncillo, E.; Carrillo, J.; Lalli, E.; Agra, N.; Gómez-López, G.; Pestaña, A.; Alonso, J. DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell-cycle progression in Ewing’s tumor cells. Oncogene 2008, 27, 6034–6043. [Google Scholar] [CrossRef] [Green Version]
- Toretsky, J.A.; Erkizan, V.; Levenson, A.; Abaan, O.D.; Parvin, J.D.; Cripe, T.P.; Rice, A.M.; Lee, S.B.; Uren, A. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 2006, 66, 5574–5581. [Google Scholar] [CrossRef] [Green Version]
- Palombo, R.; Frisone, P.; Fidaleo, M.; Mercatelli, N.; Sette, C.; Paronetto, M.P. The Promoter-Associated Noncoding RNA pncCCND1_B Assembles a Protein-RNA Complex to Regulate Cyclin D1 Transcription in Ewing Sarcoma. Cancer Res. 2019, 79, 3570–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spriano, F.; Chung, E.Y.L.; Gaudio, E.; Tarantelli, C.; Cascione, L.; Napoli, S.; Jessen, K.; Carrassa, L.; Priebe, V.; Sartori, G.; et al. The ETS Inhibitors YK-4-279 and TK-216 Are Novel Antilymphoma Agents. Clin. Cancer Res. 2019, 25, 5167–5176. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.A.; Federman, N.C.; Anderson, P.M.; Macy, M.E.; Riedel, R.F.; Davis, L.E.; Daw, N.C.; Wulff, J.; Kim, A.; Ratan, R.; et al. TK216 for relapsed/refractory Ewing sarcoma: Interim phase 1/2 results. J. Clin. Oncol. 2021, 39, 11500. [Google Scholar] [CrossRef]
- Richter, G.H.; Plehm, S.; Fasan, A.; Rössler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Löwel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef] [Green Version]
- Burdach, S.; Plehm, S.; Unland, R.; Dirksen, U.; Borkhardt, A.; Staege, M.S.; Müller-Tidow, C.; Richter, G.H. Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle 2009, 8, 1991–1996. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Fadul, J.; Bell, R.; Hoffman, L.M.; Beckerle, M.C.; Engel, M.E.; Lessnick, S.L. EWS/FLI utilizes NKX2-2 to repress mesenchymal features of Ewing sarcoma. Genes Cancer 2015, 6, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Cai, J.; Wu, Y.; Wu, R.; Lee, J.; Fu, H.; Rao, M.; Sussel, L.; Rubenstein, J.; Qiu, M. Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Development 2001, 128, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.G.; Diebold, I.; Esposito, I.; Plehm, S.; Hauer, K.; Thiel, U.; da Silva-Buttkus, P.; Neff, F.; Unland, R.; Müller-Tidow, C.; et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol. Cancer Res. 2012, 10, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Markey, F.B.; Romero, B.; Parashar, V.; Batish, M. Identification of a New Transcriptional Co-Regulator of STEAP1 in Ewing’s Sarcoma. Cells 2021, 10, 1300. [Google Scholar] [CrossRef]
- Ullah, A.; Sinkler, M.A.; Velasquez Zarate, L.; Clavijo, A.; White, J. Ewing Sarcoma and Ewing-Like Sarcoma and the Role of NKX2.2 Immunoreactivity. Cureus 2021, 13, e17391. [Google Scholar] [CrossRef]
- McCuiston, A.; Bishop, J.A. Usefulness of NKX2.2 Immunohistochemistry for Distinguishing Ewing Sarcoma from Other Sinonasal Small Round Blue Cell Tumors. Head Neck Pathol. 2018, 12, 89–94. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yamazaki, K.; Ishida, Y. Upregulation of NKX2.2, a target of EWSR1/FLI1 fusion transcript, in primary renal Ewing sarcoma. J. Cytol. 2015, 32, 30–32. [Google Scholar] [CrossRef]
- Shibuya, R.; Matsuyama, A.; Nakamoto, M.; Shiba, E.; Kasai, T.; Hisaoka, M. The combination of CD99 and NKX2.2, a transcriptional target of EWSR1-FLI1, is highly specific for the diagnosis of Ewing sarcoma. Virchows Arch. Int. J. Pathol. 2014, 465, 599–605. [Google Scholar] [CrossRef]
- Yoshida, A.; Sekine, S.; Tsuta, K.; Fukayama, M.; Furuta, K.; Tsuda, H. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am. J. Surg. Pathol. 2012, 36, 993–999. [Google Scholar] [CrossRef]
- Parrish, J.K.; Sechler, M.; Winn, R.A.; Jedlicka, P. The histone demethylase KDM3A is a microRNA-22-regulated tumor promoter in Ewing Sarcoma. Oncogene 2015, 34, 257–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedlicka, P. The potential of KDM3A as a therapeutic target in Ewing Sarcoma and other cancers. Expert Opin. Ther. Targets 2017, 21, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Sechler, M.; Parrish, J.K.; Birks, D.K.; Jedlicka, P. The histone demethylase KDM3A, and its downstream target MCAM, promote Ewing Sarcoma cell migration and metastasis. Oncogene 2017, 36, 4150–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamane, K.; Toumazou, C.; Tsukada, Y.-i.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-Containing H3K9 Demethylase, Facilitates Transcription Activation by Androgen Receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Parrish, J.K.; McCann, T.S.; Sechler, M.; Sobral, L.M.; Ren, W.; Jones, K.L.; Tan, A.C.; Jedlicka, P. The Jumonji-domain histone demethylase inhibitor JIB-04 deregulates oncogenic programs and increases DNA damage in Ewing Sarcoma, resulting in impaired cell proliferation and survival, and reduced tumor growth. Oncotarget 2018, 9, 33110–33123. [Google Scholar] [CrossRef] [Green Version]
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; May, W.A. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008, 27, 3282–3291. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [Green Version]
- Joo, J.; Christensen, L.; Warner, K.; States, L.; Kang, H.G.; Vo, K.; Lawlor, E.R.; May, W.A. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS ONE 2009, 4, e7608. [Google Scholar] [CrossRef]
- Kasper, M.; Regl, G.; Frischauf, A.M.; Aberger, F. GLI transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Guo, W.; Ren, T.T.; Lu, X.C.; Tang, G.Q.; Zhao, F.L. Arsenic trioxide inhibits Ewing’s sarcoma cell invasiveness by targeting p38(MAPK) and c-Jun N-terminal kinase. Anti-Cancer Drugs 2012, 23, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Tang, X.D.; Tang, S.; Yang, Y. Preliminary report of combination chemotherapy including Arsenic trioxide for stage III osteosarcoma and Ewing sarcoma. Zhonghua Wai Ke Za Zhi Chin. J. Surg. 2006, 44, 805–808. [Google Scholar] [PubMed]
- Lin, L.; Huang, M.; Shi, X.; Mayakonda, A.; Hu, K.; Jiang, Y.Y.; Guo, X.; Chen, L.; Pang, B.; Doan, N.; et al. Super-enhancer-associated MEIS1 promotes transcriptional dysregulation in Ewing sarcoma in co-operation with EWS-FLI1. Nucleic Acids Res. 2019, 47, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Nakatake, M.; Kuwata, T.; Couzinet, A.; Goitsuka, R.; Tsutsumi, S.; Aburatani, H.; Valk, P.J.; Delwel, R.; Nakamura, T. MEIS1-mediated transactivation of synaptotagmin-like 1 promotes CXCL12/CXCR4 signaling and leukemogenesis. J. Clin. Investig. 2016, 126, 1664–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.F.; Hughes, C.S.; Li, W.; He, J.Z.; Surdez, D.; El-Naggar, A.M.; Cheng, H.; Prudova, A.; Delaidelli, A.; Negri, G.L.; et al. Proteomic Screens for Suppressors of Anoikis Identify IL1RAP as a Promising Surface Target in Ewing Sarcoma. Cancer Discov. 2021, 11, 2884–2903. [Google Scholar] [CrossRef]
- Boraschi, D.; Tagliabue, A. The interleukin-1 receptor family. Semin. Immunol. 2013, 25, 394–407. [Google Scholar] [CrossRef]
- Hamdan, R.; Zhou, Z.; Kleinerman, E.S. Blocking SDF-1α/CXCR4 downregulates PDGF-B and inhibits bone marrow-derived pericyte differentiation and tumor vascular expansion in Ewing tumors. Mol. Cancer Ther. 2014, 13, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Fukuma, M.; Okita, H.; Hata, J.-i.; Umezawa, A. Upregulation of Id2, an oncogenic helix-loop-helix protein, is mediated by the chimeric EWS/ets protein in Ewing sarcoma. Oncogene 2003, 22, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.; Inwards, C.Y.; Janknecht, R. Vascular endothelial growth factor expression is up-regulated by EWS-ETS oncoproteins and Sp1 and may represent an independent predictor of survival in Ewing’s sarcoma. Clin. Cancer Res. 2004, 10, 1344–1353. [Google Scholar] [CrossRef] [Green Version]
- Hahm, K.B.; Cho, K.; Lee, C.; Im, Y.H.; Chang, J.; Choi, S.G.; Sorensen, P.H.; Thiele, C.J.; Kim, S.J. Repression of the gene encoding the TGF-beta type II receptor is a major target of the EWS-FLI1 oncoprotein. Nat. Genet. 1999, 23, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Niedan, S.; Kauer, M.; Aryee, D.N.; Kofler, R.; Schwentner, R.; Meier, A.; Pötschger, U.; Kontny, U.; Kovar, H. Suppression of FOXO1 is responsible for a growth regulatory repressive transcriptional sub-signature of EWS-FLI1 in Ewing sarcoma. Oncogene 2014, 33, 3927–3938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastie, C.C.; Nahlé, Z.; McLoughlin, T.; Esser, K.; Zhang, W.; Unterman, T.; Abumrad, N.A. FoxO1 stimulates fatty acid uptake and oxidation in muscle cells through CD36-dependent and -independent mechanisms. J. Biol. Chem. 2005, 280, 14222–14229. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Hu, H.M.; Zielinska-Kwiatkowska, A.; Chansky, H.A. FOXO1 is a direct target of EWS-Fli1 oncogenic fusion protein in Ewing’s sarcoma cells. Biochem. Biophys. Res. Commun. 2010, 402, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Cidre-Aranaz, F.; Grünewald, T.G.; Surdez, D.; García-García, L.; Carlos Lázaro, J.; Kirchner, T.; González-González, L.; Sastre, A.; García-Miguel, P.; López-Pérez, S.E.; et al. EWS-FLI1-mediated suppression of the RAS-antagonist Sprouty 1 (SPRY1) confers aggressiveness to Ewing sarcoma. Oncogene 2017, 36, 766–776. [Google Scholar] [CrossRef]
- Minowada, G.; Jarvis, L.A.; Chi, C.L.; Neubüser, A.; Sun, X.; Hacohen, N.; Krasnow, M.A.; Martin, G.R. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 1999, 126, 4465–4475. [Google Scholar] [CrossRef]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef]
- Stegmaier, K.; Wong, J.S.; Ross, K.N.; Chow, K.T.; Peck, D.; Wright, R.D.; Lessnick, S.L.; Kung, A.L.; Golub, T.R. Signature-based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. PLoS Med. 2007, 4, e122. [Google Scholar] [CrossRef]
- Grohar, P.J.; Woldemichael, G.M.; Griffin, L.B.; Mendoza, A.; Chen, Q.R.; Yeung, C.; Currier, D.G.; Davis, S.; Khanna, C.; Khan, J.; et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J. Natl. Cancer Inst. 2011, 103, 962–978. [Google Scholar] [CrossRef] [Green Version]
- Boro, A.; Prêtre, K.; Rechfeld, F.; Thalhammer, V.; Oesch, S.; Wachtel, M.; Schäfer, B.W.; Niggli, F.K. Small-molecule screen identifies modulators of EWS/FLI1 target gene expression and cell survival in Ewing’s sarcoma. Int. J. Cancer 2012, 131, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hwang, E.E.; Guha, R.; O’Neill, A.F.; Melong, N.; Veinotte, C.J.; Conway Saur, A.; Wuerthele, K.; Shen, M.; McKnight, C.; et al. High-throughput Chemical Screening Identifies Focal Adhesion Kinase and Aurora Kinase B Inhibition as a Synergistic Treatment Combination in Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 4552–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Gonzales, I.M.; Hagelstrom, R.T.; Beaudry, C.; Choudhary, A.; Sima, C.; Tibes, R.; Mousses, S.; Azorsa, D.O. RNAi phenotype profiling of kinases identifies potential therapeutic targets in Ewing’s sarcoma. Mol. Cancer 2010, 9, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohar, P.J.; Kim, S.; Rivera, R.G.O.; Sen, N.; Haddock, S.; Harlow, M.L.; Maloney, N.K.; Zhu, J.; O’Neill, M.; Jones, T.L.; et al. Functional Genomic Screening Reveals Splicing of the EWS-FLI1 Fusion Transcript as a Vulnerability in Ewing Sarcoma. Cell Rep. 2016, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Surdez, D.; Rantala, J.K.; Haapa-Paananen, S.; Ban, J.; Kauer, M.; Tomazou, E.; Fey, V.; Alonso, J.; Kovar, H.; et al. High-throughput RNAi screen in Ewing sarcoma cells identifies leucine rich repeats and WD repeat domain containing 1 (LRWD1) as a regulator of EWS-FLI1 driven cell viability. Gene 2017, 596, 137–146. [Google Scholar] [CrossRef]
- Stolte, B.; Iniguez, A.B.; Dharia, N.V.; Robichaud, A.L.; Conway, A.S.; Morgan, A.M.; Alexe, G.; Schauer, N.J.; Liu, X.; Bird, G.H.; et al. Genome-scale CRISPR-Cas9 screen identifies druggable dependencies in TP53 wild-type Ewing sarcoma. J. Exp. Med. 2018, 215, 2137–2155. [Google Scholar] [CrossRef]
- Seong, B.K.A.; Dharia, N.V.; Lin, S.; Donovan, K.A.; Chong, S.; Robichaud, A.; Conway, A.; Hamze, A.; Ross, L.; Alexe, G.; et al. TRIM8 modulates the EWS/FLI oncoprotein to promote survival in Ewing sarcoma. Cancer Cell 2021, 39, 1262–1278.e1267. [Google Scholar] [CrossRef]
- Tokarsky, E.J.; Crow, J.C.; Guenther, L.M.; Sherman, J.; Taslim, C.; Alexe, G.; Pishas, K.I.; Rask, G.; Justis, B.S.; Kasumova, A.; et al. Mitochondrial Dysfunction is a Driver of SP-2509 Drug Resistance in Ewing Sarcoma. Mol. Cancer Res. 2022. [Google Scholar] [CrossRef]
- Franzetti, G.A.; Laud-Duval, K.; van der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; de Pinieux, G.; Snaar-Jagalska, E.; et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef] [Green Version]
- Aynaud, M.M.; Mirabeau, O.; Gruel, N.; Grossetête, S.; Boeva, V.; Durand, S.; Surdez, D.; Saulnier, O.; Zaïdi, S.; Gribkova, S.; et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep. 2020, 30, 1767–1779.e1766. [Google Scholar] [CrossRef] [Green Version]
- Khoogar, R.; Li, F.; Chen, Y.; Ignatius, M.; Lawlor, E.R.; Kitagawa, K.; Huang, T.H.; Phelps, D.A.; Houghton, P.J. Single-cell RNA profiling identifies diverse cellular responses to EWSR1/FLI1 downregulation in Ewing sarcoma cells. Cell. Oncol. 2022, 45, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Crompton, B.D.; Carlton, A.L.; Thorner, A.R.; Christie, A.L.; Du, J.; Calicchio, M.L.; Rivera, M.N.; Fleming, M.D.; Kohl, N.E.; Kung, A.L.; et al. High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in Ewing sarcoma. Cancer Res. 2013, 73, 2873–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radic-Sarikas, B.; Tsafou, K.P.; Emdal, K.B.; Papamarkou, T.; Huber, K.V.; Mutz, C.; Toretsky, J.A.; Bennett, K.L.; Olsen, J.V.; Brunak, S.; et al. Combinatorial Drug Screening Identifies Ewing Sarcoma-specific Sensitivities. Mol. Cancer Ther. 2017, 16, 88–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chan, A.K.N.; Miyashita, K.; Delaney, C.D.; Wang, X.; Li, H.; Pokharel, S.P.; Li, S.; Li, M.; Xu, X.; et al. High-resolution characterization of gene function using single-cell CRISPR tiling screen. Nat. Commun. 2021, 12, 4063. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Chen, C.-W. Epigenetic and Transcriptional Signaling in Ewing Sarcoma—Disease Etiology and Therapeutic Opportunities. Biomedicines 2022, 10, 1325. https://doi.org/10.3390/biomedicines10061325
Li M, Chen C-W. Epigenetic and Transcriptional Signaling in Ewing Sarcoma—Disease Etiology and Therapeutic Opportunities. Biomedicines. 2022; 10(6):1325. https://doi.org/10.3390/biomedicines10061325
Chicago/Turabian StyleLi, Mingli, and Chun-Wei Chen. 2022. "Epigenetic and Transcriptional Signaling in Ewing Sarcoma—Disease Etiology and Therapeutic Opportunities" Biomedicines 10, no. 6: 1325. https://doi.org/10.3390/biomedicines10061325
APA StyleLi, M., & Chen, C. -W. (2022). Epigenetic and Transcriptional Signaling in Ewing Sarcoma—Disease Etiology and Therapeutic Opportunities. Biomedicines, 10(6), 1325. https://doi.org/10.3390/biomedicines10061325