
Molecular Pathophysiological Mechanisms in Huntington’s Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genetics of Huntington’s Disease
- -
- -
- The presence of the Δ2642 glutamic acid polymorphism (a deletion of three nucleotides in the codon encoding glutamic acid) [10].
- -
- Polymorphisms in the gene encoding for the glutamate receptor GluR6 [11].
- -
- Polymorphisms in the gene encoding the N-methyl-D-aspartate (NMDA) receptor subunit 2B [12].
- -
- Genetic variations of the PPAR-γ (peroxisome proliferator-activated receptor gamma) coactivator 1α (PGC-1α) [13].
- -
- -
- Genes that colocalize with the expanded CAG repeats, and are more likely to be replicated, such as the G-protein-coupled receptor (GPCR) 161 allele [16], which has been shown to be involved in DNA repair processes.
3. Models of HD
3.1. Chemical Models
3.2. Genetic Animal Models
3.2.1. Fragment Transgenic Mouse Models
3.2.2. Full-Length Transgenic Mouse Models
3.2.3. Knock-In Mouse Models
3.2.4. Other Animal Models of HD
3.2.5. Non-Mammalian Models of HD
3.2.6. Cell Lines for In Vitro Studies
4. Normal and Mutant Huntingtin
4.1. Normal Huntingtin Structure
4.2. Cellular Distribution
4.3. Post-Translational Modifications
4.4. Huntingtin Interacting Proteins
4.5. Functions of Huntingtin
4.5.1. Huntingtin during Embryonic Development
4.5.2. Huntingtin Protects Cells from Apoptosis
4.5.3. Huntingtin and Transcriptional Regulation
4.5.4. The Role of Huntingtin in Axonal and Vesicle Transport
4.5.5. Huntingtin and Synaptic Activity
4.6. Mutant Huntingtin
5. Mechanisms of Neurodegeneration in Huntington’s Disease
- -
- Grade 0—the brain appears normal on gross examination, but histologically 30–40% of neurons are lost in the head of the caudate nucleus.
- -
- Grade 1—a 50% neuronal loss in the head of the caudate nucleus, with neuronal loss and astrogliosis evident in the tail +/− body of the caudate nucleus.
- -
- Grade 2—striatal atrophy, with ventricular profile of the caudate nucleus less convex than normal.
- -
- Grade 3—severe striatal atrophy with flat ventricular profile of the caudate nucleus.
- -
- Grade 4—atrophy of the striatum and up to 95% neuronal loss, with concave ventricular profile of the caudate nucleus.
5.1. Excitotoxicity
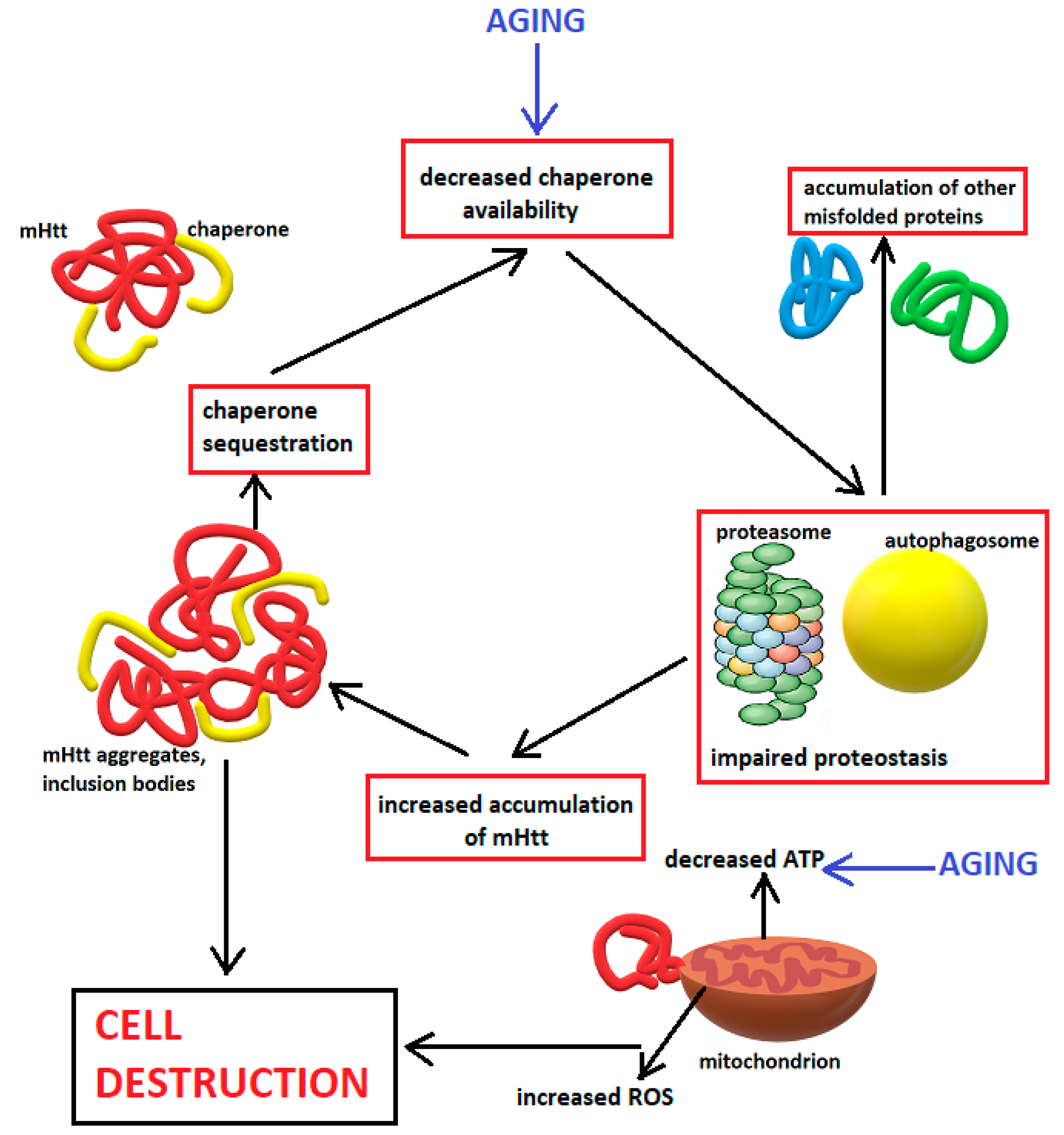
5.2. Impaired Proteostasis
- -
- Macroautophagy, in which a double-membraned vesicle (the autophagosome) forms and fuses with lysosomes, after which their content is degraded by the lysosomal enzymes.
- -
- Microautophagy, a process during which lysosomes wrap around various cytosolic compounds, followed by involution of the membrane and degradation of the vesicle content [167].
- -
- Chaperone-mediated autophagy, a process during which chaperones bind to damaged proteins and to receptors on the lysosomal membrane, leading to translocation of the protein into the lysosome and degradation [168].
5.3. Mitochondrial Dysfunction and Oxidative Stress
5.4. Transcriptional Dysregulation
5.4.1. CREB and CBP
5.4.2. Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1α
5.4.3. Specificity Protein 1
5.4.4. Nuclear Factor κ Light-Chain-Enhancer of Activated B Cells
5.4.5. Repressor Element 1-Silencing Transcription Factor (REST)
5.4.6. Other Transcription Factors
5.4.7. Dysregulation of MicroRNAs
5.5. Loss of BDNF Synthesis and Impaired BDNF Transport
5.6. Other Disturbances in Signaling
5.7. Astrocytes and Oligodendrocytes in HD
5.8. Microglial Activation and Neuroinflammation in HD
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutic targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Perandones, C.; Radrizzani, M.; Micheli, F.E. Molecular Mechanisms Involved in the Pathogenesis of Huntington’s Disease. In Huntington’s Disease: Etiology and Symptoms, Diagnosis and Treatment; Visser, T.J., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2010; pp. 1–37. ISBN 978-1-61668-984-1. [Google Scholar]
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C.; Ottina, K.; Wallace, M.R.; Sakaguchi, A.Y.; et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 1983, 306, 234–238. [Google Scholar] [CrossRef] [PubMed]
- HD Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Ranen, N.G.; Stine, O.C.; Abbott, M.H.; Sherr, M.; Codori, A.M.; Franz, M.L.; Chao, N.I.; Chung, A.S.; Pleasant, N.; Callahan, C. Anticipation and instability of IT-15 (CAG)n repeats in parent-offspring pairs with Huntington’s disease. Am. J. Hum. Genet. 1995, 57, 593–602. [Google Scholar] [PubMed]
- Pearson, C.E. Slipping while sleeping? Trinucleotide repeat expansions in germ cells. Trends Mol. Med. 2003, 9, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Barton, D.E.; Davison, B.C.; Ferguson-Smith, M.A. Analysis of the huntingtin gene reveals a trinucleotide-length polymorphism in the region of the gene that contains two CAG-rich stretches and a correlation between decreased age of onset of Huntington’s disease and CAG repeat number. Hum. Mol. Genet. 1993, 2, 1713–1715. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.E.B.; Caron, N.S.; Ng, B.; Casal, L.; Casazza, W.; Xu, X.; Ooi, J.; Pouladi, M.A.; Mostafavi, S.; Ross, C.J.D.; et al. Gene expression profiles complement the analysis of genomic modifiers of the clinical onset of Huntington disease. Hum. Mol. Genet. 2020, 29, 2788–2802. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.A.; Jurgens, C.K.; Landwehrmeyer, G.B.; van Roon-Mom, W.M.; van Ommen, G.J.; Stijnen, T.; Roos, R.A.; Orth, M.; Handley, O.J.; Schwenke, C.; et al. Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology 2009, 73, 1280–1285. [Google Scholar] [CrossRef]
- Vuillaume, I.; Vermersch, P.; Destee, A.; Petit, H.; Sablonniere, B. Genetic polymorphisms adjacent to the CAG repeat influence clinical features at onset in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 1998, 64, 758–762. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Chiano, M.; Dodge, A.; Norbury, G.; Rosser, E.; Craufurd, D. Genotypes at the GluR6 kainate receptor locus are associated with variations in the age of onset of Huntington disease. Proc. Natl. Acad. Sci. USA 1997, 94, 3872–3876. [Google Scholar] [CrossRef] [Green Version]
- Arning, L.; Kraus, P.H.; Valentin, S.; Saft, C.; Andrich, J.; Epplen, J.T. NR2A and NR2B receptor gene variations modify age at onset in Huntington disease. Neurogenetics 2005, 6, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Taherzadeh-Fard, E.; Saft, C.; Andrich, J.; Wieczorek, S.; Arning, L. PGC-1alpha as modifier of onset age in Huntington disease. Mol. Neurodegener. 2009, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arning, L.; Monte, D.; Hansen, W.; Wieczorek, S.; Jagiello, P.; Akkad, D.A.; Andrich, J.; Kraus, P.H.; Saft, C.; Epplen, J.T. ASK1 and MAP2K6 as modifiers of age at onset in Huntington’s disease. J. Mol. Med. 2008, 86, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep. 2017, 8, 619–633. [Google Scholar] [CrossRef] [Green Version]
- Shimada, I.S.; Hwang, S.H.; Somatilaka, B.N.; Wang, X.; Skowron, P.; Kim, J.; Kim, M.; Shelton, J.M.; Rajaram, V.; Xuan, Z.; et al. Basal suppression of the sonic hedgehog pathway by the G-protein-coupled receptor Gpr161 restricts medulloblastoma pathogenesis. Cell Rep. 2018, 22, 1169–1184. [Google Scholar] [CrossRef] [Green Version]
- Li, J.L.; Hayden, M.R.; Almqvist, E.W.; Brinkman, R.R.; Durr, A.; Dodé, C.; Morrison, P.J.; Suchowersky, O.; Ross, C.A.; Margolis, R.L.; et al. A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study. Am. J. Hum. Genet. 2003, 73, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Hauberg, M.E.; Zhang, W.; Giambartolomei, C.; Franzen, O.; Morris, D.L.; Vyse, T.J.; Ruusalepp, A.; CommonMind, C.; Sklar, P.; Schadt, E.E.; et al. Large-scale identification of common trait and disease variants affecting gene expression. Am. J. Hum. Genet. 2017, 100, 885–894. [Google Scholar] [CrossRef]
- Hensman Moss, D.J.; Pardinas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; TRACK-HD Investigators, REGISTRY Investigators. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Barbeira, A.N.; Dickinson, S.P.; Bonazzola, R.; Zheng, J.; Wheeler, H.E.; Torres, J.M.; Torstenson, E.S.; Shah, K.P.; Garcia, T.; Edwards, T.L.; et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018, 9, 1825. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Swartz, K.J.; Kowall, N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 1991, 11, 1649–1659. [Google Scholar] [CrossRef]
- Brouillet, E.; Conde, F.; Beal, M.F.; Hantraye, P. Replicating Huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 1999, 59, 427–468. [Google Scholar] [CrossRef]
- Raymond, L.A.; André, V.M.; Cepeda, C.; Gladding, C.M.; Milnerwood, A.J.; Levine, M.S. Pathophysiology of Huntington’s disease: Time-dependent alterations in synaptic and receptor function. Neuroscience 2011, 198, 252–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrbach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Kaye, J.; Reisine, T.; Finkbeiner, S. Huntington’s disease mouse models: Unraveling the pathology caused by CAG repeat expansion. Fac. Rev. 2021, 10, 77. [Google Scholar] [CrossRef]
- Gil, J.M.; Rego, A.C. The R6 lines of transgenic mice: A model for screening new therapies for Huntington’s disease. Brain Res. Rev. 2009, 59, 410–431. [Google Scholar] [CrossRef] [Green Version]
- Schilling, B.; Becher, M.W.; Sharp, A.H.; Jinnah, H.A.; Duan, K.; Kotzuk, J.A.; Slunt, H.H.; Ratovitski, T.; Cooper, J.K.; Jenkins, N.A.; et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant NH2-terminal fragment of huntingtin. Hum. Mol. Genet. 1999, 8, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Lucas, J.J.; Hen, R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000, 101, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.A.; Leavitt, B.R.; LePiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- van Raamsdonk, J.M.; Pearson, J.; Slow, E.J.; Hossain, S.M.; Leavitt, B.R.; Hayden, M.R. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J. Neurosci. 2005, 25, 4169–4180. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; André, V.M.; Wilburn, B.; Lu, X.H.; Tao, J.; Yamazaki, I.; Li, S.H.; Sun, Y.E.; et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Li, C.; Wei, W.; Lo, V.; Gong, S.; Li, S.H.; Iwasato, T.; Itohara, S.; Li, X.J.; Mody, I.; et al. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 2005, 46, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, S.A.; Zheng, S.; Liu, W.; Dai, Y.; Liu, Y.; Hou, Z.; Mori, S.; Tang, Y.; Cheng, J.; Duan, W.; et al. A novel and accurate full-length HTT mouse model for Huntington’s disease. eLife 2022, 11, e70217. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Tallaksen-Greene, S.; Chien, W.M.; Cearley, J.A.; Jackson, W.S.; Crouse, A.B.; Ren, S.; Li, X.J.; Albin, R.L.; Detloff, P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Horsten, S.; Schmitt, I.; Nguyen, H.P.; Holzmann, C.; Schmidt, T.; Walther, T.; Bader, M.; Pabst, R.; Kobbe, P.; Krotova, J.; et al. Transgenic rat model of Huntington’s disease. Hum. Mol. Genet. 2003, 12, 617–624. [Google Scholar] [CrossRef]
- Jacobsen, J.C.; Bawden, C.S.; Rudiger, S.R.; McLaughlan, C.J.; Reid, S.J.; Waldvogel, H.J.; Macdonald, M.E.; Gusella, J.F.; Walker, S.K.; Kelly, J.M.; et al. An ovine transgenic Huntington’s disease model. Hum. Mol. Genet. 2010, 19, 1873–1882. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Cheng, P.H.; Banta, H.; Piotrowska-Nitsche, K.; Yang, J.J.; Cheng, E.C.; Snyder, B.; Larkin, K.; Liu, J.; Orkin, F.; et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 2008, 453, 921–924. [Google Scholar] [CrossRef] [Green Version]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Connolly, J.B.; Wellington, C.; Hayden, M.; Dausset, J.; Neri, C. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 13318–13323. [Google Scholar] [CrossRef] [Green Version]
- Harding, R.J.; Tong, Y.F. Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sinica 2018, 39, 754–769. [Google Scholar] [CrossRef] [Green Version]
- Monk, R.; Lee, K.; Jones, K.S.; Connor, B. Directly reprogrammed Huntington’s disease neural precursor cells generate striatal neurons exhibiting aggregates and impaired neuronal maturation. Stem Cells 2021, 39, 1410–1422. [Google Scholar] [CrossRef]
- Yakoub, A.M. Cerebral organoids exhibit mature neurons and astrocytes and recapitulate electrophysiological activity of the human brain. Neural Regen. Res. 2019, 14, 757–761. [Google Scholar] [CrossRef]
- Eremeev, A.V.; Lebedeva, O.S.; Bogomiakova, M.E.; Lagarkova, M.A.; Bogomazova, A.N. Cerebral organoids—challenges to establish a brain prototype. Cells 2021, 10, 1790. [Google Scholar] [CrossRef]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, J.S.; Zinovyeva, A.; Yagi, K.; Makabe, K.W.; Raff, R.A. Neural expression of the Huntington’s disease gene as a chordate evolutionary novelty. J. Exp. Zool. B Mol. Dev. Evol. 2003, 297, 57–64. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.; Schwartz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.P.; Carraway, R.; Reeves, S.A. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, J.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–832. [Google Scholar] [CrossRef] [Green Version]
- Perutz, K.F.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355–5358. [Google Scholar] [CrossRef] [Green Version]
- Tartari, M.; Gissi, C.; Lo Sardo, V.; Zuccato, C.; Picardi, E.; Pesole, G.; Cattaneo, E. Phylogenetic comparison of huntingtin homologues reveals the appearance of a primitive poliQ in sea urchin. Mol. Biol. Evol. 2008, 25, 330–338. [Google Scholar] [CrossRef]
- Rockabrand, E.; Slepko, N.; Pantalone, A.; Nukala, V.N.; Kazantsev, A.; Marsh, J.L.; Sullivan, P.G.; Steffan, J.S.; Sensi, S.L.; Thompson, L.M. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol Genet. 2007, 16, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Harding, R.J.; Deme, J.C.; Hevler, J.F.; Tamara, S.; Lemak, A.; Cantle, J.P.; Szewczik, M.M.; Begeja, N.; Goss, S.; Zuo, X.; et al. Huntingtin structure is orchestrated by HAP40 and shows a polyglutamine expansion-specific interaction with exon 1. Nat. Commun. Biol. 2021, 4, 1374. [Google Scholar] [CrossRef]
- Guo, Q.; Huang, B.; Cheng, J.; Seefelder, M.; Engler, T.; Pfeifer, G.; Oeckl, P.; Otto, M.; Moser, F.; Maurer, M.; et al. The cryo-electron microscopic structure of huntingtin. Nature 2018, 555, 117–120. [Google Scholar] [CrossRef]
- Fusco, F.R.; Chen, Q.; Lamoreaux, W.J.; Figueredo-Cardenas, G.; Jiao, Y.; Coffman, J.A.; Surmeier, D.J.; Honig, M.G.; Carlock, L.R.; Reiner, A. Cellular localization of huntingtin in striatal and cortical neurons in rats: Lack of correlation with neuronal vulnerability in Huntington’s disease. J. Neurosci. 1999, 19, 1189–1202. [Google Scholar] [CrossRef] [Green Version]
- Jansen, A.H.P.; van Hall, M.; op den Kelder, I.C.; Meier, R.T.; de Ruiter, A.A.; Schut, M.H.; Smith, D.L.; Grit, C.; Brouwer, N.; Kamphuis, W.; et al. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia 2017, 65, 50–61. [Google Scholar] [CrossRef]
- Kojer, K.; Hering, T.; Bazenet, C.; Weiss, A.; Herrmann, F.; Taanman, J.W.; Orth, M. Huntingtin aggregates and mitochondrial pathology in skeletal muscle but not heart of late-stage R6/2 mice. J. Huntingtons Dis. 2019, 8, 145–159. [Google Scholar] [CrossRef]
- Didiot, M.C.; Ferguson, C.M.; Ly, S.; Coles, A.H.; Smith, A.O.; Bicknell, A.A.; Hall, L.M.; Sapp, E.; Echeverria, D.; Pai, A.A.; et al. Nuclear localization of huntingtin mRNA is specific to cells of neuronal origin. Cell Rep. 2018, 24, 2553–2560. [Google Scholar] [CrossRef] [Green Version]
- Hoffner, G.; Kahlem, P.; Dijan, P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: Relevance to Huntington’s disease. J. Cell Sci. 2002, 115, 941–948. [Google Scholar] [CrossRef]
- Strehlow, A.N.; Li, J.Z.; Myers, R.M. Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum. Mol. Genet. 2007, 16, 391–409. [Google Scholar] [CrossRef] [Green Version]
- Velier, J.; Kim, M.; Schwarz, C.; Kim, T.W.; Sapp, E.; Chase, K.; Aronin, N.; DiFiglia, M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp. Neurol. 1998, 152, 34–40. [Google Scholar] [CrossRef]
- Bassi, S.; Tripathi, T.; Monziani, A.; Di Leva, F.; Biagioli, M. Epigenetics of Huntington’s disease. Adv. Exp. Med. Biol. 2017, 978, 277–299. [Google Scholar]
- Cariulo, C.; Azzollini, L.; Verani, M.; Martufi, P.; Boggio, R.; Chiki, A.; Deguire, S.M.; Cherubini, M.; Gines, S.; Marsh, J.L.; et al. Phosphorylation of huntingtin at residue T3 is decreased in Huntington’s disease and modulates mutant huntingtin protein conformation. Proc. Natl. Acad. Sci. USA. 2017, 114, E10809–E10818. [Google Scholar] [CrossRef] [Green Version]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in the brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- DeGuire, S.; Ruggeri, F.; Fares, M.; Chiki, A.; Cendrowska, U.; Dietler, G.; Lashuel, H.A. N-terminal huntingtin (Htt) phosphorylation is a molecular switch regulating Htt aggregation, helical conformation, internalization, and nuclear targeting. J. Biol. Chem. 2018, 293, 18540–18558. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vicente, M.; Arrasate, M.; O’Rourke, J.G.; Kashwji, H.; et al. IKK phosphorylates huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef]
- Yanai, A.; Huang, K.; Kang, R.; Singaraja, R.R.; Arstikaitis, P.; Gan, L.; Orban, P.C.; Mullard, A.; Cowan, C.M.; Raymond, L.A.; et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat. Neurosci. 2006, 9, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Then, F.; Melia, T.J., Jr.; Mazzulli, J.R.; Cui, L.; Savas, J.N.; Voisine, C.; Paganetti, P.; Tanese, N.; Hart, A.C.; et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009, 137, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Stepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; et al. SUMO modification of huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Harjes, P.; Wanker, E.E. The hunt for huntingtin function. Trends Biochem. Sci. 2003, 28, 425–433. [Google Scholar] [CrossRef]
- Jiang, H.; Sandoval del Prado, L.E.; Leung, C.; Wang, D. Huntingtin-interacting protein family members have a conserved pro-viral function from Caenorhabditis elegans to humans. Proc. Natl. Acad. Sci. USA 2020, 117, 22462–22472. [Google Scholar] [CrossRef]
- Vitet, H.; Brandt, V.; Saudou, F. Traffic signaling: New functions of huntingtin and axonal transport in neurological disease. Curr. Opin. Neurobiol. 2020, 63, 122–130. [Google Scholar] [CrossRef]
- Migazzi, A.; Scaramuzzino, C.; Anderson, E.; Tripathy, D.; Hernández, I.; Grant, R.A.; Rocuzzo, M.; Tossato, L.; Virlogeux, A.; Zuccato, C.; et al. Huntingtin-mediated axonal transport requires arginine methylation by PRMT6. Cell Rep. 2021, 35, 108980. [Google Scholar] [CrossRef]
- Peng, L.; Yang, Q.; Xu, X.; Du, Y.; Wu, Y.; Shi, X.; Xu, J.; Zhu, L.; Luo, J. Huntingtin-interacting protein1-related protein plays a critical role in dendritic development and excitatory synapse formation in hippocampal neurons. Front. Mol. Neurosci. 2017, 10, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.; Peng, L.; Wu, Y.; Li, Y.; Wang, L.; Luo, J.H.; Xu, J. Endocytic adaptor protein HIP1R controls intracellular trafficking of epidermal growth factor receptor in neuronal dendritic development. Front. Mol. Neurosci. 2018, 11, 447. [Google Scholar]
- Smith, R.; Brundin, P.; Li, J.Y. Synaptic dysfunction in Huntington’s disease: A new perspective. Cell. Mol. Life Sci. 2005, 62, 1901–1912. [Google Scholar] [CrossRef]
- Su, Y.; Savanenin, A.; Reddy, P.H.; Liu, Y.F. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via postsynaptic density 95. J. Biol. Chem. 2001, 276, 24713–24718. [Google Scholar]
- Lundgren, J.L.; Vandermeulen, L.; Sandebring-Matton, A.; Ahmed, S.; Winblad, B.; Di Luca, M.; Tjernberg, L.O.; Marcello, E.; Frykman, S. Proximity ligation assay reveals both pre- and postsynaptic localization of the APP-processing enzymes ADAM10 and BACE1 in rat and human adult brain. BMC Neurosci. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, F.; Vezzoli, E.; Cheroni, C.; Besusso, D.; Conforti, P.; Valenza, M.; Iacobucci, I.; Monaco, V.; Birolini, G.; Bombaci, M.; et al. ADAM10 hyperactivation acts on piccolo to deplete synaptic vesicle stores in Huntington’s disease. Hum. Mol. Genet. 2021, 30, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, M. Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin p150 Glued. J. Biol. Chem. 2008, 283, 34880–34886. [Google Scholar] [CrossRef] [Green Version]
- Barnat, M.; Le Friec, J.; Benstaali, C.; Humbert, S. Huntingtin-mediated bipolar-multipolar transition of newborn cortical neurons is critical for their postnatal neuronal morphology. Neuron 2017, 93, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Saudou, F.; Humbert, S. The biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Dragatsis, I.; Dietrich, P.; Ren, H.; Deng, Y.P.; Del Mar, N.; Wang, H.B.; Johnson, I.M.; Jones, K.R.; Reiner, A. Effect of early embryonic deletion of huntingtin from pyramidal neurons on the development and long-term survival of neurons in cerebral cortex and striatum. Neurobiol. Dis. 2018, 111, 102–117. [Google Scholar] [CrossRef]
- Rigamonti, D.; Bauer, J.H.; De-Fraja, C.; Conti, L.; Sipione, S.; Sciorati, C.; Clementi, E.; Hackam, A.; Hayden, M.R.; Li, Y.; et al. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J. Neurosci. 2000, 20, 3705–3713. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, D.; Sipione, S.; Goffredo, D.; Zuccato, C.; Fossale, E.; Cattaneo, E. Huntingtin’s neuroprotective activity occurs via inhibition of procaspase-9 processing. J. Biol. Chem. 2001, 276, 14545–14548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervais, F.G.; Singaraja, R.; Xanthoudakis, S.; Gutekunst, C.A.; Leavitt, B.R.; Metzler, M.; Hackam, A.S.; Tam, J.; Vaillancourt, J.P.; Houtzager, V.; et al. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat. Cell Biol. 2002, 4, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Thant, C.; White, J.A., II; Banerjee, R.; Thuamsang, B.; Gunawardena, S. Excess active PI3K rescues huntingtin-mediated neuronal cell death but has no effect on axonal transport defects. Apoptosis 2019, 24, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Landles, C.; Bates, G.P. Huntingtin and the molecular pathogenesis of Huntington’s disease. EMBO Rep. 2004, 5, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Li, X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef]
- Courey, A.J.; Tjian, R. Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell 1988, 55, 887–898. [Google Scholar] [CrossRef]
- Li, S.H.; Cheng, A.L.; Zhou, H.; Lam, S.; Rao, M.; Li, H.; Li, X.J. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 2002, 22, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Nunkoo, V.S. Tau-Targeted Therapy in Alzheimer’s Disease: History and Current State. In Frontiers in Clinical Drug Research; Ibarra Arias, J.J.A., Ed.; Bentham Science Publishers: Singapore, 2021; Volume 2, pp. 56–138. [Google Scholar]
- Gunawardena, S.; Goldstein, L.S. Polyglutamine diseases and transport problems: Deadly traffic jams on neuronal highways. Arch. Neurol. 2005, 62, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchal, K.; Tiwari, A.K. Miro (mitochondrial Rho GTPase), a key player in mitochondrial axonal transport and mitochondrial dynamics in neurodegenerative diseases. Mitochondrion 2021, 56, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pages, M.; Dompierre, J.P.; Rangone, H.; Cordelieres, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehinger, Y.; Bruyère, J.; Panayotis, N.; Abada, Y.S.; Borloz, E.; Matagne, V.; Scaramuzzino, C.; Vitet, H.; Delatour, B.; Saidi, L.; et al. Huntingtin phosphorylation governs BDNF homeostasis and improves the phenotype of Mecp2 knockout mice. EMBO Mol. Med. 2020, 12, e10889. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pages, M.; Li, X.J.; Saudou, F.; Humbert, S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olenick, M.A.; Holzbaur, E.L. Dynein activators and adaptors at a glance. J. Cell Sci. 2019, 132, jcs227132. [Google Scholar] [CrossRef] [Green Version]
- White, J.A., II; Krzystek, T.J.; Hoffmar-Glennon, H.; Thant, C.; Zimmerman, K.; Iacobucci, G.; Vail, J.; Thurston, L.; Rahman, S.; Gunawardena, S. Excess Rab4 rescues synaptic and behavioral dysfunction caused by defective HTT-Rab4 axonal transport in Huntington’s disease. Acta Neuropathol. Commun. 2020, 8, 97. [Google Scholar] [CrossRef]
- Lim, A.; Rechtsteiner, A.; Saxton, W.M. Two kinesins drive anterograde neuropeptide transport. Mol. Biol. Cell 2017, 28, 3542–3553. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; Holzbaur, E.L.F. Axonal transport: Driving synaptic function. Science 2019, 366, eaaw9997. [Google Scholar] [CrossRef]
- Goldstein, A.Y.; Wang, X.; Schwarz, T.L. Axonal transport and the delivery of pre-synaptic components. Curr. Opin. Neurobiol. 2008, 18, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakakubo, Y.; Abe, S.; Yoshida, T.; Takami, C.; Isa, M.; Wojcik, S.M.; Brose, N.; Takamori, S.; Hori, T. Vesicular glutamate transporter expression ensures high-fidelity synaptic transmission at the calyx of held synapses. Cell Rep. 2020, 32, 108040. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Verstreken, P. Autophagy in the presynaptic compartment in health and disease. J. Cell Biol. 2017, 216, 1895–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, F.; Schink, K.O.; Bruns, C.; Izsvák, Z.; Hamra, F.K.; Rosenmund, C.; Garner, C.C. Critical role for Piccolo in synaptic vesicle retrieval. eLife 2019, 8, e46629. [Google Scholar] [CrossRef]
- Barron, J.C.; Hurley, E.P.; Parsons, M.P. Huntingtin and the synapse. Front. Cell. Neurosci. 2021, 15, 225. [Google Scholar] [CrossRef]
- McAdam, R.L.; Morton, A.; Gordon, S.L.; Alterman, J.F.; Khvorova, A.; Cousin, M.A.; Smillie, K.J. Loss of huntingtin function slows synaptic vesicle endocytosis in striatal neurons from the httQ140/Q140 mouse model of Huntington’s disease. Neurobiol. Dis. 2020, 134, 104637. [Google Scholar] [CrossRef]
- Skotte, N.H.; Sanders, S.S.; Ehrnhoefer, D.E.; Vaid, K.; Qiu, X.; Kannan, S.; Verma, C.; Hayden, M.R. Palmitoylation of caspase-6 by HIP14 regulates its activation. Cell Death Differ. 2017, 24, 433–444. [Google Scholar] [CrossRef]
- Cepeda, C.; Ariano, M.A.; Calvert, C.R.; Flores-Hernández, J.; Chandler, S.H.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S. NMDA receptor function in mouse models of Huntington disease. J. Neurosci. Res. 2001, 66, 525–539. [Google Scholar] [CrossRef]
- Goodman, L.; Baddeley, D.; Ambroziak, W.; Waites, C.L.; Garner, C.C.; Soeller, C.; Montgomery, J.M. N-terminal SAP97 isoforms differentially regulate synaptic structure and postsynaptic surface pools of AMPA receptors. Hippocampus 2017, 27, 668–682. [Google Scholar] [CrossRef]
- Hayashi, T. Post-translational palmitoylation of ionotropic glutamate receptors in excitatory synaptic functions. Br. J. Pharmacol. 2021, 178, 784–797. [Google Scholar] [CrossRef]
- Kang, R.; Wang, L.; Sanders, S.S.; Zuo, K.; Hayden, M.R.; Raymond, L.A. Altered regulation of striatal neuronal N-methyl-D-aspartate receptor trafficking by palmitoylation in Huntington disease mouse model. Front. Synaptic Neurosci. 2019, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kononenko, N.L.; Claßen, G.A.; Kuijpers, M.; Puchkov, D.; Maritzen, T.; Tempes, A.; Malik, A.R.; Skalecka, A.; Bera, S.; Jaworski, J.; et al. Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration. Nat. Commun. 2017, 8, 14819. [Google Scholar] [CrossRef] [PubMed]
- Matlahov, I.; van der Wel, P.C. Conformational studies of pathogenic expanded polyglutamine protein deposits from Huntington’s disease. Exp. Biol. Med. (Maywood) 2019, 244, 1584–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Rischardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Veldman, M.B.; Yang, X.W. Molecular insights into cortico-striatal miscommunications in Huntington’s disease. Curr. Opin. Neurobiol. 2018, 48, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Błaszczyk, J.W. Energy metabolism decline in the aging brain—Pathogenesis of neurodegenerative disorders. Metabolites 2020, 10, 450. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Abdulhadi, M.H.; Hussien, N.R.; Al-Niemi, M.S.; Rasheed, H.A.; Al-Gareeb, A.I. Involvement of orexinergic system in psychiatric and neurodegenerative disorders: A scoping review. Brain Circ. 2020, 6, 70–80. [Google Scholar] [CrossRef]
- Reiner, A.; Deng, Y.-P. Disrupted striatal neuron inputs and outputs in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 250–280. [Google Scholar] [CrossRef]
- Pérez-Navarro, E.; Canals, J.M.; Ginés, S.; Alberch, J. Cellular and molecular mechanisms involved in the selective vulnerability of striatal projection neurons in Huntington’s disease. Histol. Histopathol. 2006, 21, 1217–1232. [Google Scholar]
- Lee, S.T.; Chu, K.; Park, J.E.; Kang, L.; Ko, S.Y.; Jung, K.H.; Kim, M. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 2006, 1118, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Stroebel, D.; Casado, M.; Paoletti, P. Triheteromeric NMDA receptors: From structure to synaptic physiology. Curr. Opin. Physiol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurcau, A.; Ardelean, A.I. Oxidative stress in ischemia/reperfusion injuries following acute ischemic stroke. Biomedicines 2022, 10, 574. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Surmeier, D.J.; Reiner, A. NMDA and non-NMDA receptor-mediated excitotoxicity are potentiated in cultured striatal neurons by prior chronic depolarization. Exp. Neurol. 1999, 159, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.M.; Fan, M.M.; Fan, J.; Shehadeh, J.; Zhang, L.Y.; Graham, R.K.; Hayden, M.R.; Raymond, L.A. Polyglutamine-modulated striatal calpain activity in YAC transgenic Huntington disease mouse model: Impact on NMDA receptor function and toxicity. J. Neurosci. 2008, 28, 12725–12735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Zhang, Y.; Parsons, C.G.; Liu, Y.F. Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-D-aspartate receptors. J. Biol. Chem. 2003, 278, 33364–33369. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A.; Ardelean, I.A. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J. Integr. Neurosci. 2021, 20, 727–744. [Google Scholar] [PubMed]
- Raymond, L.A. Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem. Biophys. Res. Commun. 2017, 483, 1051–1062. [Google Scholar] [CrossRef]
- Braithwaite, S.P.; Paul, S.; Nairn, A.C.; Lombroso, P.J. Synaptic plasticity: One STEP at a time. Trends Neurosci. 2006, 29, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Saraf, J.; Bhattacharya, P.; Kalia, K.; Borah, A.; Sarmah, D.; Kaur, H.; Dave, K.R.; Yavagal, D.R. A friend or foe: Calcineurin across the gamut of neurological disorders. ACS Cent. Sci. 2018, 4, 805–819. [Google Scholar] [CrossRef]
- Kovalenko, M.; Milnerwood, A.; Giordano, J.; St Claire, J.; Guide, J.R.; Stromberg, M.; Gillis, T.; Sapp, E.; Di Figlia, M.; Mac Donald, M.; et al. Htt Q111/+ Huntington’s disease knock-in mice exhibit brain region-specific morphological changes and synaptic dysfunction. J. Huntingt. Dis. 2018, 7, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassel, B.; Tessler, S.; Faull, R.L.; Emson, P.C. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 2008, 33, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Petr, G.T.; Sun, Y.; Fischer, K.D.; Denovan-Wright, E.M.; Rosenberg, P.A. Huntington’s disease pattern of transcriptional dysregulation in the absence of mutant huntingtin is produced by knockout of neuronal GLT-1. Neurochem. Int. 2018, 123, 85–94. [Google Scholar] [CrossRef]
- Groc, L.; Choquet, D. Linking glutamate receptor movements and synapse function. Science 2020, 368, eaay4631. [Google Scholar] [CrossRef]
- Rebec, G.V. Corticostriatal network dysfunction in Huntington’s disease: Deficits in neural processing, glutamate transport, and ascorbate release. CNS Neurosci. Ther. 2018, 24, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, M.; Fraga, D.B.; Rodrigues, A.L.S. Preventive and therapeutic potential of ascorbic acid in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 921–929. [Google Scholar] [CrossRef]
- Hassanzadeh, P.; Atyabi, F.; Dinarvand, R. Application of modelling and nanotechnology-based approaches: The emergence of breakthroughs in theranostics of central nervous system disorders. Life Sci. 2017, 182, 93–103. [Google Scholar] [CrossRef]
- Garret, M.; Du, Z.; Chalazon, M.; Cho, Y.H.; Baufreton, J. Alteration of GABAergic neurotransmission in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 292–300. [Google Scholar] [CrossRef]
- Olsen, R.W.; Sieghart, W. GABA A receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology 2009, 56, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Ferando, I.; Mody, I. Interneuronal GABAA receptors inside and outside of synapses. Curr. Opin. Neurobiol. 2014, 26, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Tartrais, M.; Coutand, G.; Leste-Lasserre, T.; Cardoit, L.; Masmejean, F.; Halgand, C.; Cho, Y.H.; Garret, M. Differential alteration in expression of striatal GABAAR subunits in mouse models of Huntington’s disease. Front. Mol. Neurosci. 2017, 10, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Pandey, S.; Li, T.; Castellano, D.; Gu, X.; Li, J.; Tian, Q.; Lu, W. Genetic deletion of GABAA receptors reveals distinct requirements of neurotransmitter receptors for GABAergic and glutamatergic synapse development. Front. Cell. Neurosci. 2019, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, S.; Fatima, A.; Gutierrez-Garcia, R.; Vilchez, D. Proteostasis of Huntingtin in Health and Disease. Int. J. Mol. Sci. 2017, 18, 1568. [Google Scholar] [CrossRef] [Green Version]
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Berat, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the tightrope: Proteostasis and neurodegenerative disease. J. Neurochem. 2016, 137, 489–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Nillegoda, N.B.; Wenting, A.S.; Bukau, B. Protein disaggregation in multicellular organisms. Trends Biochem. Sci. 2018, 43, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, S.D.; Pinho, B.R.; Oliveira, J. Modulation of molecular chaperones in Huntington’s disease and other polyglutamine disorders. Mol. Neurobiol. 2017, 54, 5829–5854. [Google Scholar] [CrossRef] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- VerPlank, J.J.S.; Goldberg, A.L. Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 2017, 474, 3355–3371. [Google Scholar] [CrossRef] [Green Version]
- Collins, I.; Wang, H.; Caldwell, J.J.; Chopra, R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin–proteasome pathway. Biochem. J. 2017, 474, 1127–1147. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, A.B.; Gersing, S.K.; Larsen-Ledet, S.; Nielsen, S.V.; Stein, A.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Co-Chaperones in targeting and delivery of misfolded proteins to the 26S proteasome. Biomolecules 2020, 10, 1141. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.R.; Blount, J.R.; Libohova, K.; Tsou, W.L.; Joshi, G.S.; Paulson, H.L.; do Carmo Costa, M.; Scaglione, K.M.; Todi, S.V. Interaction of the polyglutamine protein ataxin-3 with Rad23 regulates toxicity in Drosophila models of Spinocerebellar Ataxia Type 3. Hum. Mol. Genet. 2017, 26, 1419–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.T.; Xue, W.; Gao, Y.G.; Hong, J.Y.; Yue, H.W.; Jiang, L.L.; Hu, H.Y. HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci. Rep. 2017, 7, 14797. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.R.; Lieberman, A.P. The ubiquitination, disaggregation and proteasomal degradation machineries in polyglutamine disease. Front. Mol. Neurosci. 2017, 10, 78. [Google Scholar] [CrossRef]
- Li, X.J.; Li, S. Proteasomal dysfunction in aging and Huntington’s disease. Neurobiol. Dis. 2011, 43, 4–8. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, N.; Venkatraman, P.; Goldberg, A.L. Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO J. 2007, 26, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.; Curley, M.; Coleman, Z.; Demontis, F. Contribution of proteases to the hallmarks of aging and to age-related neurodegeneration. Aging Cell 2022, 21, e13603. [Google Scholar] [CrossRef]
- Maheshwari, M.; Samanta, A.; Godavarthi, S.K.; Mukherjee, R.; Jana, N.R. Dysfunction of the ubiquitin ligase Ube3a may be associated with synaptic pathophysiology in a mouse model of Huntington disease. J. Biol. Chem. 2012, 287, 29949–29957. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.D.; Schmidt, M.E.; Nguyen, Y.T.; Lazic, N.; Hayden, M.R. Identification of a novel caspase cleavage site in huntingtin that regulates mutant huntingtin clearance. FASEB J. 2019, 33, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurcau, A. Insights into the pathogenesis of neurodegenerative diseases: Focus on mitochondrial dysfunction and oxidative stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays 2018, 40, 1800008. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein-protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering mutant huntingtin levels and toxicity: Autophagy-endolysosome pathways in Huntington’s disease. J. Mol. Biol. 2020, 432, 2673–2691. [Google Scholar] [CrossRef]
- Yang, J.; Chen, X.; Xu, H. SQSTM1/p62 droplet-mediated autophagosome formation: Insights into Huntington disease. Autophagy 2021, 10, 3256–3259. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, P.; Pan, Y.; Sun, X.; DiFiglia, M.; Lu, B. A toxic mutant huntingtin species is resistant to selective autophagy. Nat. Chem. Biol. 2017, 13, 1152. [Google Scholar] [CrossRef]
- Sun, X.; Fu, Y.; Pan, Y.; Lu, B. Conformation-dependent recognition of mutant HTT (huntingtin) proteins by selective autophagy. Autophagy 2017, 13, 2111–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Carabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.M.; Park, S.J.; Fernandez-Estevez, M.; Rubinsztein, D.C. Autophagy regulation by acetylation—implications for neurodegenerative diseases. Exp. Mol. Med. 2021, 53, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.G.; Sathasivam, K.; Tobaben, S.; Stahl, B.; Marber, M.; Mestril, R.; Mahel, A.; Smith, D.L.; Woodman, B.; Bates, G.P. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum. Mol. Genet. 2004, 13, 1389–1405. [Google Scholar] [CrossRef] [Green Version]
- Wanker, E.E. Protein aggregation and pathogenesis of Huntington’s disease: Mechanisms and correlations. Biol. Chem. 2000, 381, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.K.; Jayaraman, M.; Mishra, R.; Thakur, M.; Chellgren, V.M.; Byeon, I.J.; Anjum, D.H.; Kodali, R.; Creamer, T.P.; Conway, J.F.; et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009, 16, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Moily, N.S.; Ormsby, A.R.; Stojilovic, A.; Ramdzan, Y.M.; Diesch, J.; Hannan, R.D.; Zajac, M.S.; Hannan, A.J.; Oshlack, A.; Hatters, D.M. Transcriptional profiles for distinct aggregation states of mutant huntingtin exon 1 protein unmask new Huntington’s disease pathways. Mol. Cell. Neurosci. 2017, 83, 103–112. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.Y.; Uno, Y.; Sha, E.; Ikegami, K.; Ishii, N.; Dohmae, N.; Sekiguchi, H.; Sasaki, Y.C.; Yohda, M. Asymmetry in the function and dynamics of the cytosolic group II chaperonin CCT/TRIC. PLoS ONE 2017, 12, e0176054. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Tydlacka, S.; Wang, C.E.; Wang, X.; Li, S.; Li, X.J. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J. Neurosci. 2007, 28, 13285–13295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merienne, N.; Meunier, C.; Schneider, A.; Seguin, J.; Nair, S.S.; Rocher, A.B.; Le Gras, S.; Keime, C.; Faull, R.; Pellerin, L.; et al. Cell-type-specific gene expression profiling in adult mouse brain reveals normal and disease-state signatures. Cell Rep. 2019, 26, 2477–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuemmerle, S.; Gutekunst, C.A.; Klein, A.M.; Li, X.J.; Li, S.H.; Beal, M.F.; Hersch, S.M.; Ferrante, R.J. Huntingtin aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol. 1999, 46, 842–849. [Google Scholar] [CrossRef]
- Yang, J.; Yang, X. Phase transition of huntingtin: Factors and pathological relevance. Front. Genet. 2020, 11, 754. [Google Scholar] [CrossRef]
- Pandey, N.K.; Mario Isas, J.; Rawat, A.; Lee, R.V.; Langen, J.; Pandey, P.; Langen, R. The 17-residue-long N-terminus in huntingtin controls stepwise aggregation in solutions and on membranes via different mechanisms. J. Biol. Chem. 2018, 293, 2597–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crick, S.L.; Ruff, K.M.; Garai, K.; Frieden, C.; Pappu, R.V. Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 20075–20080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mario Isas, J.; Pandey, N.K.; Xu, H.; Teranishi, K.; Okada, A.K.; Fultz, E.K.; Rawat, A.; Applebaum, A.; Meier, F.; Chen, J.; et al. Huntingtin fibrils with different toxicity, structure, and seeding potential can be interconverted. Nat. Commun. 2021, 12, 4272. [Google Scholar] [CrossRef]
- Schindler, F.; Praedel, N.; Neuendorf, N.; Kunz, S.; Schnoegl, S.; Mason, M.A.; Taxy, B.A.; Bates, G.P.; Khoshnan, A.; Priller, J.; et al. Small, seeding-competent huntingtin fibrils are prominent aggregate species in brains of zQ175 Huntington’s disease knock-in mice. Front. Neurosci. 2021, 15, 766. [Google Scholar] [CrossRef]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 30, 2953–2960. [Google Scholar] [CrossRef] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Sabouny, R.; Shutt, T.E. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem. Sci. 2020, 45, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H. The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef] [PubMed]
- López-Doménech, G.; Coville-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Fenton, A.R.; Jongens, T.A.; Holzbaur, E.L.F. Mitochondrial adaptor TRAK2 activates and functionally links opposing kinesin and dynein motors. Nat. Commun. 2021, 12, 4578. [Google Scholar] [CrossRef]
- Leites, E.P.; Morais, V.A. Mitochondrial quality control pathways: PINK1acts as a gatekeeper. Biochem. Biophys. Res. Commun. 2018, 500, 45–50. [Google Scholar] [CrossRef]
- Swerdlow, N.S.; Wilkins, H.M. Mitophagy and the brain. Int. J. Mol. Sci. 2020, 21, 9661. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Agrotis, A.; Pengo, N.; Burden, J.J.; Ketteler, R. Redundancy of human Atg4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 2019, 15, 976–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurey, P.; Rieusste, J. Mitochondria-associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrin. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchitsu, Y.; Fukuda, M. Revisiting Rab7 functions in mammalian autophagy: Rab7 knockout studies. Cells 2018, 7, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Jiang, X.; Yang, Q.; Zhao, J.; Zhou, Q.; Zhou, Y. The functions, methods, and mobility of mitochondrial transfer between cells. Front. Oncol. 2021, 11, 672781. [Google Scholar] [CrossRef]
- Nicholls, D.G. Brain mitochondrial calcium transport: Origins of the set-point concept and its application to physiology and pathology. Neurochem. Int. 2017, 109, 5–12. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Reane, D.V.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Eur. J. Physiol. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [Green Version]
- Briston, T.; Selwood, D.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition: A molecular lesion with multiple drug targets. Trends Pharm. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Feigin, A.; Leenders, K.L.; Moeller, J.R.; Missimer, J.; Kuenig, G.; Spetsieris, P.; Antonini, A.; Eidelberg, D. Metabolic network abnormalities in early Huntington’s disease: An [(18)F]FDG PET study. J. Nucl. Med. 2001, 42, 1591–1595. [Google Scholar]
- The HD iPSC Consortium. Bioenergetic deficits in Huntington’s disease iPSC-derived neural cells and rescue with glycolytic metabolites. Hum. Mol. Genet. 2020, 29, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Solís-Maldonado, M.; Miró, M.P.; Acuña, A.I.; Covarrubias-Pinto, A.; Loaiza, A.; Mayorga, G.; Beltrán, F.A.; Cepeda, C.; Levine, M.S.; Concha, I.I.; et al. Altered lactate metabolism in Huntington’s disease is dependent on GLUT3 expression. CNS Neurosci. Ther. 2018, 24, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Liang, J.; Zhou, B. Glucose metabolic dysfunction in neurodegenerative diseases—New mechanistic insights and the potential of hypoxia as a prospective therapy targeting metabolic reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef] [PubMed]
- Yablonska, S.; Ganesan, V.; Ferrando, L.M.; Friedlander, R.M. Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc. Natl. Acad. Sci. USA 2019, 116, 16593–16602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.H.; Cho, H.; SEol, Y.H.; Kim, S.H.; Park, C.; Yousefian-Yazi, A.; Hyeon, S.J.; Lee, J.; Ryu, H. Power failure of mitochondria and oxidative stress in neurodegeneration and its computational model. Antioxidants 2021, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H. Mitochondrial dysfunction in aging and diseases of aging. Biology 2019, 8, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Mackay, J.P.; Nassrallah, W.B.; Raymond, L.A. Cause or compensation? Altered neuronal Ca2+ handling in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Jiang, S.; Yang, Z.; Hu, W.; Wang, Z.; Li, T.; Yang, Y. PGC-1α sparks the fire of neuroprotection against neurodegenerative disorders. Ageing Res. Rev. 2018, 44, 8–21. [Google Scholar] [CrossRef]
- Jesse, S.; Bayer, H.; Alupei, M.C.; Zügel, M.; Mulaw, M.; Tuorto, F.; Malmsheimer, S.; Singh, K.; Steinacker, J.; Schumann, U.; et al. Ribosomal transcription is regulated by PGC-1alpha and disturbed in Huntington’s disease. Sci. Rep. 2017, 7, 8513. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Ban, J.J.; Chung, J.Y.; Im, W.; Kim, M. Amelioration of Huntington’s disease phenotypes by Beta-Lapachone is associated with increases in Sirt1 expression, CREB phosphorylation and PGC-1α deacetylation. PLoS ONE 2018, 13, e0195968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czeredys, M.; Maciag, F.; Methner, A.; Kuznicki, J. Tetrahydrocarbazoes decrease elevated SOCE in medium spiny neurons from transgenic YAC128 mice, a model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1194–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimoro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2004, 47, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant, N.; Morton, H.; Kshirsagar, S.; Reddy, A.P.; Reddy, P.H. Mitochondrial abnormalities and synaptic damage in Huntington’s disease: A focus on defective mitophagy and mitochondria-targeted therapeutics. Mol. Neurobiol. 2021, 58, 6350–6377. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca2+ efflux and reactive oxygen species (ROS) homeostasis. Neurobiol. Dis. 2020, 136, 104741. [Google Scholar] [CrossRef] [PubMed]
- Roe, A.J.; Qi, X. Drp1 phosphorylation by MAPK1 causes mitochondrial dysfunction in cell culture model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2018, 496, 706–711. [Google Scholar] [CrossRef]
- Lucero, M.; Suarez, A.E.; Chambers, J.W. Phosphoregulation on mitochondria: Integration of cell and organelle responses. CNS Neurosci. Ther. 2019, 25, 837–858. [Google Scholar] [CrossRef] [Green Version]
- Aufschnaiter, A.; Kohler, V.; Büttner, S. Taking out the garbage: Cathepsin D and calcineurin in neurodegeneration. Neur. Regen. Res. 2017, 12, 1776. [Google Scholar]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Machiela, E.; Rudich, P.D.; Traa, A.; Anglas, U.; Soo, S.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Targeting mitochondrial network disorganization is protective in C. elegans models of Huntington’s disease. Aging Dis. 2021, 12, 1753. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.T.; Rintoul, G.L.; Pandipati, S.; Reynolds, I.J. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 2006, 22, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kalonia, H.; Kumar, A. Huntington’s disease: Pathogenesis to animal models. Pharmacol. Rep. 2010, 62, 1–14. [Google Scholar] [CrossRef]
- Jurcau, A.; Nunkoo, V.S. Clinical markers may identify patients at risk for early Parkinson’s disease dementia: A prospective study. Am. J. Alzheimers Dis. Other Dement. 2021, 36, 15333175211021369. [Google Scholar] [CrossRef]
- Xiang, C.; Zhang, S.; Dong, X.; Ma, S.; Cong, S. Transcriptional dysregulation and post-translational modifications in polyglutamine diseases: From pathogenesis to potential therapeutic targets. Front. Mol. Neurosci. 2018, 11, 153. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, M.; Yu, Y.; Qiu, L.; Zhang, Y.; He, L.; Zhang, J. Brain REST/NRSF is not only a silent repressor but also an active protector. Mol. Neurobiol. 2017, 54, 541–550. [Google Scholar] [CrossRef]
- Chen, G.L.; Ma, Q.; Goswami, D.; Shang, J.; Miller, G.M. Modulation of nuclear REST by alternative splicing: A potential therapeutic target for Huntington’s disease. J. Cell. Mol. Med. 2017, 21, 2974–2984. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [Green Version]
- Lloret, A.; Beal, M.F. PGC-1α, sirtuins and PARPs in Huntington’s disease and other neurodegenerative conditions: NAD+ to rule them all. Neurochem. Res. 2019, 44, 2423–2434. [Google Scholar] [CrossRef]
- Johri, A.; Starkov, A.A.; Chandra, A.; Hennessey, T.; Sharma, A.; Orobello, S.; Squitieri, F.; Yang, L.; Beal, M.F. Truncated peroxisome proliferator-activated receptor-γ coactivator 1α splice variant is severely altered in Huntington’s disease. Neurodegener. Dis. 2011, 8, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Di Cristo, F.; Finicelli, M.; Gigilio, F.A.; Paladino, S.; Valentino, A.; Scialó, F.; D’Apolito, M.; Saturnino, C.; Galderisi, U.; Giordano, A.; et al. Meldonium improves Huntington’s disease mitochondrial dysfunction by restoring peroxisome proliferator-activated receptor γ coactivator 1α expression. J. Cell. Physiol. 2019, 234, 9233–9246. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Jeong, H.; Cui, L.; Krainc, D.; Tjian, R. In vitro analysis of Huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell 2005, 123, 1241–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reijonen, S.; Kukkonen, J.P.; Hyrskyluoto, A.; Kivinen, J.; Kairisalo, M.; Takei, N.; Lindholm, D.; Korhonen, L. Downregulation of NF-κB signaling by mutant huntingtin proteins induces oxidative stress and cell death. Cell. Mol. Life Sci. 2010, 67, 1929–1941. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chen, Y.C.; Chen, H.M.; Tu, P.H.; Chern, Y. A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet. 2013, 22, 1826–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.Y.; Zukin, R.S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Kim, S.H.; Shahani, N.; Bae, B.I.; Sbodio, J.I.; Chung, Y.; Nakaso, K.; Paul, B.D.; Sawa, A. Allele-specific regulation of mutant Huntingtin by Wig1, a downstream target of p53. Hum. Mol. Genet. 2016, 25, 2514–2524. [Google Scholar]
- Khandelwal, N.; Cavalier, S.; Rybalchenko, V.; Kulkarni, A.; Anderson, A.G.; Konopka, G.; Gibson, J.R. FOXP1 negatively regulates intrinsic excitability in D2 striatal projection neurons by promoting inwardly rectifying and leak potassium currents. Mol. Psychiatry 2021, 26, 1761–1774. [Google Scholar] [CrossRef]
- Neueder, A.; Gipson, T.A.; Batterton, S.; Lazell, H.J.; Farshim, P.P.; Paganetti, P.; Housmann, D.E.; Bates, G.P. HSF1-dependent and-independent regulation of the mammalian in vivo heat shock response and its impairment in Huntington’s disease mouse models. Sci. Rep. 2017, 7, 12556. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.; Kong, G.; Tran, H.; Li, S.; Pang, T.Y.; Hannan, A.J.; Renoir, T. Small non-coding RNAs are dysregulated in Huntington’s disease transgenic mice independently of the therapeutic effects of an environmental intervention. Mol. Neurobiol. 2021, 58, 3308–3318. [Google Scholar] [CrossRef]
- Paraskevopoulos, F.; Parvizi, P.; Senger, G.; Tuncbag, N.; Rosenmund, C.; Yildirim, F. Impaired inhibitory GABAergic synaptic transmission and transcription studied in single neurons by Patch-seq in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2020293118. [Google Scholar] [CrossRef] [PubMed]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Pla, P.; Orvoen, S.; Saudou, F.; David, D.J.; Humbert, S. Mood disorders in Huntington’s disease: From behavior to cellular and molecular mechanisms. Front. Behav. Neurosci. 2014, 8, 135. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntingtin’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, F.R.; Paladino, E. Role of phosphodiesterases in Huntington’s disease. Adv. Neurobiol. 2017, 17, 285–304. [Google Scholar] [PubMed]
- Knott, E.P.; Assi, M.; Rao, S.N.; Ghosh, M.; Pearse, D.D. Phosphodiesterase inhibitors as a therapeutic approach to neuroprotection and repair. Int. J. Mol. Sci. 2017, 18, 696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinale, A.; Fusco, F.R. Inhibition of phosphodiesterases as a strategy to achieve neuroprotection in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Yousefian-Jazi, A.; Choi, S.H.; Chang, I.; Lee, J.; Ryu, H. Non-cell autonomous and epigenetic mechanisms of Huntington’s disease. Int. J. Mol. Sci. 2021, 22, 12499. [Google Scholar] [CrossRef]
- Twelvetrees, A.E.; Lesept, F.; Holzbaur, E.L.; Kittler, J.T. The adaptor proteins HAP1a and GRIP1 collaborate to activate the kinesin-1 isoform KIF5C. J. Cell Sci. 2019, 132, 215822. [Google Scholar] [CrossRef] [Green Version]
- Kalchman, M.A.; Koide, H.B.; McCutcheon, K.; Graham, R.K.; Nichol, K.; Nishiyama, K.; Kazemi-Esfarjani, P.; Lynn, F.C.; Wellington, C.; Metzler, M.; et al. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat. Genet. 1997, 16, 44–53. [Google Scholar] [CrossRef]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.-H.; Wang, Y.; Sapp, E.; Cuiffo, B.; Wanker, E.; Hayden, M.R.; Kegel, K.B.; Aronin, N.; DiFiglia, M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 2004, 24, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 2008, 18, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Ginés, S.; Paoletti, P.; Alberch, J. Impaired TrkB-mediated ERK1/2 activation in Huntington disease knock-in striatal cells involves reduced p52/p46 Shc expression. J. Biol. Chem. 2010, 285, 21537–21548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazo, O.M.; Gonzalez, A.; Ascano, M.; Kuruvilla, R.; Couve, A. BDNF regulates Rab11-mediated recycling endosome dynamics to induce dendritic branching. J. Neurosci. 2013, 33, 6112–6122. [Google Scholar] [CrossRef]
- Josephy-Hernández, S.; Jmaeff, S.; Pirvulescu, I.; Aboulkassim, T.; Saragovi, H.U. Neurotrophin receptor agonists and antagonists as therapeutic agents: An evolving paradigm. Neurobiol. Dis. 2017, 97, 139–155. [Google Scholar] [CrossRef]
- Simmons, D.A.; Longo, F.M.; Massa, S.M. Neurotrophin receptor signaling as a therapeutic target for Huntington’s disease. CNS Neurol. Disord. Drug Targets 2017, 16, 291–302. [Google Scholar] [CrossRef]
- Gómez-Pineda, V.G.; Torres-Cruz, F.M.; Vivar-Cortés, C.I.; Hernández-Echeagaray, E. Neurotrophin-3 restores synaptic plasticity in the striatum of a kouse model of Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Jamwal, S.; Kumar, P. Insight into the emerging role of striatal neurotransmitters in the pathophysiology of Parkinson’s disease and Huntington’s disease: A review. Curr. Neuropharmacol. 2019, 17, 165–175. [Google Scholar] [CrossRef]
- Tepper, J.M.; Koós, T.; Ibanez-Sandoval, O.; Tecuapetla, F.; Faust, T.W.; Assous, M. Heterogeneity and diversity of striatal GABAergic interneurons: Update 2018. Front. Neuroanat. 2018, 12, 91. [Google Scholar] [CrossRef] [Green Version]
- Castela, I.; Hernandez, L.F. Shedding light on dyskinesias. Eur. J. Neurosci. 2021, 53, 2398–2413. [Google Scholar] [CrossRef] [PubMed]
- Sebastianutto, I.; Cenci, M.A.; Fieblinger, T. Alterations of striatal indirect pathway neurons precede motor deficits in two mouse models of Huntington’s disease. Neurobiol. Dis. 2017, 105, 117–131. [Google Scholar] [CrossRef] [PubMed]
- André, V.M.; Cepeda, C.; Fisher, Y.E.; Huyhn, M.; Bardakijan, N.; Singh, S.; Yang, X.W.; Levine, M.S. Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J. Neurosci. 2011, 31, 1170–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, B.M.; Lopes, E.F.; Cragg, S.J. Axonal modulation of striatal dopamine release by local γ-aminobutyric acid (GABA) signalling. Cells 2021, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Dragunow, M.; Faull, R.L. The pattern of neurodegeneration in Huntington’s disease: A comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience 2000, 97, 505–519. [Google Scholar] [CrossRef]
- Cha, J.H.; Kosinski, C.M.; Kerner, J.A.; Alsdorf, S.A.; Mangiarini, L.; Davies, S.W.; Penney, J.B.; Bates, G.P.; Young, A.B. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc. Natl. Acad. Sci. USA 1998, 95, 6480–6485. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, C.; Hurst, R.S.; Calvert, C.R.; Hernandez-Echeagaray, E.; Nguyen, O.K.; Jocoy, E.; Christian, L.J.; Ariano, M.A.; Levine, M.S. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J. Neurosci. 2003, 23, 961–969. [Google Scholar] [CrossRef]
- Wiprich, M.T.; Bonan, C.D. Purinergic signaling in the pathophysiology and treatment of Huntington’s disease. Front. Neurosci. 2021, 15, 657338. [Google Scholar] [CrossRef]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci. Adv. 2018, 2, 2398212818817494. [Google Scholar] [CrossRef] [Green Version]
- Gundlfinger, A.; Bischofberger, J.; Johenning, F.W.; Torvinen, M.; Schmitz, D.; Breustedt, J. Adenosine modulates transmission at the hippocampal mossy fiber synapse via direct inhibition of presynaptic calcium channels. J. Physiol. 2007, 582, 263–277. [Google Scholar] [CrossRef]
- Léon-Navarro, D.A.; Albasanz, J.L.; Martin, M. Functional cross-talk between adenosine and metabotropic glutamate receptors. Curr. Neuropharmacol. 2018, 17, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, M.; Glaser, T.; Slominska, E.M.; Ulrich, H.; Smolenski, R.T. Purine nucleotide metabolism and signaling in Huntington’s disease: Search for a target for novel therapies. Int. J. Mol. Sci. 2021, 22, 6545. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Hernández, M.; Díez-Zaera, M.; Sánchez-Nogueiro, J.; Gómez-Villafuertes, R.; Canals, J.M.; Alberch, J.; Miras-Portugal, M.T.; Lucas, J.J. Altered P2X7-receptor level and function in mouse models of Huntington’s disease and therapeutic efficacy of antagonist administration. FASEB J. 2009, 23, 1893–1906. [Google Scholar] [CrossRef]
- Pini, L.; Jacquemot, C.; Cagnin, A.; Meneghello, F.; Semenza, C.; Mantini, D.; Vallesi, A. Aberrant brain network connectivity in presymptomatic and manifest Huntington’s disease: A systematic review. Hum. Brain Mapp. 2020, 41, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Brymer, K.J.; Barnes, J.R.; Parsons, M.P. Entering a new era of quantifying glutamate clearance in health and disease. J. Neurosci. Res. 2021, 99, 1598–1617. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Barry, J.; Yang, Z.; Cepeda, C.; Levine, M.S.; Gray, M. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet. 2019, 28, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 2017, 40, 422–437. [Google Scholar] [CrossRef]
- Liot, G.; Valette, J.; Pepin, J.; Flament, J.; Brouillet, E. Energy defects in Huntington’s disease: Why “in vivo” evidence matters. Biochem. Biophys. Res. Commun. 2017, 483, 1084–1095. [Google Scholar] [CrossRef]
- Zhang, X.; Wan, J.Q.; Tong, X.P. Potassium channel dysfunction in neurons and astrocytes in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Ariano, M.A.; Wagle, N.; Grissell, A.E. Neuronal vulnerability in mouse models of Huntington’s disease: Membrane channel protein changes. J. Neurosci. Res. 2005, 80, 634–645. [Google Scholar] [CrossRef]
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofroniew, M.V.; et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 2014, 17, 694–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, Y.; Kinboshi, M.; Shimizu, S. Inwardly rectifying potassium channel Kir4. 1 as a novel modulator of BDNF expression in astrocytes. Int. J. Mol. Sci. 2018, 19, 3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Skotte, N.H.; Andersen, J.V.; Santos, A.; Aldana, B.I.; Willert, C.W.; Nørremølle, A.; Waagepetersen, H.S.; Nielsen, M.L. Integrative characterization of the R6/2 mouse model of Huntington’s disease reveals dysfunctional astrocyte metabolism. Cell Rep. 2018, 23, 2211–2224. [Google Scholar] [CrossRef] [PubMed]
- Dvorzhak, A.; Helassa, N.; Török, K.; Schmitz, D.; Grantyn, R. Single synapse indicators of impaired glutamate clearance derived from fast iGluu imaging of cortical afferents in the striatum of normal and Huntington (Q175) mice. J. Neurosci. 2019, 39, 3970–3982. [Google Scholar] [CrossRef] [Green Version]
- Valtcheva, S.; Venance, L. Astrocytes gate Hebbian synaptic plasticity in the striatum. Nat. Commun. 2016, 7, 13845. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Diaz-Castro, B.; Tong, X.; Looger, L.L.; Khakh, B.S. Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.J.; Rushton, D.J.; Tom, C.M.; Allen, N.D.; Kemp, P.J.; Svendsen, C.N.; Mattis, V.B. Huntington’s disease patient-derived astrocytes display electrophysiological impairments and reduced neuronal support. Front. Neurosci. 2019, 13, 669. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Cognition, statins, and cholesterol in elderly ischemic stroke patients: A neurologist’s perspective. Medicina 2021, 57, 616. [Google Scholar] [CrossRef]
- Leoni, V.; Mariotti, C.; Tabrizi, S.J.; Valenza, M.; Wild, E.J.; Henley, S.M.; Hobbs, N.Z.; Mandelli, M.L.; Grisoli, M.; Björkhelm, I.; et al. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain 2008, 131, 2851–2859. [Google Scholar] [CrossRef]
- Phillips, O.; Squitieri, F.; Sanchez-Castaneda, C.; Elifani, F.; Caltagirone, C.; Sabatini, U.; Di Paola, M. Deep white matter in Huntington’s disease. PLoS ONE 2014, 9, e109676. [Google Scholar] [CrossRef] [PubMed]
- Bardile, C.F.; Garcia-Miralles, M.; Caron, N.S.; Rayan, N.A.; Langley, S.R.; Harmston, N.; Rondelli, A.M.; Teo, R.T.Y.; Waltl, S.; Anderson, L.M.; et al. Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc. Natl. Acad. Sci. USA 2019, 116, 9622–9627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Valenza, M.; Cui, L.; Leoni, V.; Jeong, H.K.; Brilli, E.; Zhang, J.; Peng, Q.; Duan, W.; Reeves, S.A.; et al. Peroxisome proliferator-activated receptor gamma coactivator 1 α contributes to dysmyelination in experimental models of Huntington’s disease. J. Neurosci. 2011, 31, 9544–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarette, L.P.; Pastor, M.G.; Ramos-Escobar, N. neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurcau, A.; Simion, A. Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: From pathophysiology to therapeutic strategies. Int. J. Mol. Sci. 2022, 23, 14. [Google Scholar] [CrossRef]
- Ollà, I.; Santos-Galindo, M.; Elorza, A.; Lucas, J.J. P2X7 receptor upregulation in Huntington’s disease brains. Front. Mol. Neurosci. 2020, 13, 567430. [Google Scholar] [CrossRef]
- Savage, J.C.; St-Pierre, M.K.; Carrier, M.; El Hajj, H.; Novak, S.W.; Sanchez, M.G.; Cicchetti, F.; Tremblay, M.È. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflamm. 2020, 17, 98. [Google Scholar] [CrossRef]
- Silvestroni, A.; Faull, R.L.; Strand, A.D.; Möller, T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport 2009, 20, 1098–1103. [Google Scholar] [CrossRef]
- Rocha, N.P.; Charron, O.; Latham, L.B.; Colpo, G.D.; Zanotti-Fregonara, P.; Yu, M.; Freeman, L.; Furr Stimming, E.; Teixeira, A.L. Microglia activation in basal ganglia is a late event in Huntington disease pathophysiology. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e984. [Google Scholar] [CrossRef]
- O’Regan, G.C.; Farag, S.H.; Casey, C.S.; Wood-Kaczmar, A.; Pocock, J.M.; Tabrizi, S.J.; Andre, R. Human Huntington’s disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species. J. Neuroinflamm. 2021, 18, 94. [Google Scholar] [CrossRef]
- Yang, H.M.; Yang, S.; Huang, S.S.; Tang, B.S.; Guo, J.F. Microglial activation in the pathogenesis of Huntington’s disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Möller, T. Neuroinflammation in Huntington’s disease. J. Neural Transm. 2010, 117, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Chiarlone, A.; Bellocchio, L.; Resel, E.; Pruunsild, P.; García-Rincón, D.; Sendtner, M.; Timmusk, T.; Lutz, B.; Galve-Roperh, I. The CB(1) receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015, 22, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Komorowska-Müller, J.A.; Schmöle, A.C. CB2 receptor in microglia: The guardian of self-control. Int. J. Mol. Sci. 2020, 22, 19. [Google Scholar] [CrossRef]
- Young, A.P.; Denovan-Wright, E.M. The dynamic role of microglia and the endocannabinoid system in neuroinflammation. Front. Pharmacol. 2022, 12, 806417. [Google Scholar] [CrossRef]
- Valdeolivas, S.; Nevarette, C.; Cantarero, I.; Ballido, M.L.; Muñoz, E.; Sagredo, O. Neuroprotective properties of cannabigerol in Huntington’s disease: Studies in R6/2 mice and 3-nitropropionate-lesioned mice. Neurotherapeutics 2015, 12, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The role of microglia and astrocytes in Huntington’s disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurcau, A. Molecular Pathophysiological Mechanisms in Huntington’s Disease. Biomedicines 2022, 10, 1432. https://doi.org/10.3390/biomedicines10061432
Jurcau A. Molecular Pathophysiological Mechanisms in Huntington’s Disease. Biomedicines. 2022; 10(6):1432. https://doi.org/10.3390/biomedicines10061432
Chicago/Turabian StyleJurcau, Anamaria. 2022. "Molecular Pathophysiological Mechanisms in Huntington’s Disease" Biomedicines 10, no. 6: 1432. https://doi.org/10.3390/biomedicines10061432
APA StyleJurcau, A. (2022). Molecular Pathophysiological Mechanisms in Huntington’s Disease. Biomedicines, 10(6), 1432. https://doi.org/10.3390/biomedicines10061432