
Targeting the Tumor Microenvironment in Acute Myeloid Leukemia: The Future of Immunotherapy and Natural Products

 , , ,
, , ,
Abstract
:1. Introduction
2. Composition of the AML Tumor Microenvironment
2.1. Mesenchymal Stromal Cells
2.2. Conventional and Regulatory T Cells
2.3. Natural Killer Cells
2.4. Myeloid-Derived Suppressor Cells and Tumor- Associated Macrophages
2.5. Soluble Environmental Factors
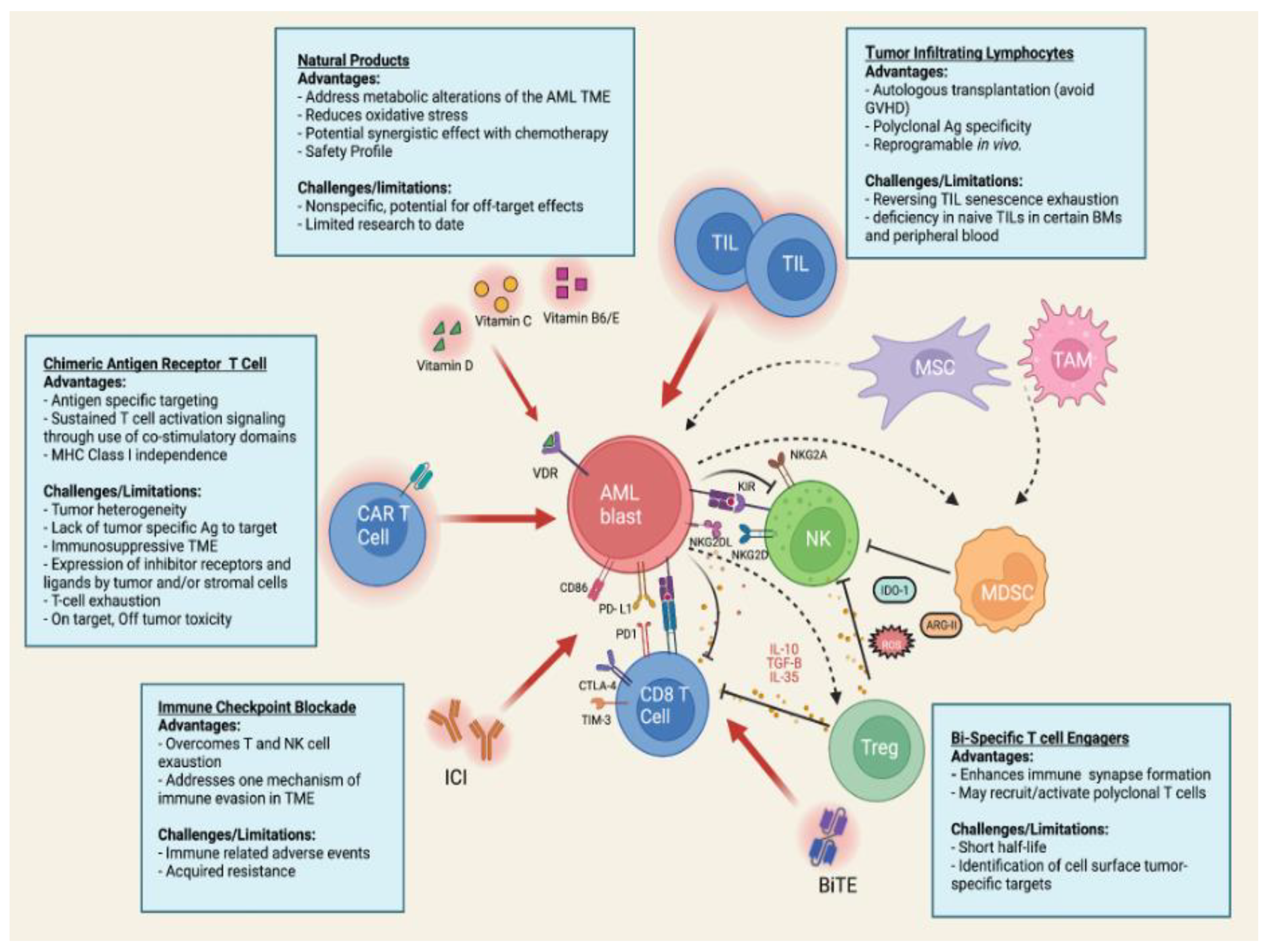
3. Targeting the Tumor Microenvironment Using Immunotherapy
3.1. Specific Alterations in TME by AML Blasts
3.2. Strategies to Overcome AML Resistance to Immunotherapy
3.3. Immunomodulation as a Hallmark of Cancer
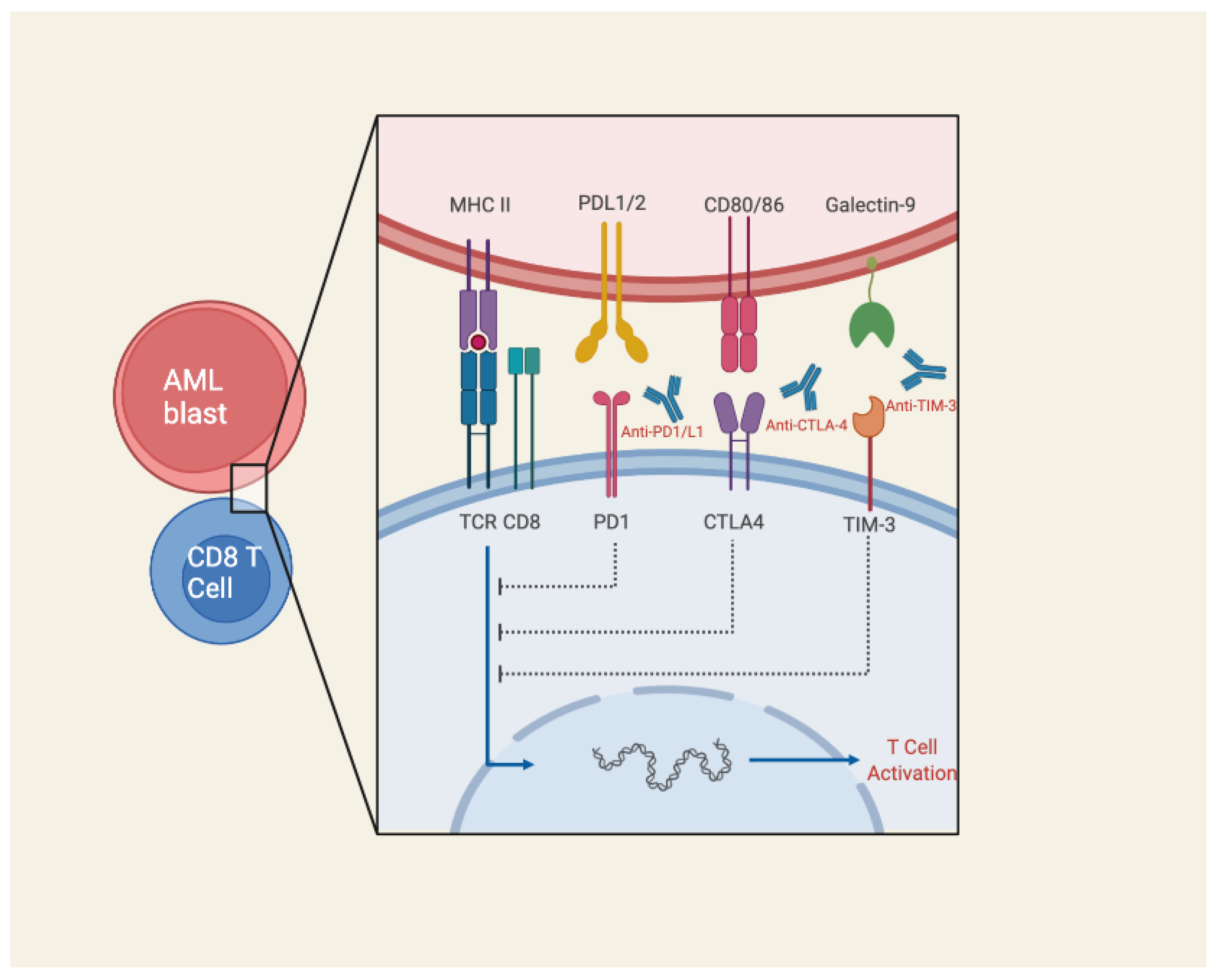
3.4. Immune Checkpoint Blockade
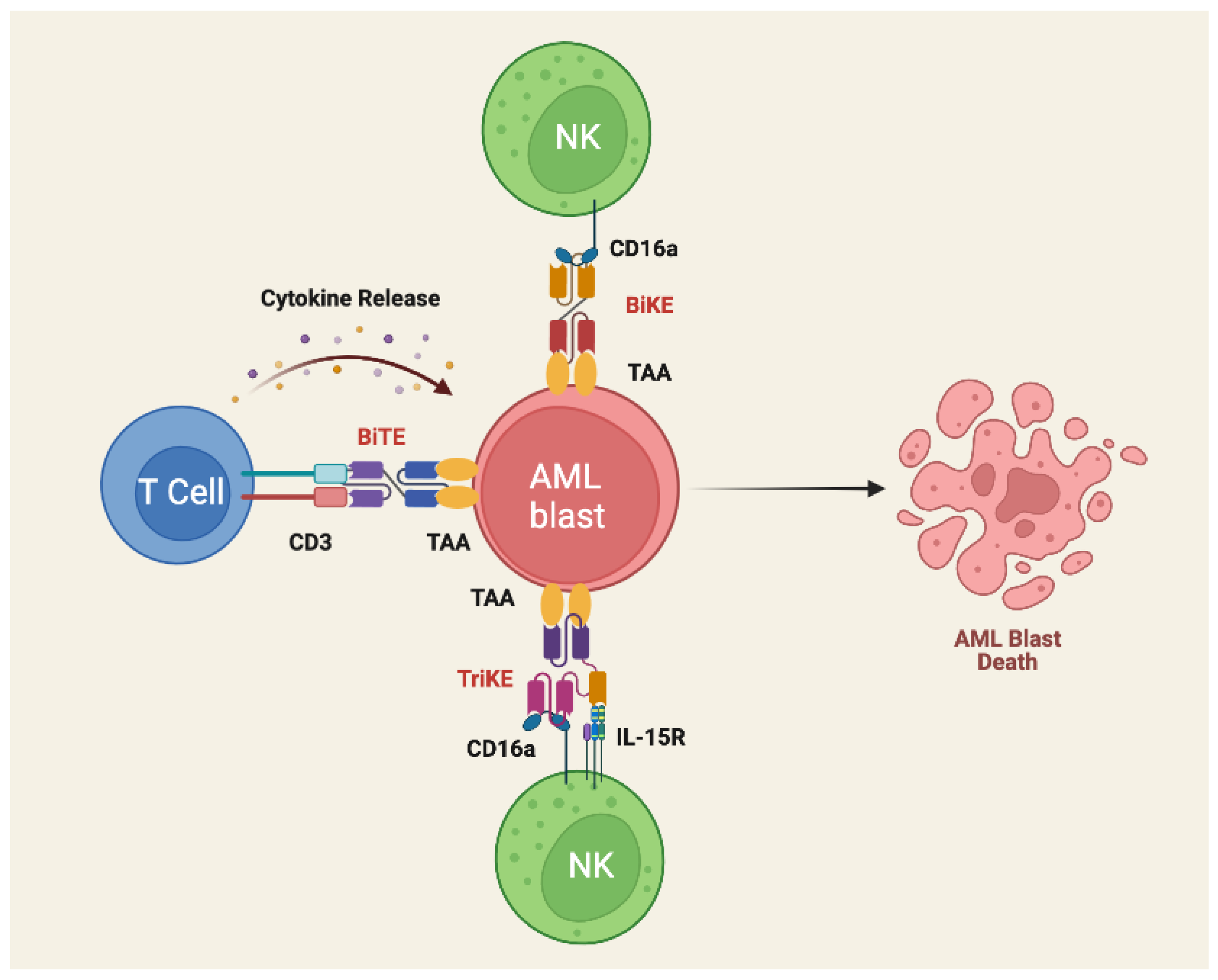
3.5. Bispecific T- Cell Engagers
3.6. Tumor Infiltrating Lymphocytes
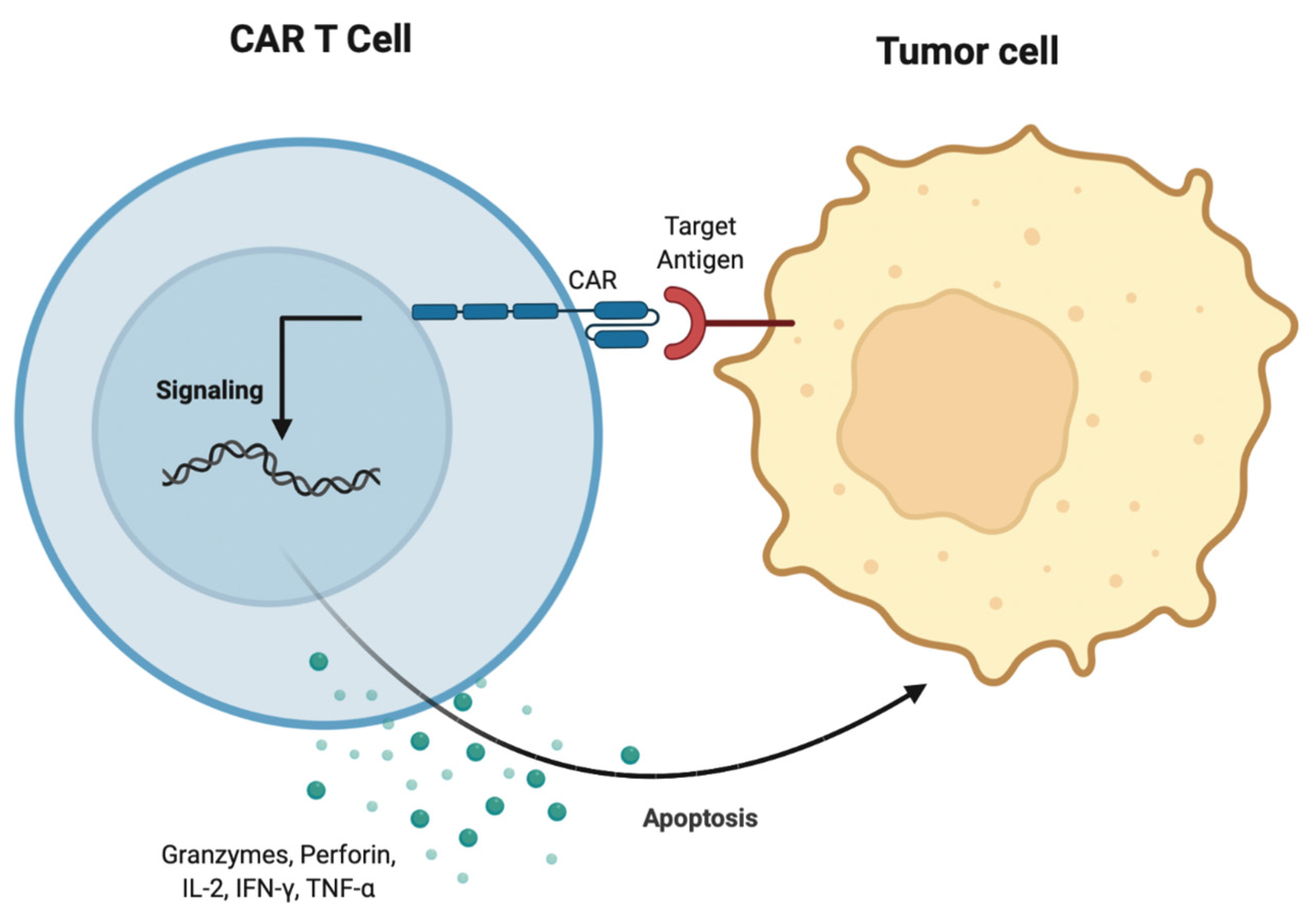
3.7. CAR T Cell Therapy
4. Application of Natural Products in AML Therapy
4.1. Vitamin C
4.2. Vitamin D
4.3. Vitamin B6 and Vitamin E
4.4. Other Natural Products
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Lowenberg, B.; Downing, J.R.; Burnett, A. Acute Myeloid Leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute Myeloid Leukemia: Current Progress and Future Directions. Blood Cancer J. 2021, 11, 1–25. [Google Scholar] [CrossRef]
- Casey, S.C.; Amedei, A.; Aquilano, K.; Azmi, A.S.; Benencia, F.; Bhakta, D.; Bilsland, A.E.; Boosani, C.S.; Chen, S.; Ciriolo, M.R.; et al. Cancer Prevention and Therapy through the Modulation of the Tumor Microenvironment. Semin. Cancer Biol. 2015, 35, S199–S223. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Dorshkind, K. Regulation of Hemopoiesis by Bone Marrow Stromal Cells and Their Products. Annu. Rev. Immunol. 1990, 8, 111–137. [Google Scholar] [CrossRef]
- Dexter, T.M. Regulation of Hemopoietic Cell Growth and Development: Experimental and Clinical Studies. Leukemia 1989, 3, 469–474. [Google Scholar]
- Saleh, M.; Shamsasanjan, K.; Movassaghpourakbari, A.; Akbarzadehlaleh, P.; Molaeipour, Z. The Impact of Mesenchymal Stem Cells on Differentiation of Hematopoietic Stem Cells. Adv. Pharm. Bull. 2015, 5, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Pittenger, M.F. Human Mesenchymal Stem Cells Modulate Allogeneic Immune Cell Responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Muguruma, Y.; Yahata, T.; Miyatake, H.; Sato, T.; Uno, T.; Itoh, J.; Kato, S.; Ito, M.; Hotta, T.; Ando, K. Reconstitution of the Functional Human Hematopoietic Microenvironment Derived from Human Mesenchymal Stem Cells in the Murine Bone Marrow Compartment. Blood 2006, 107, 1878–1887. [Google Scholar] [CrossRef]
- Garrido, S.M.; Appelbaum, F.R.; Willman, C.L.; Banker, D.E. Acute Myeloid Leukemia Cells Are Protected from Spontaneous and Drug-Induced Apoptosis by Direct Contact with a Human Bone Marrow Stromal Cell Line (HS-5). Exp. Hematol. 2001, 29, 448–457. [Google Scholar] [CrossRef]
- Chen, P.; Jin, Q.; Fu, Q.; You, P.; Jiang, X.; Yuan, Q.; Huang, H. Induction of Multidrug Resistance of Acute Myeloid Leukemia Cells by Cocultured Stromal Cells via Upregulation of the PI3K/Akt Signaling Pathway. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2016, 24, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Moshaver, B.; van der Pol, M.A.; Westra, A.H.; Ossenkoppele, G.J.; Zweegman, S.; Schuurhuis, G.J. Chemotherapeutic Treatment of Bone Marrow Stromal Cells Strongly Affects Their Protective Effect on Acute Myeloid Leukemia Cell Survival. Leuk. Lymphoma 2008, 49, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Nepstad, I.; Bruserud, Ø. Mesenchymal Stem Cells Support Survival and Proliferation of Primary Human Acute Myeloid Leukemia Cells through Heterogeneous Molecular Mechanisms. Front. Immunol. 2017, 8, 1331. [Google Scholar] [CrossRef] [Green Version]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The Yin and Yang of the Bone Marrow Microenvironment: Pros and Cons of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef] [Green Version]
- Civini, S.; Jin, P.; Ren, J.; Sabatino, M.; Castiello, L.; Jin, J.; Wang, H.; Zhao, Y.; Marincola, F.; Stroncek, D. Leukemia Cells Induce Changes in Human Bone Marrow Stromal Cells. J. Transl. Med. 2013, 11, 298. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Yu, Y.; Perlaky, L.; Man, T.-K.; Redell, M.S. Stromal CYR61 Confers Resistance to Mitoxantrone via Spleen Tyrosine Kinase Activation in Human Acute Myeloid Leukaemia. Br. J. Haematol. 2015, 170, 704–718. [Google Scholar] [CrossRef]
- Konopleva, M.; Konoplev, S.; Hu, W.; Zaritskey, A.; Afanasiev, B.; Andreeff, M. Stromal Cells Prevent Apoptosis of AML Cells by Up-Regulation of Anti-Apoptotic Proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef] [Green Version]
- Gynn, L.E.; Anderson, E.; Robinson, G.; Wexler, S.A.; Upstill-Goddard, G.; Cox, C.; May, J.E. Primary Mesenchymal Stromal Cells in Co-Culture with Leukaemic HL-60 Cells Are Sensitised to Cytarabine-Induced Genotoxicity, While Leukaemic Cells Are Protected. Mutagenesis 2021, 36, 419–428. [Google Scholar] [CrossRef]
- Buggins, A.G.S.; Milojkovic, D.; Arno, M.J.; Lea, N.C.; Mufti, G.J.; Thomas, N.S.B.; Hirst, W.J.R. Microenvironment Produced by Acute Myeloid Leukemia Cells Prevents T Cell Activation and Proliferation by Inhibition of NF-ΚB, c-Myc, and PRb Pathways. J. Immunol. 2001, 167, 6021–6030. [Google Scholar] [CrossRef] [Green Version]
- Le Dieu, R.; Taussig, D.C.; Ramsay, A.G.; Mitter, R.; Miraki-Moud, F.; Fatah, R.; Lee, A.M.; Lister, T.A.; Gribben, J.G. Peripheral Blood T Cells in Acute Myeloid Leukemia (AML) Patients at Diagnosis Have Abnormal Phenotype and Genotype and Form Defective Immune Synapses with AML Blasts. Blood 2009, 114, 3909–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanski, M.J.; Szajnik, M.; Czystowska, M.; Mandapathil, M.; Strauss, L.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Increased Frequency and Suppression by Regulatory T Cells in Patients with Acute Myelogenous Leukemia. Clin. Cancer Res. 2009, 15, 3325–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Xu, Y. Clinical Significance of Treg Cell Frequency in Acute Myeloid Leukemia. Int. J. Hematol. 2013, 98, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Shenghui, Z.; Yixiang, H.; Jianbo, W.; Kang, Y.; Laixi, B.; Yan, Z.; Xi, X. Elevated Frequencies of CD4+CD25+CD127lo Regulatory T Cells Is Associated to Poor Prognosis in Patients with Acute Myeloid Leukemia. Int. J. Cancer 2011, 129, 1373–1381. [Google Scholar] [CrossRef]
- Zhou, Q.; Bucher, C.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Munn, D.H.; Levine, B.L.; Riddle, M.; June, C.H.; Vallera, D.A.; et al. Depletion of Endogenous Tumor-Associated Regulatory T Cells Improves the Efficacy of Adoptive Cytotoxic T-Cell Immunotherapy in Murine Acute Myeloid Leukemia. Blood 2009, 114, 3793–3802. [Google Scholar] [CrossRef]
- Tao, Q.; Pan, Y.; Wang, Y.; Wang, H.; Xiong, S.; Li, Q.; Wang, J.; Tao, L.; Wang, Z.; Wu, F.; et al. Regulatory T Cells-Derived IL-35 Promotes the Growth of Adult Acute Myeloid Leukemia Blasts: IL-35 in the Pathogenesis of AML. Int. J. Cancer 2015, 137, 2384–2393. [Google Scholar] [CrossRef]
- Carlsten, M.; Järås, M. Natural Killer Cells in Myeloid Malignancies: Immune Surveillance, NK Cell Dysfunction, and Pharmacological Opportunities to Bolster the Endogenous NK Cells. Front. Immunol. 2019, 10, 2357. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Mancusi, A.; Burchielli, E.; Aversa, F.; Martelli, M.F.; Velardi, A. Natural Killer Cell Alloreactivity in Allogeneic Hematopoietic Transplantation. Curr. Opin. Oncol. 2007, 19, 142–147. [Google Scholar] [CrossRef]
- Khaznadar, Z.; Boissel, N.; Agaugué, S.; Henry, G.; Cheok, M.; Vignon, M.; Geromin, D.; Cayuela, J.-M.; Castaigne, S.; Pautas, C.; et al. Defective NK Cells in Acute Myeloid Leukemia Patients at Diagnosis Are Associated with Blast Transcriptional Signatures of Immune Evasion. J. Immunol. 2015, 195, 2580–2590. [Google Scholar] [CrossRef] [Green Version]
- Chretien, A.-S.; Fauriat, C.; Orlanducci, F.; Galseran, C.; Rey, J.; Bouvier Borg, G.; Gautherot, E.; Granjeaud, S.; Hamel-Broza, J.-F.; Demerle, C.; et al. Natural Killer Defective Maturation Is Associated with Adverse Clinical Outcome in Patients with Acute Myeloid Leukemia. Front. Immunol. 2017, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Cianga, V.A.; Campos Catafal, L.; Cianga, P.; Pavel Tanasa, M.; Cherry, M.; Collet, P.; Tavernier, E.; Guyotat, D.; Rusu, C.; Aanei, C.M. Natural Killer Cell Subpopulations and Inhibitory Receptor Dynamics in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Front. Immunol. 2021, 12, 665541. [Google Scholar] [CrossRef] [PubMed]
- Fauriat, C.; Just-Landi, S.; Mallet, F.; Arnoulet, C.; Sainty, D.; Olive, D.; Costello, R.T. Deficient Expression of NCR in NK Cells from Acute Myeloid Leukemia: Evolution during Leukemia Treatment and Impact of Leukemia Cells in NCRdull Phenotype Induction. Blood 2007, 109, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Mastaglio, S.; Wong, E.; Perera, T.; Ripley, J.; Blombery, P.; Smyth, M.J.; Koldej, R.; Ritchie, D. Natural Killer Receptor Ligand Expression on Acute Myeloid Leukemia Impacts Survival and Relapse after Chemotherapy. Blood Adv. 2018, 2, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D Ligands Defines Leukaemia Stem Cells and Mediates Their Immune Evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Zhang, Z.; Ju, Y.; Li, L.; Zhang, B.; Liu, B. Increase in Myeloid-Derived Suppressor Cells (MDSCs) Associated with Minimal Residual Disease (MRD) Detection in Adult Acute Myeloid Leukemia. Int. J. Hematol. 2015, 102, 579–586. [Google Scholar] [CrossRef]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1-Mediated Induction of Myeloid-Derived Suppressor Cells in Patients with Acute Myeloid Leukemia. Blood 2017, 129, 1791–1801. [Google Scholar] [CrossRef] [Green Version]
- Jitschin, R.; Saul, D.; Braun, M.; Tohumeken, S.; Völkl, S.; Kischel, R.; Lutteropp, M.; Dos Santos, C.; Mackensen, A.; Mougiakakos, D. CD33/CD3-Bispecific T-Cell Engaging (BiTE®) Antibody Construct Targets Monocytic AML Myeloid-Derived Suppressor Cells. J. Immunother. Cancer 2018, 6, 116. [Google Scholar] [CrossRef]
- Wang, L.; Jia, B.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Naik, S.; Khawaja, M.R.; Sivik, J.; Han, J.; et al. VISTA Is Highly Expressed on MDSCs and Mediates an Inhibition of T Cell Response in Patients with AML. OncoImmunology 2018, 7, e1469594. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Miari, K.E.; Guzman, M.L.; Wheadon, H.; Williams, M.T.S. Macrophages in Acute Myeloid Leukaemia: Significant Players in Therapy Resistance and Patient Outcomes. Front. Cell Dev. Biol. 2021, 9, 692800. [Google Scholar] [CrossRef] [PubMed]
- Al-Matary, Y.S.; Botezatu, L.; Opalka, B.; Hones, J.M.; Lams, R.F.; Thivakaran, A.; Schutte, J.; Koster, R.; Lennartz, K.; Schroeder, T.; et al. Acute Myeloid Leukemia Cells Polarize Macrophages towards a Leukemia Supporting State in a Growth Factor Independence 1 Dependent Manner. Haematologica 2016, 101, 1216–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Walker, M.R.; Kasprowicz, D.J.; Gersuk, V.H.; Bènard, A.; Van Landeghen, M.; Buckner, J.H.; Ziegler, S.F. Induction of FoxP3 and Acquisition of T Regulatory Activity by Stimulated Human CD4+CD25– T Cells. J. Clin. Investig. 2003, 112, 1437–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cools, N.; Van Tendeloo, V.F.I.; Smits, E.L.J.M.; Lenjou, M.; Nijs, G.; Van Bockstaele, D.R.; Berneman, Z.N.; Ponsaerts, P. Immunosuppression Induced by Immature Dendritic Cells Is Mediated by TGF-Beta/IL-10 Double-Positive CD4+ Regulatory T Cells. J. Cell. Mol. Med. 2008, 12, 690–700. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Tadros, V.; Hiramoto, B.; Leeper, K.; Hino, C.; Xiao, J.; Pham, B.; Kim, D.H.; Reeves, M.E.; Chen, C.-S.; et al. Targeting TKI-Activated NFKB2-MIF/CXCLs-CXCR2 Signaling Pathways in FLT3 Mutated Acute Myeloid Leukemia Reduced Blast Viability. Biomedicines 2022, 10, 1038. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, X.; Shen, J.; Yao, J. Macrophage Migration Inhibitory Factor in the Pathogenesis of Leukemia (Review). Int. J. Oncol. 2021, 59, 1–10. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Shafat, M.S.; Mehta, T.K.; Di Palma, F.; Lawes, M.J.; Rushworth, S.A.; Bowles, K.M. MIF-Induced Stromal PKCβ/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017, 77, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Pievani, A.; Biondi, M.; Tomasoni, C.; Biondi, A.; Serafini, M. Location First: Targeting Acute Myeloid Leukemia Within Its Niche. J. Clin. Med. 2020, 9, 1513. [Google Scholar] [CrossRef]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute Myeloid Leukemia Creates an Arginase-Dependent Immunosuppressive Microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Mussai, F.; Wheat, R.; Sarrou, E.; Booth, S.; Stavrou, V.; Fultang, L.; Perry, T.; Kearns, P.; Cheng, P.; Keeshan, K.; et al. Targeting the Arginine Metabolic Brake Enhances Immunotherapy for Leukaemia. Int. J. Cancer 2019, 145, 2201–2208. [Google Scholar] [CrossRef]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do Reactive Oxygen Species Play a Role in Myeloid Leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.J.; Davies, S.; Darley, R.L.; Tonks, A. Reactive Oxygen Species Rewires Metabolic Activity in Acute Myeloid Leukemia. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Sillar, J.R.; Germon, Z.P.; DeIuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [Green Version]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch Me If You Can: How AML and Its Niche Escape Immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Vasold, J.; Wagner, M.; Drolle, H.; Deniffel, C.; Kütt, A.; Oostendorp, R.; Sironi, S.; Rieger, C.; Fiegl, M. The Bone Marrow Microenvironment Is a Critical Player in the NK Cell Response against Acute Myeloid Leukaemia In Vitro. Leuk. Res. 2015, 39, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the Hallmarks of Cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Taghiloo, S.; Asgarian-Omran, H. Immune Evasion Mechanisms in Acute Myeloid Leukemia: A Focus on Immune Checkpoint Pathways. Crit. Rev. Oncol. Hematol. 2021, 157, 103164. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.N.; Pu, J.J. PD-1 Pathway and Its Clinical Application: A 20year Journey after Discovery of the Complete Human PD-1 Gene. Gene 2018, 638, 20–25. [Google Scholar] [CrossRef]
- Daver, N.; Basu, S.; Garcia-Manero, G.; Cortes, J.E.; Ravandi, F.; Ning, J.; Xiao, L.; Juliana, L.; Kornblau, S.M.; Konopleva, M.; et al. Defining the Immune Checkpoint Landscape in Patients (Pts) with Acute Myeloid Leukemia (AML). Blood 2016, 128, 2900. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Basu, S.; Garcia-Manero, G.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Hendrickson, S.; Pierce, S.; Ning, J.; Konopleva, M.; et al. Phase IB/II Study of Nivolumab in Combination with Azacytidine (AZA) in Patients (Pts) with Relapsed Acute Myeloid Leukemia (AML). Blood 2016, 128, 763. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson, P.A.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, Cytarabine, and Nivolumab in Patients with Newly Diagnosed Acute Myeloid Leukaemia or High-Risk Myelodysplastic Syndrome: A Single-Arm, Phase 2 Study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef]
- Schnorfeil, F.M.; Lichtenegger, F.S.; Emmerig, K.; Schlueter, M.; Neitz, J.S.; Draenert, R.; Hiddemann, W.; Subklewe, M. T Cells Are Functionally Not Impaired in AML: Increased PD-1 Expression Is Only Seen at Time of Relapse and Correlates with a Shift towards the Memory T Cell Compartment. J. Hematol. Oncol. 2015, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Goswami, M.; Oetjen, K.; Mulé, M.P.; Sheela, S.; Wong, H.Y.; Liu, Q.; Calvo, K.R.; Lai, C.E.; Hourigan, C.S. Increased Frequencies of PD-1+ CD8+ Marrow-Infiltrating Lymphocytes Associated with Highly Clonal T-Lymphocyte Expansions in Relapsed and Refractory AML Patients but Not Healthy Adults. Blood 2016, 128, 1644. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated Results from Phase I Dose-Escalation Study of AMG 330, a Bispecific T-Cell Engager Molecule, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134, 833. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as Salvage Immunotherapy for Refractory Acute Myeloid Leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Stock, W.; Foran, J.M.; Mawad, R.; Egan, D.; Blum, W.; Yang, A.; Pastore, A.; Johnson, C.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Watts, J.M.; Lin, T.L.; Mims, A.S.; Patel, P.; Shami, P.J.; Cull, E.H.; Cogle, C.R.; Lee, C.; Uckun, F. Tolerability and Single Agent Anti-Neoplastic Activity of the CD3xCD123 Bispecific Antibody APVO436 in Patients with Relapsed/Refractory AML or MDS. Blood 2021, 138, 3415. [Google Scholar] [CrossRef]
- Guy, D.G.; Uy, G.L. Bispecific Antibodies for the Treatment of Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2018, 13, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Zeidan, A.M.; Bewersdorf, J.P. BiTEs, DARTS, BiKEs and TriKEs—Are Antibody Based Therapies Changing the Future Treatment of AML? Life 2021, 11, 465. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 Bispecific Killer Cell Engager (BiKE) Activates NK Cells against Primary MDS and MDSC CD33+ Targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A New Approach to the Adoptive Immunotherapy of Cancer with Tumor-Infiltrating Lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Balch, C.M.; Riley, L.B.; Bae, Y.J.; Salmeron, M.A.; Platsoucas, C.D.; von Eschenbach, A.; Itoh, K. Patterns of Human Tumor-Infiltrating Lymphocytes in 120 Human Cancers. Arch. Surg. Chic. Ill 1960 1990, 125, 200–205. [Google Scholar] [CrossRef]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Müller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-Associated Lymphocytes as an Independent Predictor of Response to Neoadjuvant Chemotherapy in Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Gooden, M.J.M.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The Prognostic Influence of Tumour-Infiltrating Lymphocytes in Cancer: A Systematic Review with Meta-Analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Hanson, H.L.; Donermeyer, D.L.; Ikeda, H.; White, J.M.; Shankaran, V.; Old, L.J.; Shiku, H.; Schreiber, R.D.; Allen, P.M. Eradication of Established Tumors by CD8+ T Cell Adoptive Immunotherapy. Immunity 2000, 13, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The Immune Contexture in Cancer Prognosis and Treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The Prognostic Value of Infiltrating Immune Cells in Cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.-C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune Recognition of Somatic Mutations Leading to Complete Durable Regression in Metastatic Breast Cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevanović, S.; Helman, S.R.; Wunderlich, J.R.; Langhan, M.M.; Doran, S.L.; Kwong, M.L.M.; Somerville, R.P.T.; Klebanoff, C.A.; Kammula, U.S.; Sherry, R.M.; et al. A Phase II Study of Tumor-Infiltrating Lymphocyte Therapy for Human Papillomavirus–Associated Epithelial Cancers. Clin. Cancer Res. 2019, 25, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Noonan, K.A.; Huff, C.A.; Davis, J.; Lemas, M.V.; Fiorino, S.; Bitzan, J.; Ferguson, A.; Emerling, A.; Luznik, L.; Matsui, W.; et al. Adoptive Transfer of Activated Marrow-Infiltrating Lymphocytes Induces Measurable Antitumor Immunity in the Bone Marrow in Multiple Myeloma. Sci. Transl. Med. 2015, 7, 288ra78. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Reinoso, A.; Nehme-Álvarez, D.; Domínguez-Alonso, C.; Álvarez-Vallina, L. Synthetic TILs: Engineered Tumor-Infiltrating Lymphocytes with Improved Therapeutic Potential. Front. Oncol. 2021, 10, 593848. [Google Scholar] [CrossRef]
- Lizée, G.; Overwijk, W.W.; Radvanyi, L.; Gao, J.; Sharma, P.; Hwu, P. Harnessing the Power of the Immune System to Target Cancer. Annu. Rev. Med. 2013, 64, 71–90. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive Immunotherapy for Cancer: Harnessing the T Cell Response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef]
- Alonso-Camino, V.; Harwood, S.L.; Álvarez-Méndez, A.; Alvarez-Vallina, L. Efficacy and Toxicity Management of CAR-T-Cell Immunotherapy: A Matter of Responsiveness Control or Tumour-Specificity? Biochem. Soc. Trans. 2016, 44, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Kim, D.H.; Howard, A.; Moz, H.; Wasnik, S.; Baylink, D.J.; Chen, C.-S.; Reeves, M.E.; Mirshahidi, S.; Xiao, J.; et al. Ex Vivo Isolation, Expansion and Bioengineering of CCR7+CD95-/or CD62L+CD45RA+ Tumor Infiltrating Lymphocytes from Acute Myeloid Leukemia Patients’ Bone Marrow. Neoplasia 2021, 23, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- van den Broek, T.; Borghans, J.A.M.; van Wijk, F. The Full Spectrum of Human Naive T Cells. Nat. Rev. Immunol. 2018, 18, 363–373. [Google Scholar] [CrossRef] [PubMed]
- García-Guerrero, E.; Sánchez-Abarca, L.I.; Domingo, E.; Ramos, T.L.; Bejarano-García, J.A.; Gonzalez-Campos, J.A.; Caballero-Velázquez, T.; Pérez-Simón, J.A. Selection of Tumor-Specific Cytotoxic T Lymphocytes in Acute Myeloid Leukemia Patients Through the Identification of T-Cells Capable to Establish Stable Interactions with the Leukemic Cells: “Doublet Technology.”. Front. Immunol. 2018, 9, 1971. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Wang, Z.; Zhang, Z.; Li, Y.; Fan, S.; Zhao, Y.; Liu, Z.; Ye, X.; Zhang, F.; Yu, Y.; et al. Assessment of the Presence and Anti-Tumor Potential of Tumor-Infiltrating Lymphocytes in Patients with Acute Myeloid Leukemia. Cancer Manag. Res. 2019, 11, 3187–3196. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and Efficacy of Second Generation CAR T Cell Against the LeY Antigen in Acute Myeloid Leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Qian, C.; Xu, N.; Kang, L.; Dai, H.; Cui, W.; Song, B.; Yin, J.; Li, Z.; Zhu, X.; et al. CD38-Directed CAR-T Cell Therapy: A Novel Immunotherapy Strategy for Relapsed Acute Myeloid Leukemia after Allogeneic Hematopoietic Stem Cell Transplantation. J. Hematol. Oncol. J. Hematol. Oncol. 2021, 14, 82. [Google Scholar] [CrossRef]
- Peinert, S.; Prince, H.M.; Guru, P.M.; Kershaw, M.H.; Smyth, M.J.; Trapani, J.A.; Gambell, P.; Harrison, S.; Scott, A.M.; Smyth, F.E.; et al. Gene-Modified T Cells as Immunotherapy for Multiple Myeloma and Acute Myeloid Leukemia Expressing the Lewis Y Antigen. Gene Ther. 2010, 17, 678–686. [Google Scholar] [CrossRef] [Green Version]
- Marofi, F.; Rahman, H.S.; Al-Obaidi, Z.M.J.; Jalil, A.T.; Abdelbasset, W.K.; Suksatan, W.; Dorofeev, A.E.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; et al. Novel CAR T Therapy Is a Ray of Hope in the Treatment of Seriously Ill AML Patients. Stem Cell Res. Ther. 2021, 12, 465. [Google Scholar] [CrossRef]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and Levels of Cell Surface Expression of CD33 and CD123 in Acute Myeloid Leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical Targeting of Human Acute Myeloid Leukemia and Myeloablation Using Chimeric Antigen Receptor–Modified T Cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.Y.; Yu, K.-R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.-H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargett, T.; Brown, M.P. The Inducible Caspase-9 Suicide Gene System as a “Safety Switch” to Limit on-Target, off-Tumor Toxicities of Chimeric Antigen Receptor T Cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Mardiana, S.; Lai, J.; House, I.G.; Beavis, P.A.; Darcy, P.K. Switching on the Green Light for Chimeric Antigen Receptor T-cell Therapy. Clin. Transl. Immunol. 2019, 8, e1046. [Google Scholar] [CrossRef]
- Goswami, M.; Hourigan, C. Novel Antigen Targets for Immunotherapy of Acute Myeloid Leukemia. Curr. Drug Targets 2017, 18, 296–303. [Google Scholar] [CrossRef]
- Adamia, S.; Haibe-Kains, B.; Pilarski, P.M.; Bar-Natan, M.; Pevzner, S.; Avet-Loiseau, H.; Lode, L.; Verselis, S.; Fox, E.A.; Burke, J.; et al. A Genome-Wide Aberrant RNA Splicing in Patients with Acute Myeloid Leukemia Identifies Novel Potential Disease Markers and Therapeutic Targets. Clin. Cancer Res. 2014, 20, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Adamia, S.; Bar-Natan, M.; Haibe-Kains, B.; Pilarski, P.M.; Bach, C.; Pevzner, S.; Calimeri, T.; Avet-Loiseau, H.; Lode, L.; Verselis, S.; et al. NOTCH2 and FLT3 Gene Mis-Splicings Are Common Events in Patients with Acute Myeloid Leukemia (AML): New Potential Targets in AML. Blood 2014, 123, 2816–2825. [Google Scholar] [CrossRef] [Green Version]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-Targeted T Cells Mediate Potent Antitumor Effects against Acute Myeloid Leukemia and Multiple Myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef]
- Park, S. The Effects of High Concentrations of Vitamin C on Cancer Cells. Nutrients 2013, 5, 3496–3505. [Google Scholar] [CrossRef] [Green Version]
- Monti, D.A.; Mitchell, E.; Bazzan, A.J.; Littman, S.; Zabrecky, G.; Yeo, C.J.; Pillai, M.V.; Newberg, A.B.; Deshmukh, S.; Levine, M. Phase I Evaluation of Intravenous Ascorbic Acid in Combination with Gemcitabine and Erlotinib in Patients with Metastatic Pancreatic Cancer. PLoS ONE 2012, 7, e29794. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C Induces Tet-Dependent DNA Demethylation and a Blastocyst-like State in ES Cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhu, H.; Huang, J.; Zhu, Y.; Hong, M.; Zhu, H.; Zhang, J.; Li, S.; Yang, L.; Lian, Y.; et al. The Synergy of Vitamin C with Decitabine Activates TET2 in Leukemic Cells and Significantly Improves Overall Survival in Elderly Patients with Acute Myeloid Leukemia. Leuk. Res. 2018, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, L.; Ørskov, A.D.; Nasif, A.; Ohtani, H.; Madaj, Z.; Hansen, J.W.; Rapin, N.; Mogensen, J.B.; Liu, M.; Dufva, I.H.; et al. Oral Vitamin C Supplementation to Patients with Myeloid Cancer on Azacitidine Treatment: Normalization of Plasma Vitamin C Induces Epigenetic Changes. Clin. Epigenetics 2019, 11, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Díaz, C.C.; Magon, N.J.; McKenzie, J.L.; Hampton, M.B.; Vissers, M.C.M.; Das, A.B. Ascorbate Inhibits Proliferation and Promotes Myeloid Differentiation in TP53-Mutant Leukemia. Front. Oncol. 2021, 11, 709543. [Google Scholar] [CrossRef]
- Hmama, Z.; Nandan, D.; Sly, L.; Knutson, K.L.; Herrera-Velit, P.; Reiner, N.E. 1alpha,25-Dihydroxyvitamin D(3)-Induced Myeloid Cell Differentiation Is Regulated by a Vitamin D Receptor-Phosphatidylinositol 3-Kinase Signaling Complex. J. Exp. Med. 1999, 190, 1583–1594. [Google Scholar] [CrossRef] [Green Version]
- Marcinkowska, E.; Kutner, A. Side-Chain Modified Vitamin D Analogs Require Activation of Both PI 3-K and Erk1,2 Signal Transduction Pathways to Induce Differentiation of Human Promyelocytic Leukemia Cells. Acta Biochim. Pol. 2002, 49, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.J.; Lee, J.S.; Reiner, N.E.; Brown, G. The Vitamin D Receptor-Mediated Activation of Phosphatidylinositol 3-Kinase (PI3Kalpha) Plays a Role in the 1alpha,25-Dihydroxyvitamin D3-Stimulated Increase in Steroid Sulphatase Activity in Myeloid Leukaemic Cell Lines. J. Cell. Biochem. 2008, 103, 1551–1572. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Studzinski, G.P. AKT Pathway Is Activated by 1,25-Dihydroxyvitamin D3 and Participates in Its Anti-Apoptotic Effect and Cell Cycle Control in Differentiating HL60 Cells. Cell Cycle Georget. Tex 2006, 5, 447–451. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Lee, M.H.; Cohen, M.; Bommakanti, M.; Freedman, L.P. Transcriptional Activation of the Cdk Inhibitor P21 by Vitamin D3 Leads to the Induced Differentiation of the Myelomonocytic Cell Line U937. Genes Dev. 1996, 10, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.J.; Marcinkowska, E.; Gocek, E.; Studzinski, G.P.; Brown, G. Vitamin D3-Driven Signals for Myeloid Cell Differentiation--Implications for Differentiation Therapy. Leuk. Res. 2010, 34, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-MTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocek, E.; Studzinski, G.P. The Potential of Vitamin D-Regulated Intracellular Signaling Pathways as Targets for Myeloid Leukemia Therapy. J. Clin. Med. 2015, 4, 504–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Xu, Y.; de Necochea-Campion, R.; Baylink, D.J.; Payne, K.J.; Tang, X.; Ratanatharathorn, C.; Ji, Y.; Mirshahidi, S.; Chen, C.-S. Application of Vitamin D and Vitamin D Analogs in Acute Myelogenous Leukemia. Exp. Hematol. 2017, 50, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paubelle, E.; Zylbersztejn, F.; Maciel, T.T.; Carvalho, C.; Mupo, A.; Cheok, M.; Lieben, L.; Sujobert, P.; Decroocq, J.; Yokoyama, A.; et al. Vitamin D Receptor Controls Cell Stemness in Acute Myeloid Leukemia and in Normal Bone Marrow. Cell Rep. 2020, 30, 739–754.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Payne, K.; Pham, L.H.G.; Eunwoo, P.; Xiao, J.; Chi, D.; Lyu, J.; Campion, R.; Wasnik, S.; Jeong, I.S.; et al. A Novel Vitamin D Gene Therapy for Acute Myeloid Leukemia. Transl. Oncol. 2020, 13, 100869. [Google Scholar] [CrossRef]
- Shabtay, A.; Sharabani, H.; Barvish, Z.; Kafka, M.; Amichay, D.; Levy, J.; Sharoni, Y.; Uskokovic, M.R.; Studzinski, G.P.; Danilenko, M. Synergistic Antileukemic Activity of Carnosic Acid-Rich Rosemary Extract and the 19-nor Gemini Vitamin D Analogue in a Mouse Model of Systemic Acute Myeloid Leukemia. Oncology 2008, 75, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Sabatier, M.; Boet, E.; Zaghdoudi, S.; Guiraud, N.; Hucteau, A.; Polley, N.; Cognet, G.; Saland, E.; Lauture, L.; Farge, T.; et al. Activation of Vitamin D Receptor Pathway Enhances Differentiating Capacity in Acute Myeloid Leukemia with Isocitrate Dehydrogenase Mutations. Cancers 2021, 13, 5243. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Norman, A.W.; Chen, D.L.; Sun, G.W.; Uskokovic, M.; Koeffler, H.P. 1,25-Dihydroxy-16-Ene-23-Yne-Vitamin D3 Prolongs Survival Time of Leukemic Mice. Proc. Natl. Acad. Sci. USA 1990, 87, 3929–3932. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.S.; Bershadskiy, A. Clinical Experience Using Vitamin d and Analogs in the Treatment of Myelodysplasia and Acute Myeloid Leukemia: A Review of the Literature. Leuk. Res. Treat. 2012, 2012, 125814. [Google Scholar] [CrossRef]
- Paubelle, E.; Zylbersztejn, F.; Alkhaeir, S.; Suarez, F.; Callens, C.; Dussiot, M.; Isnard, F.; Rubio, M.-T.; Damaj, G.; Gorin, N.-C.; et al. Deferasirox and Vitamin D Improves Overall Survival in Elderly Patients with Acute Myeloid Leukemia after Demethylating Agents Failure. PLoS ONE 2013, 8, e65998. [Google Scholar] [CrossRef] [PubMed]
- Radujkovic, A.; Schnitzler, P.; Ho, A.D.; Dreger, P.; Luft, T. Low Serum Vitamin D Levels Are Associated with Shorter Survival after First-Line Azacitidine Treatment in Patients with Myelodysplastic Syndrome and Secondary Oligoblastic Acute Myeloid Leukemia. Clin. Nutr. Edinb. Scotl. 2017, 36, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Seyedalipour, F.; Mansouri, A.; Vaezi, M.; Gholami, K.; Heidari, K.; Hadjibabaie, M.; Ghavamzadeh, A. High Prevalence of Vitamin D Deficiency in Newly Diagnosed Acute Myeloid Leukemia Patients and Its Adverse Outcome. Int. J. Hematol.-Oncol. Stem Cell Res. 2017, 11, 209–216. [Google Scholar]
- Koeffler, H.P.; Hirji, K.; Itri, L. 1,25-Dihydroxyvitamin D3: In Vivo and in Vitro Effects on Human Preleukemic and Leukemic Cells. Cancer Treat. Rep. 1985, 69, 1399–1407. [Google Scholar] [PubMed]
- Petrich, A.; Kahl, B.; Bailey, H.; Kim, K.; Turman, N.; Juckett, M. Phase II Study of Doxercalciferol for the Treatment of Myelodysplastic Syndrome. Leuk. Lymphoma 2008, 49, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Oguma, S.; Uchino, H.; Maekawa, T.; Nomura, T. A Randomized Study of Alfacalcidol in the Refractory Myelodysplastic Anaemias. A Japanese Cooperative Study. Int. J. Clin. Pharmacol. Res. 1993, 13, 21–27. [Google Scholar]
- Pezeshki, S.M.S.; Asnafi, A.A.; Khosravi, A.; Shahjahani, M.; Azizidoost, S.; Shahrabi, S. Vitamin D and Its Receptor Polymorphisms: New Possible Prognostic Biomarkers in Leukemias. Oncol. Rev. 2018, 12, 366. [Google Scholar] [CrossRef]
- Ghanem, P.; Zouein, A.; Mohamad, M.; Hodroj, M.H.; Haykal, T.; Abou Najem, S.; Naim, H.Y.; Rizk, S. The Vitamin E Derivative Gamma Tocotrienol Promotes Anti-Tumor Effects in Acute Myeloid Leukemia Cell Lines. Nutrients 2019, 11, 2808. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.; Safah, H.; Lobelle-Rich, P.; Whaley, S.; Campbell, S.; Saba, N.S. Reduction in Cell Viability and in Homeobox Protein Levels Following in Vitro Exposure to δ-Tocopherol in Acute Myeloid Leukemia. Nutr. Cancer 2016, 68, 530–534. [Google Scholar] [CrossRef]
- Yang, W.; Liu, S.; Li, Y.; Wang, Y.; Deng, Y.; Sun, W.; Huang, H.; Xie, J.; He, A.; Chen, H.; et al. Pyridoxine Induces Monocyte-Macrophages Death as Specific Treatment of Acute Myeloid Leukemia. Cancer Lett. 2020, 492, 96–105. [Google Scholar] [CrossRef]
- Chen, C.C.; Li, B.; Millman, S.E.; Chen, C.; Li, X.; Morris, J.P.I.; Mayle, A.; Ho, Y.J.; Loizou, E.; Liu, H.; et al. Vitamin B6 Addiction in Acute Myeloid Leukemia. Cancer Cell 2020, 37, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Cotoraci, C.; Ciceu, A.; Sasu, A.; Miutescu, E.; Hermenean, A. The Anti-Leukemic Activity of Natural Compounds. Molecules 2021, 26, 2709. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Kim, M.; Park, H.; Jeong, M.I.; Jung, W.; Kim, B. Natural Products and Acute Myeloid Leukemia: A Review Highlighting Mechanisms of Action. Nutrients 2019, 11, 1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.-N.; Jiang, Y.-L.; Zhang, S.-G.; Huang, S.-Y.; Li, H. Grape Seed Proanthocyanidin Extract Reverses Multidrug Resistance in HL-60/ADR Cells via Inhibition of the PI3K/Akt Signaling Pathway. Biomed. Pharmacother. 2020, 125, 109885. [Google Scholar] [CrossRef] [PubMed]
- Papiez, M.A.; Bukowska-Straková, K.; Krzysciak, W.; Baran, J. (-)-Epicatechin Enhances Etoposide-Induced Antileukaemic Effect in Rats with Acute Myeloid Leukaemia. Anticancer Res. 2012, 32, 2905–2913. [Google Scholar] [PubMed]
- Maso, V.; Calgarotto, A.K.; Franchi, G.C.; Nowill, A.E.; Filho, P.L.; Vassallo, J.; Saad, S.T.O. Multitarget Effects of Quercetin in Leukemia. Cancer Prev. Res. Phila. Pa 2014, 7, 1240–1250. [Google Scholar] [CrossRef] [Green Version]
- Calgarotto, A.K.; Maso, V.; Junior, G.C.F.; Nowill, A.E.; Filho, P.L.; Vassallo, J.; Saad, S.T.O. Antitumor Activities of Quercetin and Green Tea in Xenografts of Human Leukemia HL60 Cells. Sci. Rep. 2018, 8, 3459. [Google Scholar] [CrossRef] [Green Version]
- Papież, M.A.; Krzyściak, W.; Szade, K.; Bukowska-Straková, K.; Kozakowska, M.; Hajduk, K.; Bystrowska, B.; Dulak, J.; Jozkowicz, A. Curcumin Enhances the Cytogenotoxic Effect of Etoposide in Leukemia Cells through Induction of Reactive Oxygen Species. Drug Des. Devel. Ther. 2016, 10, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M.L.; Rossi, R.M.; Karnischky, L.; Li, X.; Peterson, D.R.; Howard, D.S.; Jordan, C.T. The Sesquiterpene Lactone Parthenolide Induces Apoptosis of Human Acute Myelogenous Leukemia Stem and Progenitor Cells. Blood 2005, 105, 4163–4169. [Google Scholar] [CrossRef]
- Chen, Y.; Gan, D.; Huang, Q.; Luo, X.; Lin, D.; Hu, J. Emodin and Its Combination with Cytarabine Induce Apoptosis in Resistant Acute Myeloid Leukemia Cells in Vitro and in Vivo. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 2061–2073. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, Q.; Liang, L.; Ding, L.; Liang, Y.; Zhang, D.; Wu, B.; Yang, T.; Liu, H.; Huang, T.; et al. Compound Kushen Injection Suppresses Human Acute Myeloid Leukaemia by Regulating the Prdxs/ROS/Trx1 Signalling Pathway. J. Exp. Clin. Cancer Res. 2018, 37, 277. [Google Scholar] [CrossRef] [PubMed]
- McGill, C.M.; Tomco, P.L.; Ondrasik, R.M.; Belknap, K.C.; Dwyer, G.K.; Quinlan, D.J.; Kircher, T.A.; Andam, C.P.; Brown, T.J.; Claxton, D.F.; et al. Therapeutic Effect of Northern Labrador Tea Extracts for Acute Myeloid Leukemia. Phytother. Res. PTR 2018, 32, 1636–1641. [Google Scholar] [CrossRef] [PubMed]
- Calgarotto, A.K.; Longhini, A.L.; Pericole de Souza, F.V.; Duarte, A.S.S.; Ferro, K.P.; Santos, I.; Maso, V.; Olalla Saad, S.T.; Torello, C.O. Immunomodulatory Effect of Green Tea Treatment in Combination with Low-Dose Chemotherapy in Elderly Acute Myeloid Leukemia Patients with Myelodysplasia-Related Changes. Integr. Cancer Ther. 2021, 20, 15347354211002648. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Quan, R.; Liu, W.; Xiao, H.; Tang, X.; Liu, C.; Li, L.; Lv, Y.; Wang, H.; Xu, Y.; et al. Arsenic-Containing Qinghuang Powder Is an Alternative Treatment for Elderly Acute Myeloid Leukemia Patients Refusing Low-Intensity Chemotherapy. Chin. J. Integr. Med. 2020, 26, 339–344. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Phase | Intervention | Patient Population | Disease State | Outcomes |
|---|---|---|---|---|---|
| Davis et al. 2016 [61] | I/IIb | Ipilimumab | AML, NHL, HL, CML, CLL, MM, MPN, AL | Relapsed after Allo-HSCT | CR: 23% (5/28) PR: 9% (2/22) |
| Daver et al. 2016 [62] | I/IIb | Nivolumab + Azacitidine | AML | Relapsed after prior therapy | CR: 18% (6/51) HI: 15% (5/51) |
| Lindblad et al. 2018 | I/II | Pembrolizumab + decitabine | AML | Relapsed after prior therapy | CR: 10% (1/10) SD:40% (4/10) |
| Daver et al. 2018 [63] | II | Nivolumab + Azacitidine + Ipilimumab | AML | R/R | CR/CRi: 36% (6/20) Prolonged SD:10% (2/20) |
| Ravandi et al. 2019 [64] | II | Idarubicin + Cytarabine + Nivolumab | AML and high risk MDS | Newly diagnosed | CR:/CRi 78% (34/44) Negative MRD: 41% (18/34) |
| Target | Author | Drug (Antibody Construct) | Patient Population | Outcomes |
|---|---|---|---|---|
| CD33 | Ravandi et al. 2018/2020 [67] | AMG330 (anti-CD3 × CD33 BiTE) | 55 patients with R/R-AML | Efficacy: 19% ORR (7% CR, 9% CR with incomplete hematologic recovery, 2% with morphological leukemia free state) Safety: 90% AE rate; 67% CRS (13% ≥ grade 3), nausea (20%) |
| CD33 (HLE *) | Subklewe et al. 2019 [68] | AMG673 (Half-Life Extended Anti-CD3 × CD33 BiTE) | 30 patients with R/R-AML | Efficacy: (12/27) 44% with bone marrow blast reduction, 6 of which had >50% reduction in blasts; 1 patient with complete remission with 85% reduction Safety: 50% patients had CRS (13% ≥ grade 3), transaminitis (17%), leukopenia (13%), thrombocytopenia (7%), febrile neutropenia (7%) |
| CD123 | Uy et al. 2021 [69] | Flotetuzumab (anti CD3 × CD123 DART) | 92 R/R-AML patients | Primary induction failure or early relapse cohort (n = 30): Efficacy: 27% with CR/CRh; median OS 10.2 months among responders Safety: 100% CRS (13% ≥ grade) |
| CD123 | Ravandi et al. 2020 [70] | Vibecotamab (XmAb14045; anti CD3 × CD123 BiTE) | 104 R/R-AML, 1 B-cell ALL, and 1 CML | Efficacy: 14% ORR (4% CR); 71% SD Safety: 59% CRS (15% ≥ grade 3) |
| CD123 | Watts et al. 2021 [71] | APVO436 (anti CD3 × CD123 BiTE) | 22 R/R-AML and 6 R/R-MDS | Efficacy: 2 patients with blast reduction Safety: edema (32%), febrile neutropenia (29%), infusion reaction (21%), CRS (18%) |
| Target | Drug (Antibody Construct) | Patient Population | NCT | Phase |
|---|---|---|---|---|
| CD33 | AMV564 (CD3 × CD33 bispecific antibody) | R/R AML | NCT03144245 | 1 |
| AMG673 (CD3 × CD33 bispecific antibody) | R/R AML | NCT03224819 | 1 | |
| GEM333 (CD3 × CD33 bispecific antibody) | R/R AML | NCT03516760 | 1 | |
| JNJ-67571244 (CD3 × CD33 bispecific antibody) | R/R AML, MDS | NCT03915379 | 1 | |
| AMG330 (CD3 × CD33 bispecific antibody) | R/R AML, Minimal Residual Disease Positive AML, MDS | NCT02520427 | 1 | |
| AMV564 (CD3 × CD33 bispecific antibody) | MDS | NCT03516591 | 1 | |
| CD123 | JNJ-63709178 (CD3 × CD123 bispecific antibody) | R/R AML | NCT02715011 | 1 |
| APVO436 (CD3 × CD123 bispecific antibody) | R/R AML, MDS | NCT03647800 | 1 | |
| MGD006 (CD3 × CD123 DART) | R/R AML, MDS | NCT02152956 | 1 and 2 | |
| SAR440234 (CD3 × CD123 bispecific antibody) | R/R AML, MDS, B-ALL | NCT03594955 | 1 and 2 | |
| XmAb14045 (CD3 × CD123 bispecific antibody) | CD123 Expressing hematologic malignancies | NCT02730312 | 1 | |
| CD16/CD33 | GTB-3550 (CD16/IL-15/CD33 TriKE) | R/R AML, MDS, Advanced Systemic Mastocytosis | NCT03214666 | 1 and 2 |
| CD135 | AMG427 (CD3 × CD135(FLT3) bispecific antibody) | R/R AML | NCT03541369 | 1 |
| CLEC12A | MCLA-117 (CD3 × CLEC12A bispecific antibody) | R/R AML and newly diagnosed elderly AML | NCT03038230 | 1 |
| Target Antigen | Population | NCT ID | Phase |
|---|---|---|---|
| CD33 | R/R AML | NCT03126864 | I |
| R/R AML | NCT02799680 | I | |
| R/R AML | NCT01864902 | I/II | |
| R/R AML | NCT02944162 | I/II | |
| R/R AML, MDS; ALL | NCT03291444 | I | |
| R/R AML | NCT03473457 | ||
| AML | NCT03222674 | I/II | |
| CD123 | AML | NCT03585517 | I |
| Recurred AML after allo-HSCT | NCT03114670 | I | |
| R/R AML | NCT03556982 | I/II | |
| R/R AML | NCT02623582 | I | |
| R/R AML | NCT02159495 | I | |
| R/R AML | NCT03672851 | I | |
| R/R AML | NCT03766126 | I | |
| R/R AML, MDS; ALL | NCT03291444 | I | |
| R/R AML | NCT03473457 | n/a | |
| R/R AML | NCT03796390 | I | |
| AML | NCT03222674 | I/II | |
| CD38 | R/R AML, MDS; ALL | NCT03291444 | I |
| R/R AML | NCT03473457 | ||
| AML | NCT03222674 | I/II | |
| UCART23 | R/R AML | NCT03190278 | I |
| R/R AML, high-risk AML | NCT01864902 | I | |
| CD/123/CLL1 | R/R AML | NCT03631576 | II/III |
| CD33/CLL1 | R/R AML, MDS, MPN, CML | NCT03795779 | I |
| CCL1 | AML | NCT03222674 | I/II |
| NKG2D | AML, MDS-RAEB, MM | NCT02203825 | I |
| R/R AML, AML, Myeloma | NCT03018405 | I/II | |
| Lewis Y | Myeloma, AML, MDS | NCT01716364 | I |
| WT1 | R/R AML, ALL, MDS | NCT03291444 | I |
| CD7/NK92 | R/R AML | NCT03018405 | I/II |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hino, C.; Pham, B.; Park, D.; Yang, C.; Nguyen, M.H.K.; Kaur, S.; Reeves, M.E.; Xu, Y.; Nishino, K.; Pu, L.; et al. Targeting the Tumor Microenvironment in Acute Myeloid Leukemia: The Future of Immunotherapy and Natural Products. Biomedicines 2022, 10, 1410. https://doi.org/10.3390/biomedicines10061410
Hino C, Pham B, Park D, Yang C, Nguyen MHK, Kaur S, Reeves ME, Xu Y, Nishino K, Pu L, et al. Targeting the Tumor Microenvironment in Acute Myeloid Leukemia: The Future of Immunotherapy and Natural Products. Biomedicines. 2022; 10(6):1410. https://doi.org/10.3390/biomedicines10061410
Chicago/Turabian StyleHino, Christopher, Bryan Pham, Daniel Park, Chieh Yang, Michael H.K. Nguyen, Simmer Kaur, Mark E. Reeves, Yi Xu, Kevin Nishino, Lu Pu, and et al. 2022. "Targeting the Tumor Microenvironment in Acute Myeloid Leukemia: The Future of Immunotherapy and Natural Products" Biomedicines 10, no. 6: 1410. https://doi.org/10.3390/biomedicines10061410
APA StyleHino, C., Pham, B., Park, D., Yang, C., Nguyen, M. H. K., Kaur, S., Reeves, M. E., Xu, Y., Nishino, K., Pu, L., Kwon, S. M., Zhong, J. F., Zhang, K. K., Xie, L., Chong, E. G., Chen, C. -S., Nguyen, V., Castillo, D. R., & Cao, H. (2022). Targeting the Tumor Microenvironment in Acute Myeloid Leukemia: The Future of Immunotherapy and Natural Products. Biomedicines, 10(6), 1410. https://doi.org/10.3390/biomedicines10061410