
Modulatory Effects of Estradiol and Its Mixtures with Ligands of GPER and PPAR on MAPK and PI3K/Akt Signaling Pathways and Tumorigenic Factors in Mouse Testis Explants and Mouse Tumor Leydig Cells

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Testis Explants’ Culture Conditions
2.2. Mouse Leydig Cell Line (MA-10) Culture Conditions
2.3. Testis Explants and MA-10 Cells Treatments
2.4. Western Blot Analysis
2.5. Immunofluorescence
2.6. Statistical Analysis
3. Results
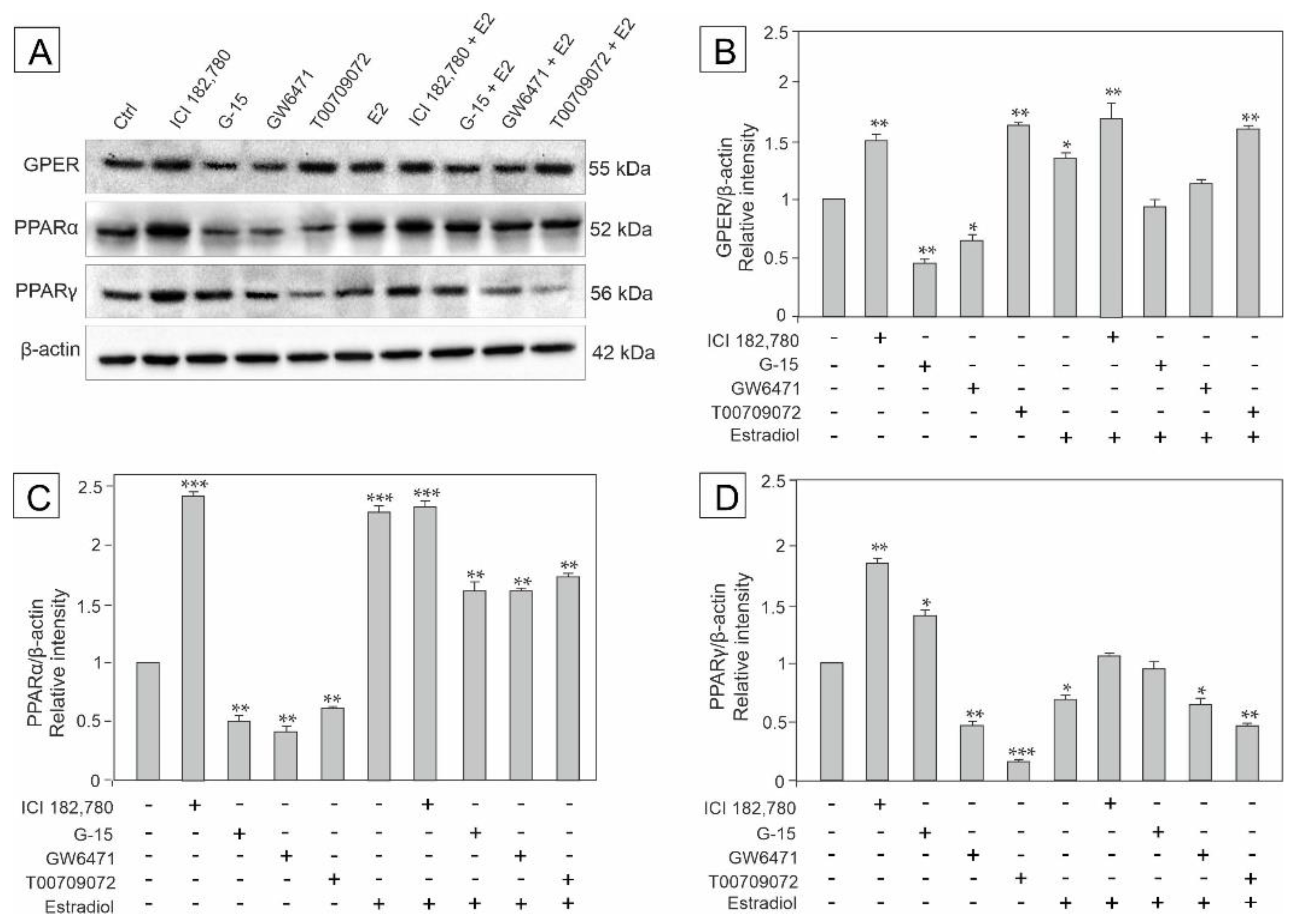
3.1. Effect of GPER Agonist, GPER and PPAR Antagonists, and Estradiol on the Expression of GPER, PPARα and PPARγ in Mouse Testis Explants
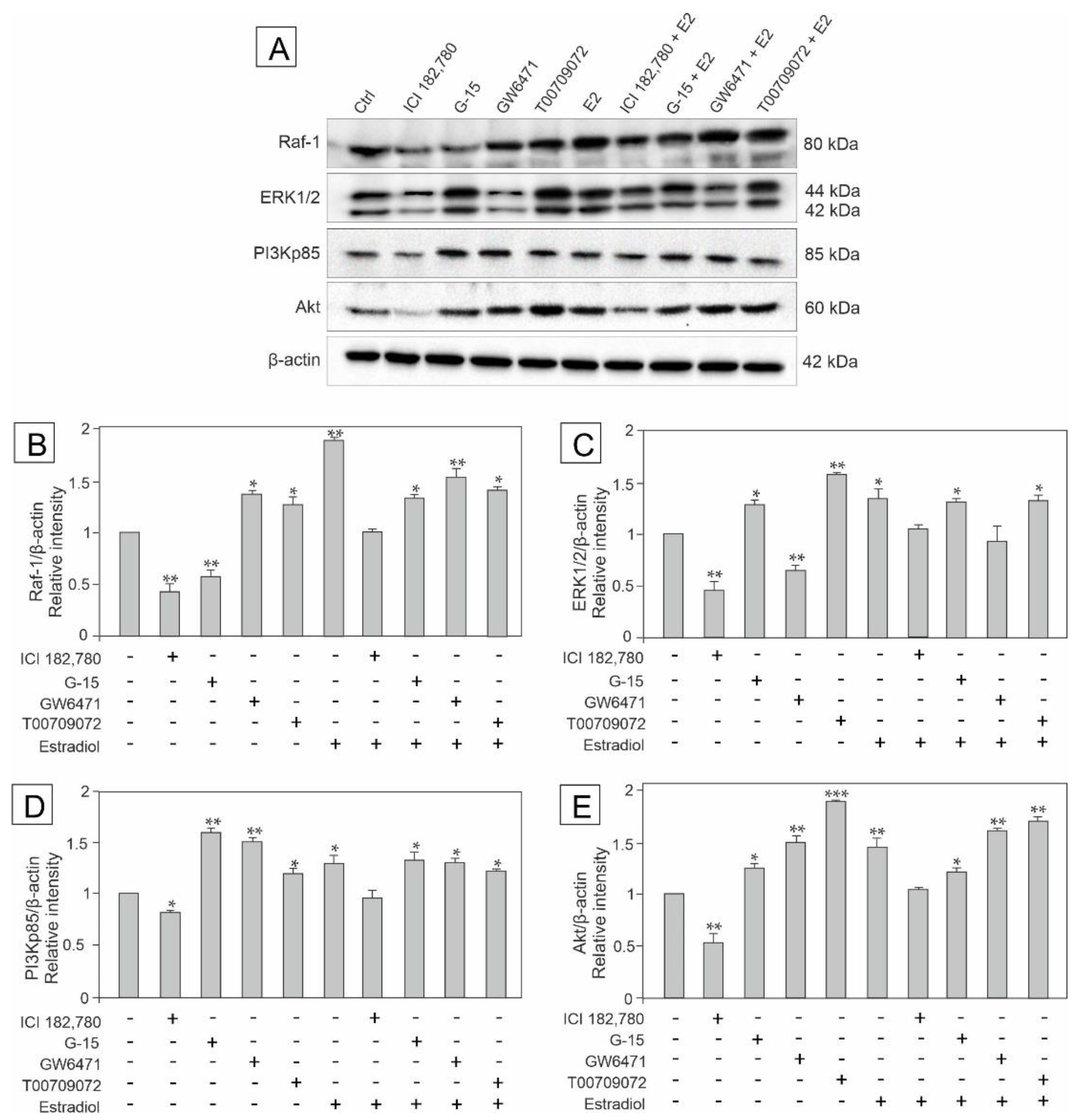
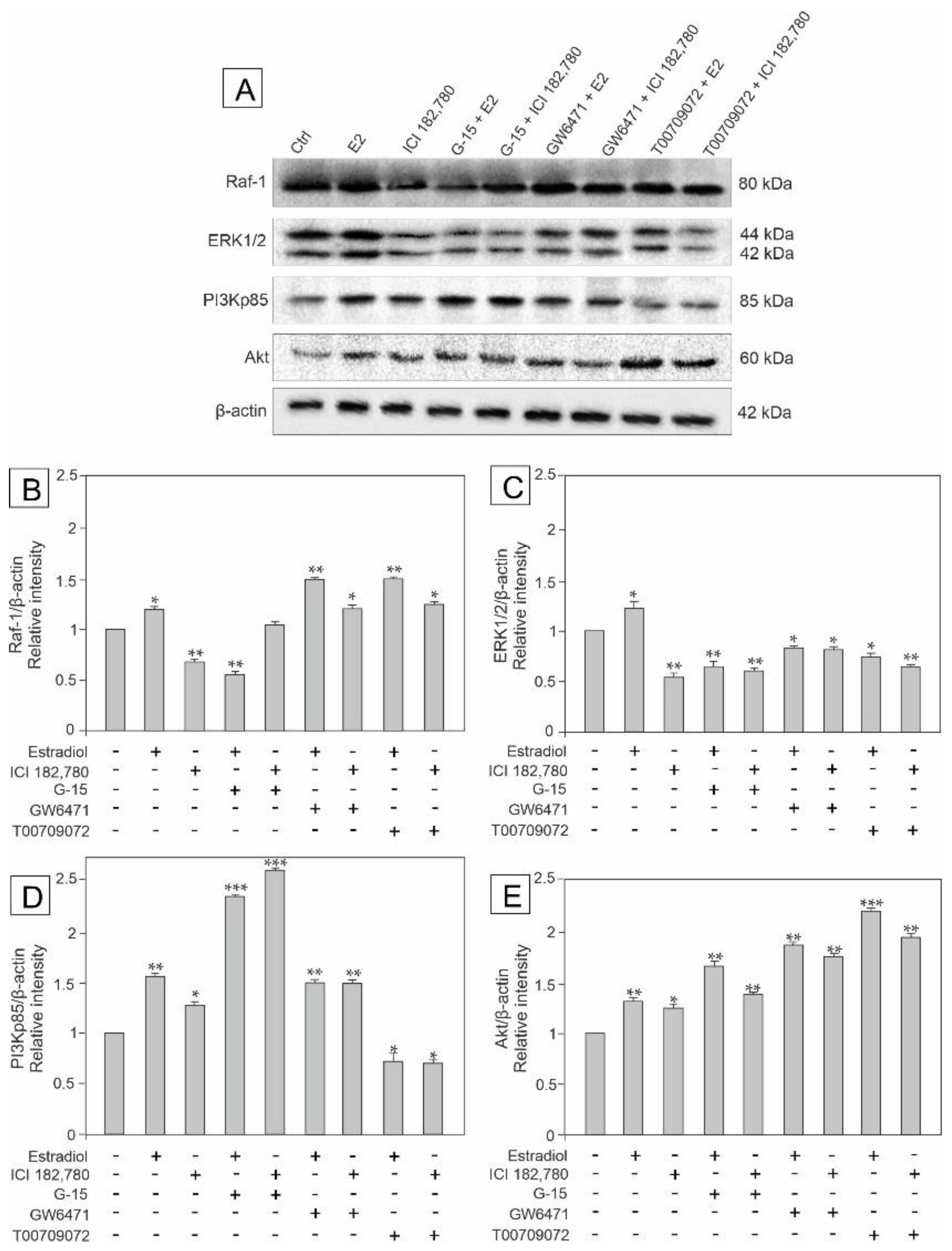
3.2. Effect of GPER Agonist, GPER and PPAR Antagonists, and Estradiol on the Expression of Raf, ERK1/2, PI3Kp85, and Akt in Mouse Testis Explants
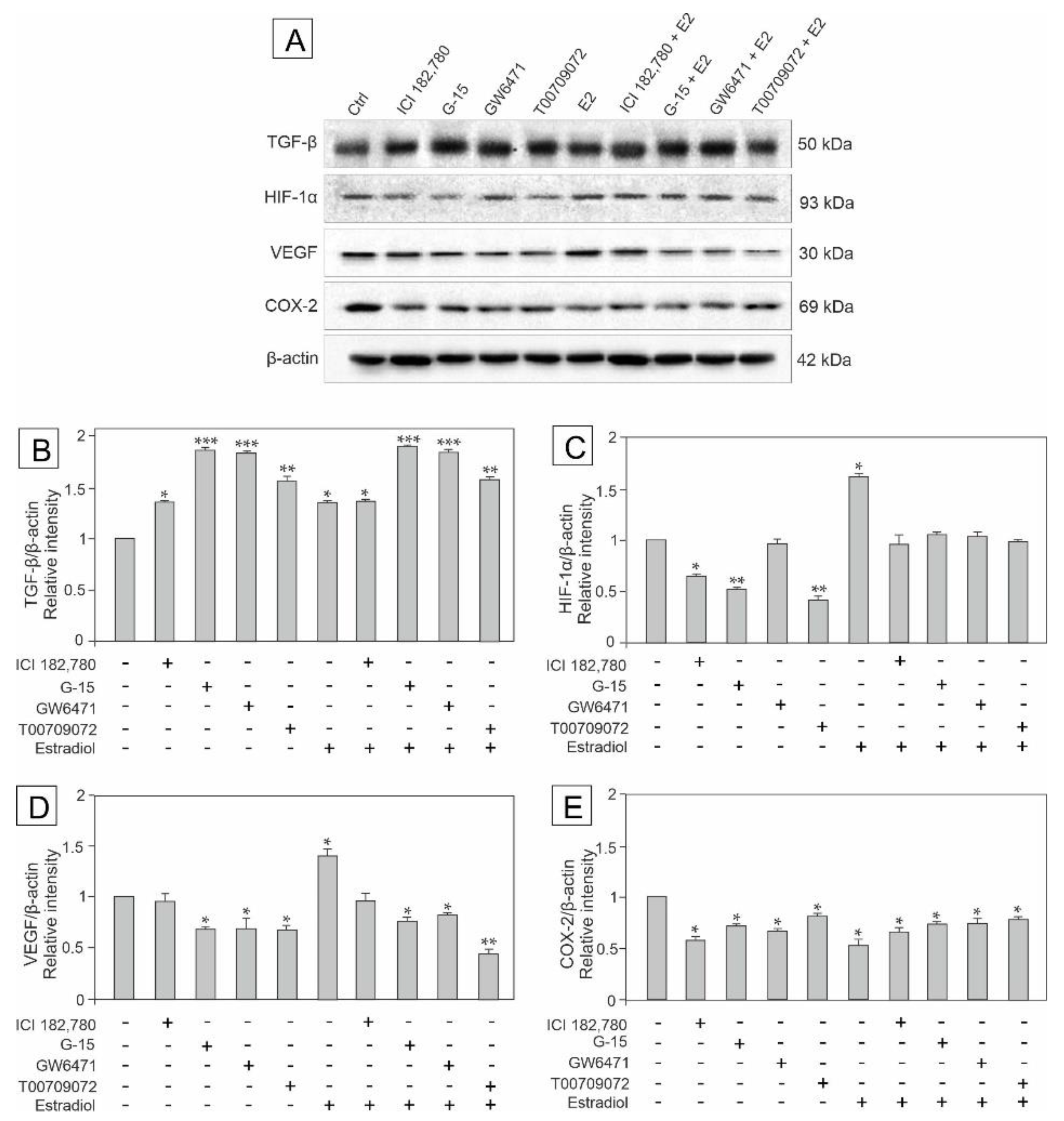
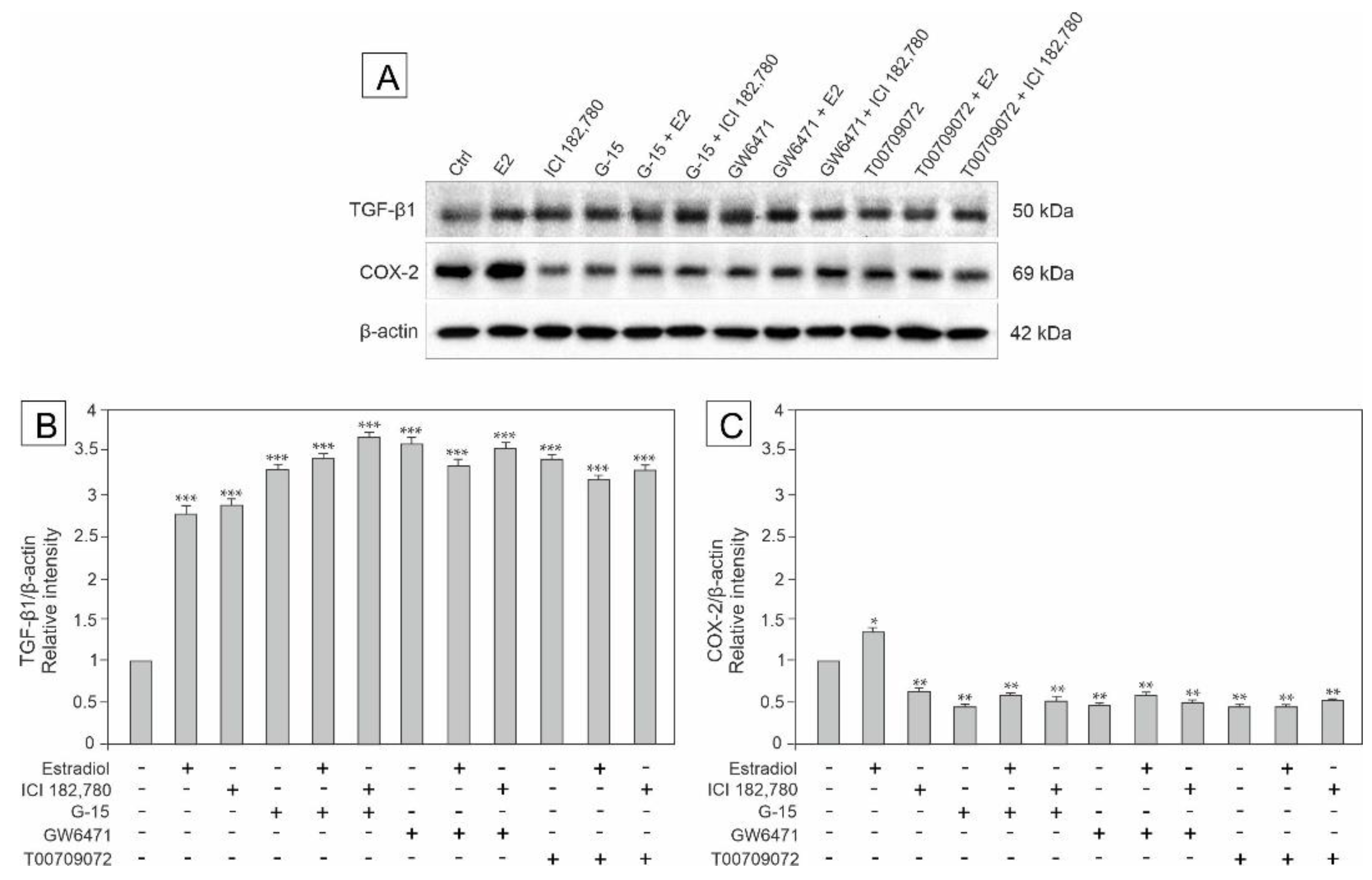
3.3. Effect of GPER Agonist, GPER and PPAR Antagonists, and Estradiol on the Expression of TGF-β, HIF-1α, VEGF and COX-2 in Mouse Testis Explants
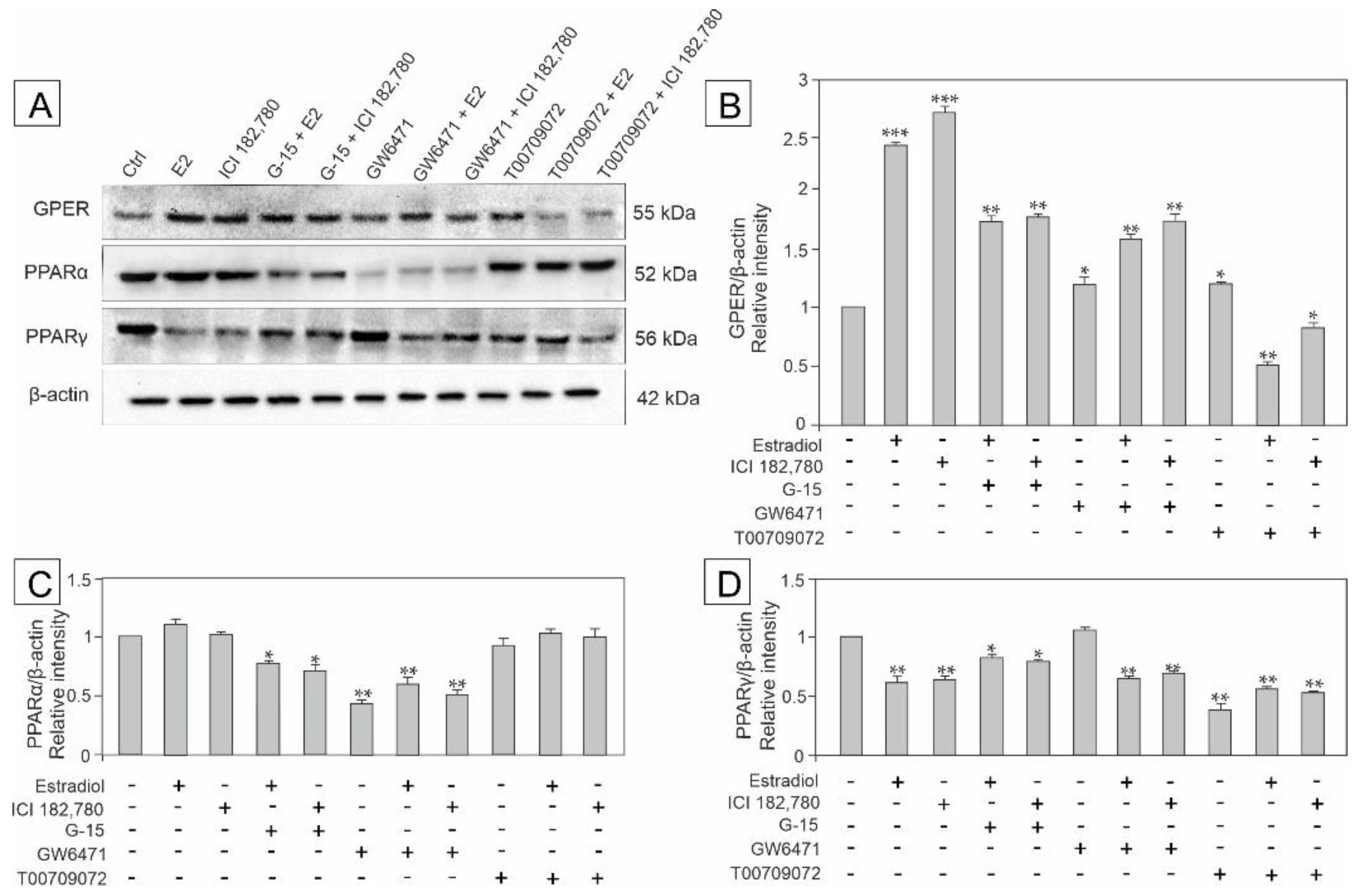
3.4. Effect of Estradiol, GPER Agonist, and GPER and PPAR Antagonists on the Expression of GPER, PPARα and PPARγ in MA-10 Cells
3.5. Effect of Estradiol, GPER Agonist, GPER and PPAR Antagonists on the Expression of Raf-1, ERK1/2, PI3Kp85, and Akt Signaling Kinases in MA-10 Cells
3.6. Effect of Estradiol, GPER Agonist, and GPER and PPAR Antagonists on the Expression of TGF-β and COX-2 in MA-10 Cells
3.7. Effect of Estradiol, GPER Agonist, and GPER and PPAR Antagonists on the Expression of Aromatase (P450arom) in MA-10 Cells
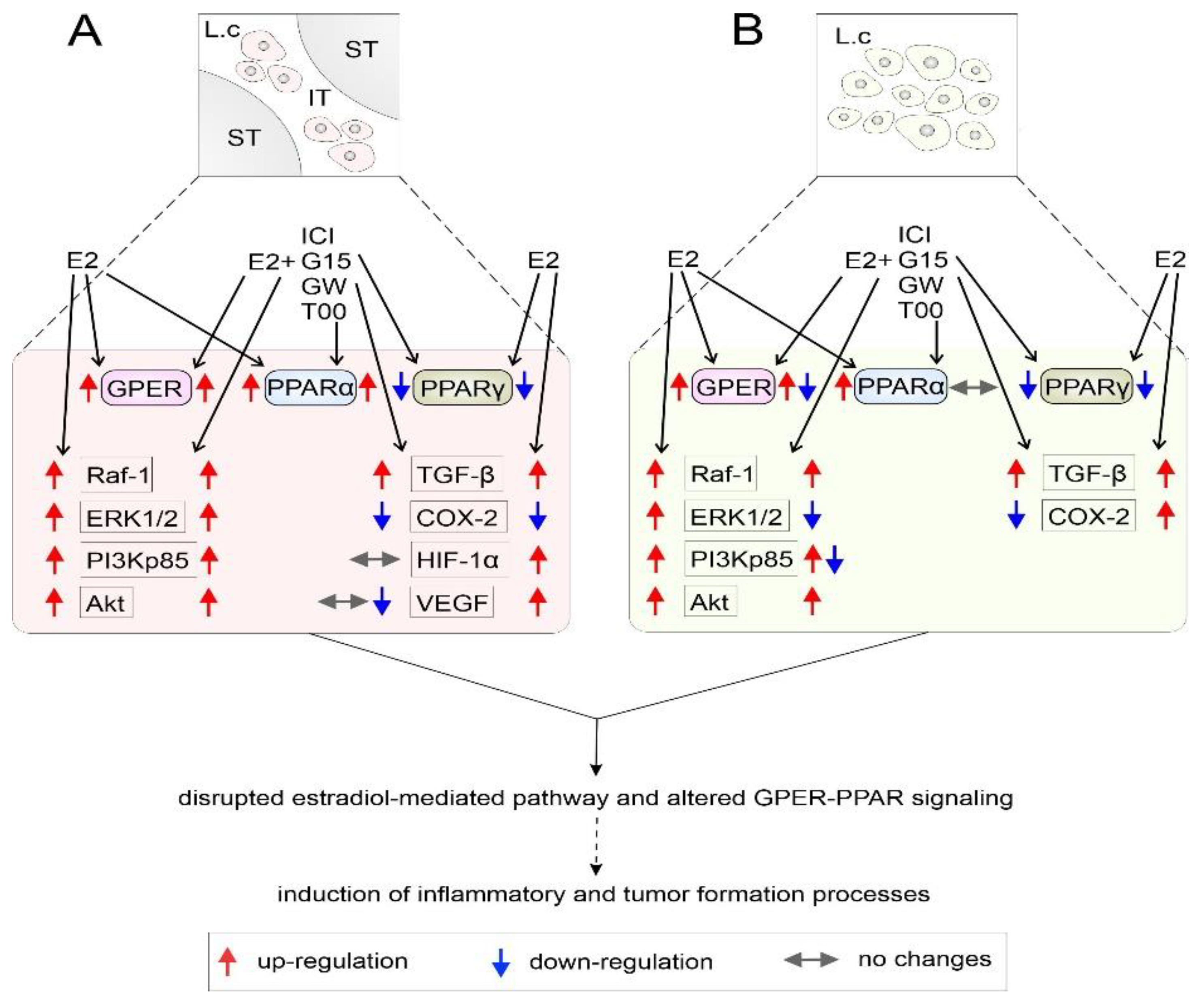
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faltas, C.L.; LeBron, A.K.; Holz, M.K. Unconventional Estrogen Signaling in Health and Disease. Endocrinology 2020, 161, bqaa030. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, A.; Mulder, E.; Lamers-Stahlhofen, G.; Mechielsen, M.; Van Der Molen, H. An Oestradiol receptor in rat testis interstitial tissue. FEBS Lett. 1972, 26, 301–305. [Google Scholar] [CrossRef]
- Abney, O.T. The potential roles of estrogens in regulating Leydig cell development and function: A review. Steroids 1999, 64, 610–617. [Google Scholar] [CrossRef]
- Ramasamy, R.; Schulster, M.; Bernie, A.M. The role of estradiol in male reproductive function. Asian J. Androl. 2016, 18, 435–440. [Google Scholar] [CrossRef]
- Bilińska, B.; Schmalz-Frączek, B.; Sadowska, J.; Carreau, S. Localization of cytochrome P450 aromatase and estrogen receptors α and β in testicular cells—An immunohistochemical study of the bank vole. Acta Histochem. 2000, 102, 167–181. [Google Scholar] [CrossRef]
- Lazari, M.F.M.; Lucas, T.F.G.; Yasuhara, F.; Gomes, G.R.O.; Siu, E.R.; Royer, C.; Fernandes, S.A.F.; Porto, C.S. Estrogen receptors and function in the male reproductive system. Arq. Bras. Endocrinol. Metabol. 2009, 53, 923–933. [Google Scholar] [CrossRef]
- Chimento, A.; Sirianni, R.; Casaburi, I.; Pezzi, V. GPER signaling in spermatogenesis and testicular tumors. Front. Endocrinol. 2014, 5, 30. [Google Scholar] [CrossRef]
- Chen, B.; Chen, D.; Jiang, Z.; Li, J.; Liu, S.; Dong, Y.; Yao, W.; Akingbemi, B.; Ge, R.; Li, X. Effects of Estradiol and Methoxychlor on Leydig Cell Regeneration in the Adult Rat Testis. Int. J. Mol. Sci. 2014, 15, 7812–7826. [Google Scholar] [CrossRef]
- Tamer, S.A.; Yıldırım, A.; Arabacı, Ş.; Çiftçi, S.; Akın, S.; Sarı, E.; Köroğlu, M.K.; Ercan, F.; Yüksel, M.; Çevik, Ö. Treatment with estrogen receptor agonist ERβ improves torsion-induced oxidative testis injury in rats. Life Sci. 2019, 222, 203–211. [Google Scholar] [CrossRef]
- Zanatta, A.P.; Gonçalves, R.; da Silva, F.O.; Pedrosa, R.C.; Zanatta, L.; Bouraïma-Lelong, H.; Delalande, C.; Silva, F.R.M.B. Estradiol and 1α,25(OH)2 vitamin D3 share plasma membrane downstream signal transduction through calcium influx and genomic activation in immature rat testis. Theriogenology 2021, 172, 36–46. [Google Scholar] [CrossRef]
- Owman, C.; Blay, P.; Nilsson, C.; Lolait, S.J. Cloning of Human cDNA Encoding a Novel Heptahelix Receptor Expressed in Burkitt’s Lymphoma and Widely Distributed in Brain and Peripheral Tissues. Biochem. Biophys. Res. Commun. 1996, 228, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R. Identification of a Gene (GPR30) with Homology to the G-Protein-Coupled Receptor Superfamily Associated with Estrogen Receptor Expression in Breast Cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Kvingedal, A.M.; Smeland, E.B. A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 1997, 407, 59–62. [Google Scholar] [CrossRef]
- Olde, B.; Leeb-Lundberg, L.F. GPR30/GPER1: Searching for a role in estrogen physiology. Trends Endocrinol. Metab. 2009, 20, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Vaucher, L.; Funaro, M.G.; Mehta, A.; Mielnik, A.; Bolyakov, A.; Prossnitz, E.R.; Schlegel, P.N.; Paduch, D. Activation of GPER-1 Estradiol Receptor Downregulates Production of Testosterone in Isolated Rat Leydig Cells and Adult Human Testis. PLoS ONE 2014, 9, e92425. [Google Scholar] [CrossRef] [PubMed]
- Randeva, H.S.; Patel, V.H.; Chen, J.; Ramanjaneya, M.; Karteris, E.; Zachariades, E.; Thomas, P.; Been, M. G-protein coupled estrogen receptor 1 expression in rat and human heart: Protective role during ischaemic stress. Int. J. Mol. Med. 2010, 26, 193–199. [Google Scholar] [CrossRef]
- Recchia, A.G.; De Francesco, E.M.; Vivacqua, A.; Sisci, D.; Panno, M.L.; Andò, S.; Maggiolini, M. The G Protein-coupled Receptor 30 Is Up-regulated by Hypoxia-inducible Factor-1α (HIF-1α) in Breast Cancer Cells and Cardiomyocytes. J. Biol. Chem. 2011, 286, 10773–10782. [Google Scholar] [CrossRef]
- Hazell, G.G.; Yao, S.T.; Roper, J.A.; Prossnitz, E.R.; O’Carroll, A.-M.; Lolait, S.J. Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. J. Endocrinol. 2009, 202, 223–236. [Google Scholar] [CrossRef]
- Cheng, S.-B.; Dong, J.; Pang, Y.; LaRocca, J.; Hixon, M.; Thomas, P.; Filardo, E.J. Anatomical location and redistribution of G protein-coupled estrogen receptor-1 during the estrus cycle in mouse kidney and specific binding to estrogens but not aldosterone. Mol. Cell. Endocrinol. 2013, 382, 950–959. [Google Scholar] [CrossRef]
- Rago, V.; Romeo, F.; Giordano, F.; Maggiolini, M.; Carpino, A. Identification of the estrogen receptor GPER in neoplastic and non-neoplastic human testes. Reprod. Biol. Endocrinol. 2011, 9, 135. [Google Scholar] [CrossRef]
- Kotula-Balak, M.; Pawlicki, P.; Milon, A.; Tworzydlo, W.; Sekula, M.; Pacwa, A.; Gorowska-Wojtowicz, E.; Bilinska, B.; Pawlicka, B.; Wiater, J.; et al. The role of G-protein-coupled membrane estrogen receptor in mouse Leydig cell function—In vivo and in vitro evaluation. Cell Tissue Res. 2018, 374, 389–412. [Google Scholar] [CrossRef] [PubMed]
- Rago, V.; Giordano, F.; Brunelli, E.; Zito, D.; Aquila, S.; Carpino, A. Identification of G protein-coupled estrogen receptor in human and pig spermatozoa. J. Anat. 2014, 224, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Xiao, L.; Duan, H.; Jiang, Y.; Lv, J.; Ding, Z.; Hu, J.; Zhao, X.; Zhang, Y. Androgen receptor, aromatase, oestrogen receptor α/β and G protein-coupled receptor 30 expression in the testes and epididymides of adult sheep. Reprod. Domest. Anim. 2020, 55, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Haas, E.; Prossnitz, E.R.; Barton, M. Non-genomic regulation of vascular cell function and growth by estrogen. Mol. Cell. Endocrinol. 2009, 308, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Scaling, A.L.; Prossnitz, E.R.; Hathaway, H.J. GPER Mediates Estrogen-Induced Signaling and Proliferation in Human Breast Epithelial Cells and Normal and Malignant Breast. Horm. Cancer 2014, 5, 146–160. [Google Scholar] [CrossRef]
- Funder, J.W. GPR30, Mineralocorticoid Receptors, and the Rapid Vascular Effects of Aldosterone. Hypertension 2011, 57, 370–372. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER Mediates Activation of HIF1α/VEGF Signaling by Estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Hathaway, H.J. What have we learned about GPER function in physiology and disease from knockout mice? J. Steroid Biochem. Mol. Biol. 2015, 153, 114–126. [Google Scholar] [CrossRef]
- Gorowska-Wojtowicz, E.; Dutka, P.; Kudrycka, M.; Pawlicki, P.; Milon, A.; Plachno, B.J.; Tworzydlo, W.; Pardyak, L.; Kaminska, A.; Hejmej, A.; et al. Regulation of steroidogenic function of mouse Leydig cells: G-coupled membrane estrogen receptor and peroxisome proliferator-activated receptor partnership. J. Physiol. Pharmacol. 2018, 69, 373–390. [Google Scholar] [CrossRef]
- Gorowska-Wojtowicz, E.; Duliban, M.; Kudrycka, M.; Dutka, P.; Pawlicki, P.; Milon, A.; Zarzycka, M.; Placha, W.; Kotula-Balak, M.; Ptak, A.; et al. Leydig cell tumorigenesis—Implication of G-protein coupled membrane estrogen receptor, peroxisome proliferator-activated receptor and xenoestrogen exposure. In vivo and in vitro appraisal. Tissue Cell 2019, 61, 51–60. [Google Scholar] [CrossRef]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Latini, G.; Scoditti, E.; Verrotti, A.; De Felice, C.; Massaro, M. Peroxisome Proliferator-Activated Receptors as Mediators of Phthalate-Induced Effects in the Male and Female Reproductive Tract: Epidemiological and Experimental Evidence. PPAR Res. 2008, 2008, 359267. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Minireview: Extranuclear Steroid Receptors: Roles in Modulation of Cell Functions. Mol. Endocrinol. 2011, 25, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Kotula-Balak, M.; Gorowska-Wojtowicz, E.; Milon, A.; Pawlicki, P.; Tworzydlo, W.; Płachno, B.J.; Krakowska, I.; Hejmej, A.; Wolski, J.K.; Bilinska, B. Towards understanding leydigioma: Do G protein-coupled estrogen receptor and peroxisome proliferator–activated receptor regulate lipid metabolism and steroidogenesis in Leydig cell tumors? Protoplasma 2020, 257, 1149–1163. [Google Scholar] [CrossRef]
- Lee, S.J.; Yang, E.K.; Kim, S.G. Peroxisome Proliferator-Activated Receptor-γ and Retinoic Acid X Receptor α Represses theTGFβ1Gene via PTEN-Mediated p70 Ribosomal S6 Kinase-1 Inhibition: Role for Zf9 Dephosphorylation. Mol. Pharmacol. 2006, 70, 415–425. [Google Scholar] [CrossRef]
- Breit, S.N.; Johnen, H.; Cook, A.D.; Tsai, V.W.W.; Mohammad, M.G.; Kuffner, T.; Zhang, H.P.; Marquis, C.P.; Jiang, L.; Lockwood, G.; et al. The TGF-β superfamily cytokine, MIC-1/GDF15: A pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth Factors 2011, 29, 187–195. [Google Scholar] [CrossRef]
- Sanjabi, S.; Zenewicz, L.; Kamanaka, M.; Flavell, A.R. Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Bae, S.-C. TGF-β-dependent Cell Growth Arrest and Apoptosis. BMB Rep. 2002, 35, 47–53. [Google Scholar] [CrossRef]
- Neeraja, S.; Sreenath, A.; Reddy, P.; Reddanna, P. Expression of cyclooxygenase-2 in rat testis. Reprod. Biomed. Online 2003, 6, 302–309. [Google Scholar] [CrossRef]
- Singh, B.; Berry, A.J.; Shoher, A.; Ayers, G.D.; Wei, C.; Lucci, A. COX-2 involvement in breast cancer metastasis to bone. Oncogene 2007, 26, 3789–3796. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; Schilsky, R.L.; Dubois, R.N. Cyclooxygenase-2 and Cancer Treatment: Understanding the Risk Should Be Worth the Reward. Clin. Cancer Res. 2010, 16, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-M.; Sun, Y.-Z.; Sun, J.-M.; Ma, J.-Q.; Cheng, C. Protective role of quercetin against lead-induced inflammatory response in rat kidney through the ROS-mediated MAPKs and NF-κB pathway. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Echizen, K.; Hirose, O.; Maeda, Y.; Oshima, M. Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E 2 and Toll-like receptor/MyD88 pathways. Cancer Sci. 2016, 107, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.; Bologa, C.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef]
- An, G.; Li, W.; Yan, T.; Li, S. Estrogen Rapidly Enhances Incisional Pain of Ovariectomized Rats Primarily through the G Protein-Coupled Estrogen Receptor. Int. J. Mol. Sci. 2014, 15, 10479–10491. [Google Scholar] [CrossRef]
- Kang, W.-B.; Cong, Y.; Ru, J.-Y.; Ying, S.-Q.; Zhu, T.; Wang, N.-S.; Liu, X.-W.; Liu, G.; Zhao, J.-N. Osteoprotective effect of combination therapy of low-dose oestradiol with G15, a specific antagonist of GPR30/GPER in ovariectomy-induced osteoporotic rats. Biosci. Rep. 2015, 35, e00239. [Google Scholar] [CrossRef]
- Bilinska, B.; Hejmej, A.; Kotula-Balak, M. Preparation of Testicular Samples for Histology and Immunohistochemistry. In Sertoli Cells; Humana Press: New York, NY, USA, 2018; Volume 1748, pp. 17–36. [Google Scholar] [CrossRef]
- Kamińska, A.; Pardyak, L.; Marek, S.; Wróbel, K.; Kotula-Balak, M.; Bilinska, B.; Hejmej, A. Notch signaling regulates nuclear androgen receptor AR and membrane androgen receptor ZIP 9 in mouse Sertoli cells. Andrology 2020, 8, 457–472. [Google Scholar] [CrossRef]
- Pérez-Martínez, C.; Garcia-Iglesias, M.J.; Estrada, M.D.C.F.; Bravo-Moral, A.; Espinosa-Alvarez, J.; Escudero-Díez, A. Effects of in-utero exposure to zeranol or diethylstilboestrol on morphological development of the fetal testis in mice. J. Comp. Pathol. 1996, 114, 407–418. [Google Scholar] [CrossRef]
- Li, X.; Strauss, L.; Mäkelä, S.; Streng, T.; Huhtaniemi, I.; Santti, R.; Poutanen, M. Multiple Structural and Functional Abnormalities in the P450 Aromatase Expressing Transgenic Male Mice Are Ameliorated by a P450 Aromatase Inhibitor. Am. J. Pathol. 2004, 164, 1039–1048. [Google Scholar] [CrossRef]
- Maeda, T.; Itoh, N.; Kobayashi, K.; Takahashi, A.; Masumori, N.; Tsukamoto, T. Elevated serum estradiol suggesting recurrence of Leydig cell tumor nine years after radical orchiectomy. Int. J. Urol. 2002, 11, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Ruf, C.G.; Sanatgar, N.; Isbarn, H.; Ruf, B.; Simon, J.; Fankhauser, C.D.; Dieckmann, K.-P. Leydig-cell tumour of the testis: Retrospective analysis of clinical and therapeutic features in 204 cases. World J. Urol. 2020, 38, 2857–2862. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.A.; De Jong, F.H.; Teerds, K.; De Rooij, D.G.; Dieleman, S.J.; Van Sluijs, F.J. Ageing, testicular tumours and the pituitary-testis axis in dogs. J. Endocrinol. 2000, 166, 153–161. [Google Scholar] [CrossRef]
- Chimento, A.; De Luca, A.; Nocito, M.C.; Avena, P.; La Padula, D.; Zavaglia, L.; Pezzi, V. Role of GPER-Mediated Signaling in Testicular Functions and Tumorigenesis. Cells 2020, 9, 2115. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Graeber, C.T.; Frackelton, A.R.; Kim, M.; Schwarzbauer, J.E.; Filardo, E.J. Coordinate Regulation of Estrogen-Mediated Fibronectin Matrix Assembly and Epidermal Growth Factor Receptor Transactivation by the G Protein-Coupled Receptor, GPR30. Mol. Endocrinol. 2009, 23, 1052–1064. [Google Scholar] [CrossRef]
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 2019, 174, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Permezel, M.; Georgiou, H.; Rice, G. Nuclear Factor Kappa B Regulation of Proinflammatory Cytokines in Human Gestational Tissues In Vitro1. Biol. Reprod. 2002, 67, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ren, Y.; Dai, Z.-J.; Wu, C.-J.; Ji, Y.-H.; Xu, J. IL-6, IL-8 and TNF-α levels correlate with disease stage in breast cancer patients. Adv. Clin. Exp. Med. 2017, 26, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Yanase, T.; Morinaga, H.; Mu, Y.-M.; Nomura, M.; Okabe, T.; Goto, K.; Harada, N.; Nawata, H. Activation of Peroxisome Proliferator-Activated Receptor-γ and Retinoid X Receptor Inhibits Aromatase Transcription via Nuclear Factor-κB. Endocrinology 2005, 146, 85–92. [Google Scholar] [CrossRef]
- Roberts-Thomson, S.J.; Snyderwine, E.G. Characterization of peroxisome proliferator-activated receptor alpha in normal rat mammary gland and 2-amino-l-methyl-6-phenylimidazo[4,5-b]pyridine-induced mammary gland tumors from rats fed high and low fat diets. Toxicol. Lett. 2000, 118, 79–86. [Google Scholar] [CrossRef]
- Suchanek, K.M.; May, F.; Robinson, J.A.; Lee, W.J.; Holman, N.A.; Monteith, G.; Roberts-Thomson, S. Peroxisome proliferator-activated receptor α in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol. Carcinog. 2002, 34, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Tanaka, N.; Ichikawa, M.; Kamijo, Y.; Sugiyama, E.; Gonzalez, F.J.; Aoyama, T. PPARα-dependent cholesterol/testosterone disruption in Leydig cells mediates 2,4-dichlorophenoxyacetic acid-induced testicular toxicity in mice. Arch. Toxicol. 2016, 90, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Biegel, L.B.; Hurtt, M.E.; Frame, S.R.; O’Connor, J.C.; Cook, J.C. Mechanisms of Extrahepatic Tumor Induction by Peroxisome Proliferators in Male CD Rats. Toxicol. Sci. 2001, 60, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Balemans, W.; Van Hul, W. Extracellular regulation of BMP signaling in vertebrates: A cocktail of modulators. Dev. Biol. 2002, 250, 231–250. [Google Scholar] [CrossRef]
- Ingman, W.V.; Robertson, S.A. Defining the actions of transforming growth factor beta in reproduction. BioEssays 2002, 24, 904–914. [Google Scholar] [CrossRef]
- Itman, C.; Mendis, S.; Barakat, B.; Loveland, K.L. All in the family: TGF-β family action in testis development. Reproduction 2006, 132, 233–246. [Google Scholar] [CrossRef]
- Salama, N.; Tsuji, M.; Tamura, M.; Kagawa, S. Transforming growth factor (β1) in testes of aged and diabetic rats: Correlation with testicular function. Arch. Androl. 2001, 47, 217–226. [Google Scholar] [CrossRef]
- Dobashi, M.; Fujisawa, M.; Yamazaki, T.; Okada, H.; Kamidono, S. Distribution of intracellular and extracellular expression of transforming growth factor-beta1 (TGF- beta1) in human testis and their association with spermatogenesis. Asian J. Androl. 2002, 4, 105–109. [Google Scholar] [CrossRef]
- Richardsen, E.; Uglehus, R.D.; Due, J.; Busch, C.; Busund, L.-T. COX-2 is overexpressed in primary prostate cancer with metastatic potential and may predict survival. A comparison study between COX-2, TGF-β, IL-10 and Ki67. Cancer Epidemiol. 2010, 34, 316–322. [Google Scholar] [CrossRef]
- Jarrar, M.H.; Baranova, A. PPARγ activation by thiazolidinediones (TZDs) may modulate breast carcinoma outcome: The importance of interplay with TGFβ signalling. J. Cell. Mol. Med. 2007, 11, 71–87. [Google Scholar] [CrossRef]
- LeCarpentier, Y.; Gourrier, E.; Gobbert, V.; Vallée, A. Bronchopulmonary Dysplasia: Crosstalk Between PPARγ, WNT/β-Catenin and TGF-β Pathways; The Potential Therapeutic Role of PPARγ Agonists. Front. Pediatr. 2019, 7, 176. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-L.; Wang, H.; Wang, Z.-R.; Zhang, Y.-F.; Chen, Y.-Q.; Zhu, F.-H.; Zhang, Y.-Q.; Ma, J.; Li, Z. TGF-β1 Regulation of Estrogen Production in Mature Rat Leydig Cells. PLoS ONE 2013, 8, e60197. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.R.; Gonzalez, B.; Rulli, S.B.; dos Santos, M.L.; Costa, G.M.J.; França, L.R.; Calandra, R.S.; Gonzalez-Calvar, S.I. TGF-beta1 system in Leydig cells. part II: TGF-beta1 and progesterone, through Smad1/5, are involved in the hyperplasia/hypertrophy of Leydig cells. J. Reprod. Dev. 2010, 56, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.R.; Matzkin, E.M.; Frungieri, M.B.; Terradas, C.; Ponzio, R.; Puigdomenech, E.; Levalle, O.; Calandra, R.S.; Gonzalez-Calvar, I.S. Expression of the TGF-beta1 system in human testicular pathologies. Reprod. Biol. Endocrinol. 2010, 8, 148. [Google Scholar] [CrossRef]
- Bellone, G.; Gramigni, C.; Vizio, B.; Mauri, F.A.; Prati, A.; Solerio, D.; Dughera, L.; Ruffini, E.; Gasparri, G.; Camandona, M. Abnormal expression of Endoglin and its receptor complex (TGF-β1 and TGF-β receptor II) as early angiogenic switch indicator in premalignant lesions of the colon mucosa. Int. J. Oncol. 2010, 37, 1153–1165. [Google Scholar] [CrossRef]
- Soufla, G.; Sifakis, S.; Baritaki, S.; Zafiropoulos, A.; Koumantakis, E.; Spandidos, D.A. VEGF, FGF2, TGFB1 and TGFBR1 mRNA expression levels correlate with the malignant transformation of the uterine cervix. Cancer Lett. 2005, 221, 105–118. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Qu, Y.; Wang, L.; Geng, D.; Chen, W.; Li, L.; Tian, Y.; Chang, S.; Zhao, C.; et al. The roles of p38 MAPK → COX2 and NF-κB → COX2 signal pathways in age-related testosterone reduction. Sci. Rep. 2019, 9, 10556. [Google Scholar] [CrossRef]
- Ishikawa, T.; Hwang, K.; Lazzarino, D.; Morris, P.L. Sertoli Cell Expression of Steroidogenic Acute Regulatory Protein-Related Lipid Transfer 1 and 5 Domain-Containing Proteins and Sterol Regulatory Element Binding Protein-1 Are Interleukin-1β Regulated by Activation of c-Jun N-Terminal Kinase and Cyclooxygenase-2 and Cytokine Induction. Endocrinology 2005, 146, 5100–5111. [Google Scholar] [CrossRef]
- Onorato, T.M.; Brown, P.W.; Morris, P.L. Mono-(2-ethylhexyl) Phthalate Increases Spermatocyte Mitochondrial Peroxiredoxin 3 and Cyclooxygenase 2. J. Androl. 2008, 29, 293–303. [Google Scholar] [CrossRef]
- Sirianni, R.; Chimento, A.; De Luca, A.; Zolea, F.; Carpino, A.; Rago, V.; Maggiolini, M.; Andò, S.; Pezzi, V. Inhibition of Cyclooxygenase-2 Down-regulates Aromatase Activity and Decreases Proliferation of Leydig Tumor Cells. J. Biol. Chem. 2009, 284, 28905–28916. [Google Scholar] [CrossRef]
- Hermenegildo, P.J.O.A.A.C.C.; Oviedo, P.; Cano, A. Cyclooxygenases Regulation by Estradiol on Endothelium. Curr. Pharm. Des. 2006, 12, 205–215. [Google Scholar] [CrossRef]
- Kirkpatrick, K.; Ogunkolade, W.; Elkak, A.; Bustin, S.; Jenkins, P.; Ghilchik, M.; Mokbel, K. The mRNA Expression of Cyclo-oxygenase-2 (COX-2) and Vascular Endothelial Growth Factor (VEGF) in Human Breast Cancer. Curr. Med. Res. Opin. 2002, 18, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.A.; Molitoris, K.H.; Koos, R.D. Estrogen Rapidly Activates the PI3K/AKT Pathway and Hypoxia-Inducible Factor 1 and Induces Vascular Endothelial Growth Factor A Expression in Luminal Epithelial Cells of the Rat Uterus1. Biol. Reprod. 2009, 81, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.D.; Vouret-Craviari, V.; Pouysségur, J. Angiogenesis and G-protein-coupled receptors: Signals that bridge the gap. Oncogene 2001, 20, 1556–1562. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15, R64. [Google Scholar] [CrossRef]
- Zhang, L.; Xiong, W.; Li, N.; Liu, H.; He, H.; Du, Y.; Zhang, Z.; Liu, Y. Estrogen stabilizes hypoxia-inducible factor 1α through G protein-coupled estrogen receptor 1 in eutopic endometrium of endometriosis. Fertil. Steril. 2017, 107, 439–447. [Google Scholar] [CrossRef]
- Bustos, V.; Nolan, Á.M.; Nijhuis, A.; Harvey, H.; Parker, A.; Poulsom, R.; McBryan, J.; Thomas, W.; Silver, A.; Harvey, B. GPER mediates differential effects of estrogen on colon cancer cell proliferation and migration under normoxic and hypoxic conditions. Oncotarget 2017, 8, 84258–84275. [Google Scholar] [CrossRef]
- Wei, W.; Chen, Z.-J.; Zhang, K.-S.; Yang, X.; Wu, Y.-M.; Chen, X.-H.; Huang, H.-B.; Liu, H.-L.; Cai, S.-H.; Du, J.; et al. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1428. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef]
- Guan, B.-Z.; Yan, R.-L.; Huang, J.-W.; Li, F.-L.; Zhong, Y.-X.; Chen, Y.; Liu, F.-N.; Hu, B.; Huang, S.-B.; Yin, L.-H. Activation of G protein coupled estrogen receptor (GPER) promotes the migration of renal cell carcinoma via the PI3K/AKT/MMP-9 signals. Cell Adhes. Migr. 2018, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Lee, M.Y.; Ryu, J.M.; Song, C.H.; Han, H.J. Role of HIF-1α and VEGF in human mesenchymal stem cell proliferation by 17β-estradiol: Involvement of PKC, PI3K/Akt, and MAPKs. Am. J. Physiol. Physiol. 2009, 296, C317–C326. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.A.; Koos, R.D. Estrogen-Induced Activation of Hypoxia-Inducible Factor-1α, Vascular Endothelial Growth Factor Expression, and Edema in the Uterus Are Mediated by the Phosphatidylinositol 3-Kinase/Akt Pathway. Endocrinology 2007, 148, 2363–2374. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host Species | Dilution | Vendor | Cat. No. |
|---|---|---|---|---|
| GPER | Rabbit | 1:500 (WB) | Abcam (Cambridge, UK) | 39742 |
| PPARα | Mouse | 1:500 (WB) | Thermo Fisher Scientific (Waltham, MA, USA) | MA1–822 |
| PPARγ | Rabbit | 1:500 (WB) | Abcam | 209350 |
| Raf-1 | Rabbit | 1:200 (WB) | Santa Cruz Biotechnology (Dallas, TX, USA) | sc-133 |
| ERK1/2 | Rabbit | 1:500 (WB) | Cell Signaling Technology (Danvers, MA, USA) | 9102 |
| PI3Kp85 | Rabbit | 1:500 (WB) | Cell Signaling Technology | 4292 |
| Akt | Rabbit | 1:500 (WB) | Cell Signaling Technology | 9272S |
| TGF-β | Mouse | 1:500 (WB) | Thermo Fisher Scientific | MA5-15065 |
| COX-2 | Rabbit | 1:500 (WB) | Abcam | ab15191 |
| VEGF | Rabbit | 1:200 (WB) | Thermo Fisher Scientific | PA1-21796 |
| HIF-1α | Mouse | 1:500 (WB) | Thermo Fisher Scientific | PA1-16601 |
| P450arom | Mouse | 1:500 (WB) 1:50 (IF) | Bio Rad Labs | MCA2077S |
| β-actin | Mouse | 1:3000 (WB) | Sigma–Aldrich | A2228 |
| β-tubulin | Rabbit | 1:500 (WB) | Sigma–Aldrich | T2200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorowska-Wojtowicz, E.; Duliban, M.; Kotula-Balak, M.; Bilinska, B. Modulatory Effects of Estradiol and Its Mixtures with Ligands of GPER and PPAR on MAPK and PI3K/Akt Signaling Pathways and Tumorigenic Factors in Mouse Testis Explants and Mouse Tumor Leydig Cells. Biomedicines 2022, 10, 1390. https://doi.org/10.3390/biomedicines10061390
Gorowska-Wojtowicz E, Duliban M, Kotula-Balak M, Bilinska B. Modulatory Effects of Estradiol and Its Mixtures with Ligands of GPER and PPAR on MAPK and PI3K/Akt Signaling Pathways and Tumorigenic Factors in Mouse Testis Explants and Mouse Tumor Leydig Cells. Biomedicines. 2022; 10(6):1390. https://doi.org/10.3390/biomedicines10061390
Chicago/Turabian StyleGorowska-Wojtowicz, Ewelina, Michal Duliban, Malgorzata Kotula-Balak, and Barbara Bilinska. 2022. "Modulatory Effects of Estradiol and Its Mixtures with Ligands of GPER and PPAR on MAPK and PI3K/Akt Signaling Pathways and Tumorigenic Factors in Mouse Testis Explants and Mouse Tumor Leydig Cells" Biomedicines 10, no. 6: 1390. https://doi.org/10.3390/biomedicines10061390
APA StyleGorowska-Wojtowicz, E., Duliban, M., Kotula-Balak, M., & Bilinska, B. (2022). Modulatory Effects of Estradiol and Its Mixtures with Ligands of GPER and PPAR on MAPK and PI3K/Akt Signaling Pathways and Tumorigenic Factors in Mouse Testis Explants and Mouse Tumor Leydig Cells. Biomedicines, 10(6), 1390. https://doi.org/10.3390/biomedicines10061390