
Therapeutic Aspects and Molecular Targets of Autophagy to Control Pancreatic Cancer Management

 ,
,  ,
,  , and
, and 
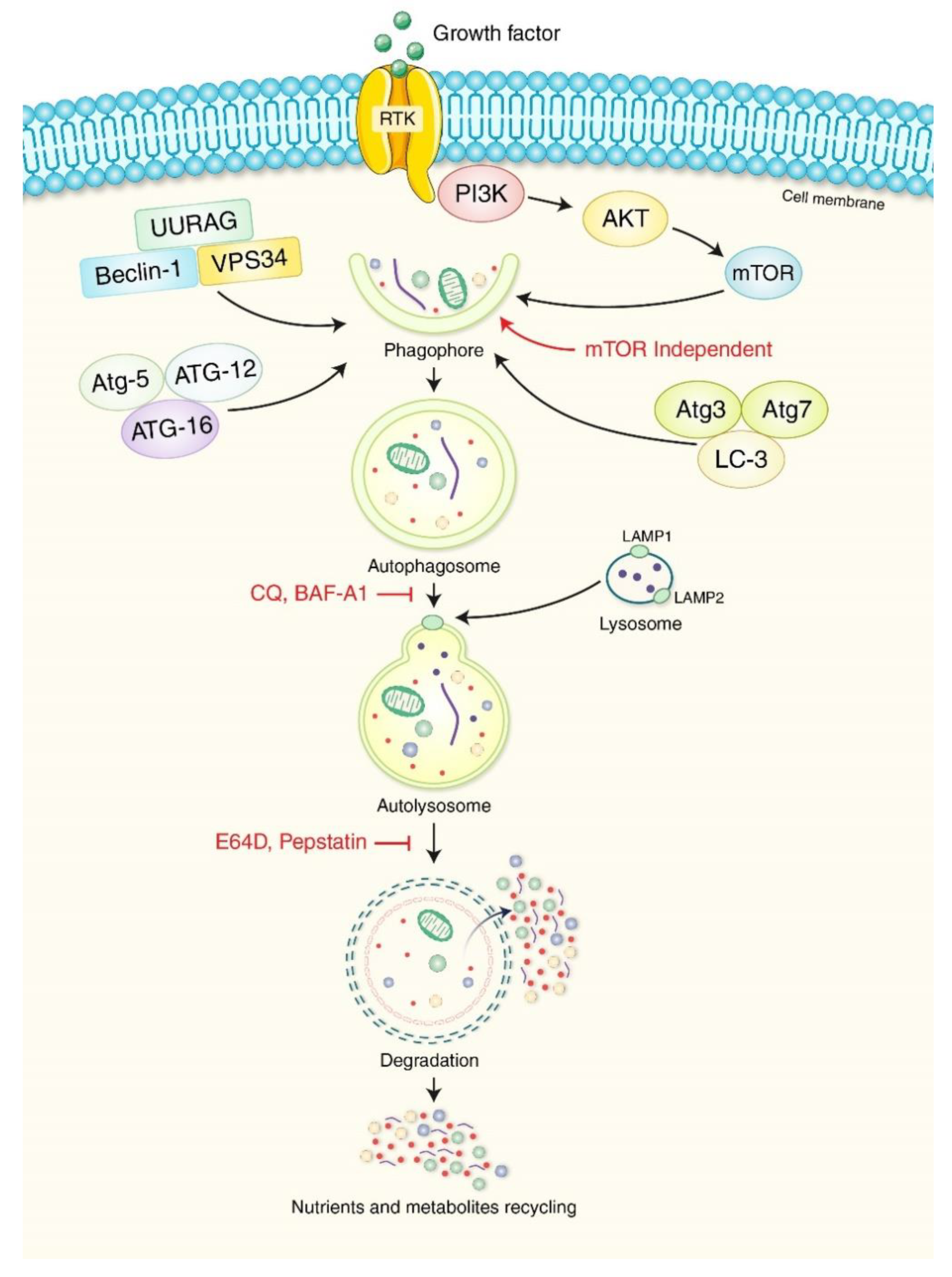{kind=link}
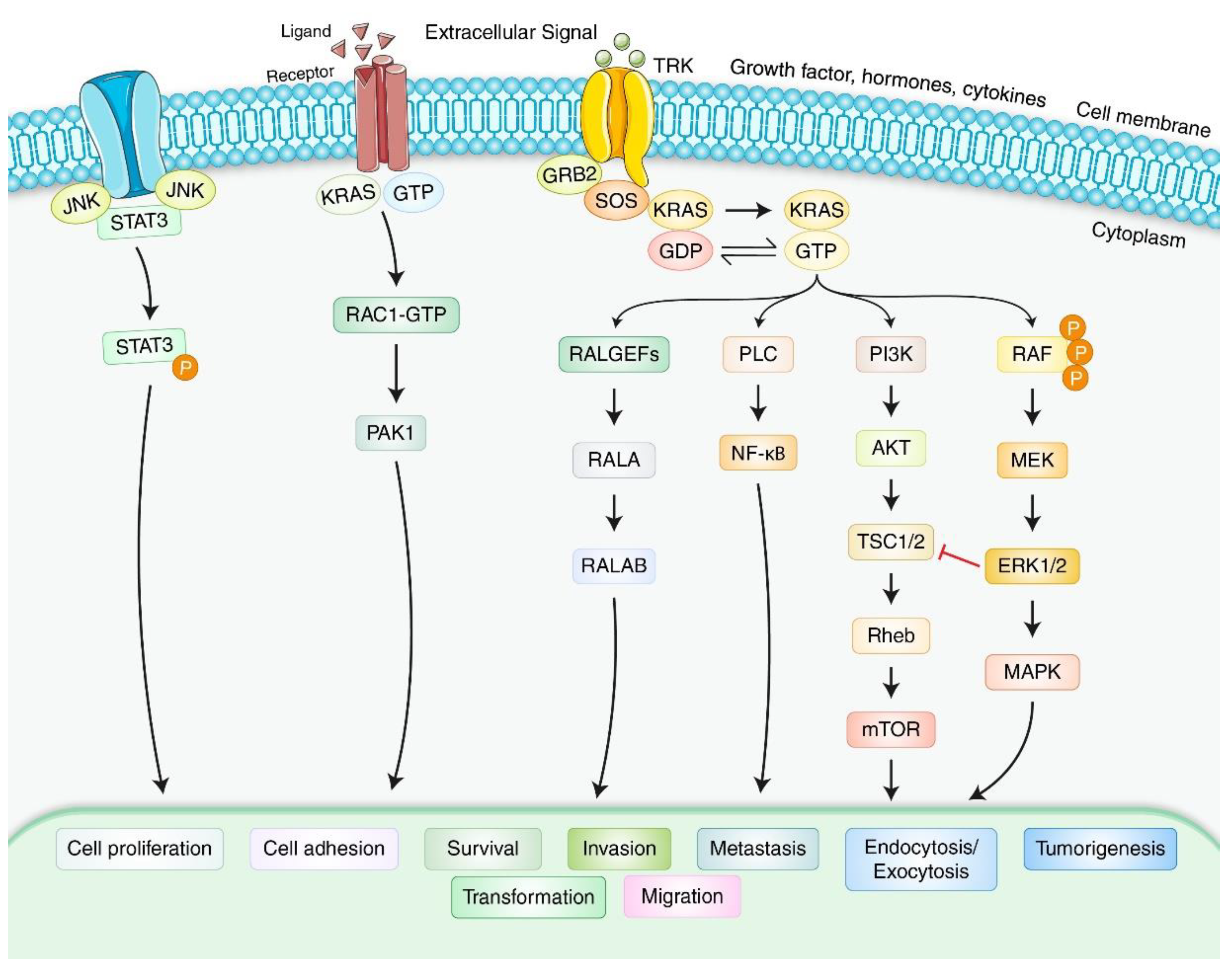{kind=link}
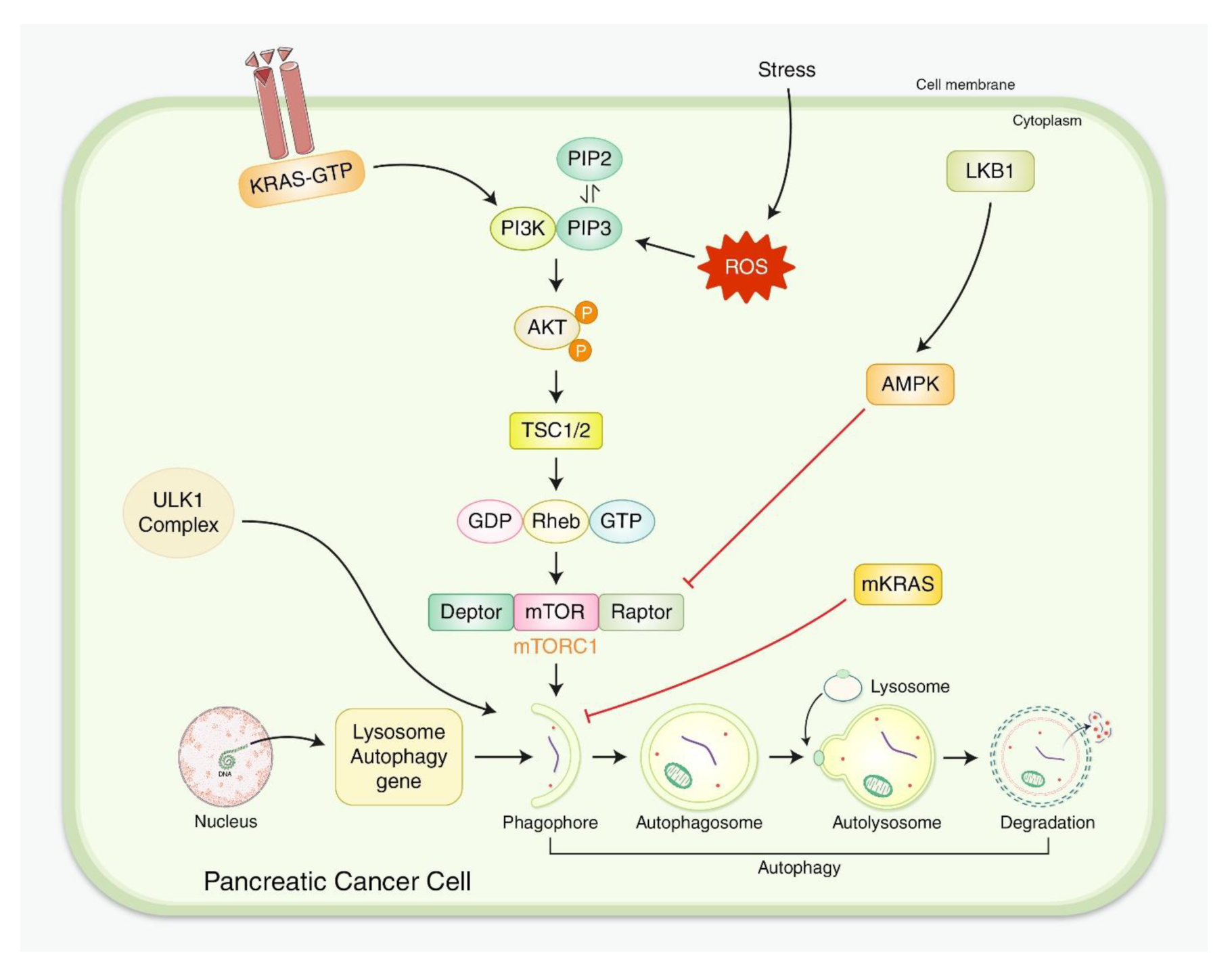{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Molecular Pathway of the Autophagy Process
4. Molecular Mechanism and Formation of Pancreatic Adenocarcinoma
5. Interactions for Both Autophagy and PDAC
5.1. Positive Role of Autophagy in PDAC Development
5.2. Adverse Role of Autophagy in PDAC Development
6. Autophagy-Based Treatment Strategy for PDAC
7. Pharmacological Modulation of Autophagy in PDAC Regulation
7.1. Autophagy Inhibition in PDAC Regulation
7.2. Autophagy Activation in PDAC Regulation
8. Limitations and Prospective Regulation of PDAC via Autophagy
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Naudin, S.; Viallon, V.; Hashim, D.; Freisling, H.; Jenab, M.; Weiderpass, E.; Perrier, F.; McKenzie, F.; Bueno-de-Mesquita, H.B.; Olsen, A.; et al. Healthy lifestyle and the risk of pancreatic cancer in the EPIC study. Eur. J. Epidemiol. 2020, 35, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.; Conroy, T.; Ducreux, M. Future directions in drug development in pancreatic cancer. Semin. Oncol. 2021, 48, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.H.; Cho, Y.S.; Choi, J.Y.; Lee, K.-H.; Lee, J.K.; Min, J.H.; Hyun, S.H. Imaging phenotype using 18 F-fluorodeoxyglucose positron emission tomography-based radiomics and genetic alterations of pancreatic ductal adenocarcinoma. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Ji, Y.; Jiang, H.; Qiu, G. Clinical Effect of Driver Mutations of KRAS, CDKN2A/P16, TP53, and SMAD4 in Pancreatic Cancer: A Meta-Analysis. Genet. Test. Mol. Biomark. 2020, 24, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Görgülü, K.; Diakopoulos, K.N.; Kaya-Aksoy, E.; Ciecielski, K.J.; Ai, J.; Lesina, M.; Algül, H. The role of autophagy in pancreatic cancer: From bench to the dark bedside. Cells 2020, 9, 1063. [Google Scholar] [CrossRef]
- Guo, Q.Q.; Wang, S.S.; Zhang, S.S.; Xu, H.D.; Li, X.M.; Guan, Y.; Yi, F.; Zhou, T.T.; Jiang, B.; Bai, N. ATM-CHK 2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rhim, H. Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep. 2017, 50, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Pavlakis, E.; Neumann, M.; Stiewe, T. Extracellular Vesicles: Messengers of p53 in Tumor–Stroma Communication and Cancer Metastasis. Int. J. Mol. Sci. 2020, 21, 9648. [Google Scholar] [CrossRef]
- Uddin, M.S.; Rahman, M.A.; Kabir, M.T.; Behl, T.; Mathew, B.; Perveen, A.; Barreto, G.E.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Multifarious roles of mTOR signaling in cognitive aging and cerebrovascular dysfunction of Alzheimer’s disease. IUBMB Life 2020, 72, 1843–1855. [Google Scholar] [CrossRef]
- Jangra, A.; Arora, M.K.; Kisku, A.; Sharma, S. The multifaceted role of mangiferin in health and diseases: A review. Adv. Tradit. Med. 2020, 21, 619–643. [Google Scholar] [CrossRef]
- Sato, M.; Ueda, E.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H.; Seki, T. Glucocorticoids negatively regulates chaperone mediated autophagy and microautophagy. Biochem. Biophys. Res. Commun. 2020, 528, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Park, M.N.; Rahman, M.H.; Rashid, M.M.; Islam, R.; Uddin, M.J.; Hannan, M.A.; Kim, B. p53 Modulation of Autophagy Signaling in Cancer Therapies: Perspectives Mechanism and Therapeutic Targets. Front. Cell Dev. Biol. 2022, 10, 761080. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Ahmed, K.R.; Rahman, M.H.; Park, M.N.; Kim, B. Potential Therapeutic Action of Autophagy in Gastric Cancer Managements: Novel Treatment Strategies and Pharmacological Interventions. Front. Pharmacol. 2021, 12, 813703. [Google Scholar] [CrossRef]
- Reyes-Castellanos, G.; Abdel Hadi, N.; Carrier, A. Autophagy Contributes to Metabolic Reprogramming and Therapeutic Resistance in Pancreatic Tumors. Cells 2022, 11, 426. [Google Scholar] [CrossRef]
- Zhang, S.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Liu, J.; He, G.; Zheng, H.; Yang, L.; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis 2022, 13, 132. [Google Scholar] [CrossRef]
- Wang, X.; Lee, J.; Xie, C. Autophagy Regulation on Cancer Stem Cell Maintenance, Metastasis, and Therapy Resistance. Cancers 2022, 14, 381. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, C.; Yang, G.; Zhao, B.; Wang, W. The role of autophagy in pancreatic cancer progression. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188592. [Google Scholar] [CrossRef]
- Kiruthiga, C.; Devi, K.P.; Nabavi, S.M.; Bishayee, A. Autophagy: A potential therapeutic target of polyphenols in hepatocellular carcinoma. Cancers 2020, 12, 562. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, Y.; Hu, W.; Wang, X.; Hu, J.; Yuan, C.; Zhou, C.; Wang, H.; Du, J.; Wang, Y.; et al. TEOA Promotes Autophagic Cell Death via ROS-Mediated Inhibition of mTOR/p70S6k Signaling Pathway in Pancreatic Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 734818. [Google Scholar] [CrossRef]
- Abernathy, E.; Mateo, R.; Majzoub, K.; van Buuren, N.; Bird, S.W.; Carette, J.E.; Kirkegaard, K. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 2019, 17, e2006926. [Google Scholar] [CrossRef] [Green Version]
- Shabaninia, M.; Tourchi, A.; Di Carlo, H.; Gearhart, J.P. Autophagy, apoptosis, and cell proliferation in exstrophy-epispadias complex. Urology 2018, 111, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, C.; Li, R.; Zhang, L.; Tian, J. TEOA, a triterpenoid from Actinidia eriantha, induces autophagy in SW620 cells via endoplasmic reticulum stress and ROS-dependent mitophagy. Arch. Pharm. Res. 2017, 40, 579–591. [Google Scholar] [CrossRef]
- Cordani, M.; Sánchez-Álvarez, M.; Strippoli, R.; Bazhin, A.V.; Donadelli, M. Sestrins at the interface of ROS control and autophagy regulation in health and disease. Oxidative Med. Cell. Longev. 2019, 2019, 1283075. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Jia, J.; Zhang, X.; Dai, H. Selective binding of mitophagy receptor protein Bcl-rambo to LC3/GABARAP family proteins. Biochem. Biophys. Res. Commun. 2020, 530, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Chmurska, A.; Matczak, K.; Marczak, A. Two Faces of Autophagy in the Struggle against Cancer. Int. J. Mol. Sci. 2021, 22, 2981. [Google Scholar] [CrossRef]
- Rahman, M.A.; Cho, Y.; Nam, G.; Rhim, H. Antioxidant Compound, Oxyresveratrol, Inhibits APP Production through the AMPK/ULK1/mTOR-Mediated Autophagy Pathway in Mouse Cortical Astrocytes. Antioxidants 2021, 10, 408. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hannan, M.A.; Dash, R.; Rahman, M.H.; Islam, R.; Uddin, M.J.; Sohag, A.A.; Rahman, M.H.; Rhim, H. Phytochemicals as a Complement to Cancer Chemotherapy: Pharmacological Modulation of the Autophagy-Apoptosis Pathway. Front. Pharmacol. 2021, 12, 639628. [Google Scholar] [CrossRef]
- Li, X.; Sun, L.; Yan, G.; Yan, X. PFKP facilitates ATG4B phosphorylation during amino acid deprivation-induced autophagy. Cell. Signal. 2021, 82, 109956. [Google Scholar] [CrossRef]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Akter, S.; Rahman, M.A.; Hasan, M.N.; Akhter, H.; Noor, P.; Islam, R.; Shin, Y.; Rahman, M.D.H.; Gazi, M.S.; Huda, M.N.; et al. Recent Advances in Ovarian Cancer: Therapeutic Strategies, Potential Biomarkers, and Technological Improvements. Cells 2022, 11, 650. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Rahman, M.H.; Rashid, A.N.M.M.; Hwang, H.; Chung, S.; Kim, B.; Rhim, H. Autophagy Modulation in Aggresome Formation: Emerging Implications and Treatments of Alzheimer’s Disease. Biomedicines 2022, 10, 1027. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Liu, X.; Wang, H.; Yang, X.; Gu, Y. Glycolysis in the progression of pancreatic cancer. Am. J. Cancer Res. 2022, 12, 861. [Google Scholar] [PubMed]
- Li, F.-J.; Xu, Z.-S.; Soo, A.D.; Lun, Z.-R.; He, C.Y. ATP-driven and AMPK-independent autophagy in an early branching eukaryotic parasite. Autophagy 2017, 13, 715–729. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Li, Q.; Qin, X.; Zhang, M.; Du, Q.; Luan, Y. An Injectable Hydrogel Reshaping Adenosinergic Axis for Cancer Therapy. Adv. Funct. Mater. 2022, 32, 2200801. [Google Scholar] [CrossRef]
- Eng, C.; Abraham, R. The autophagy conundrum in cancer: Influence of tumorigenic metabolic reprogramming. Oncogene 2011, 30, 4687–4696. [Google Scholar] [CrossRef] [Green Version]
- Webster, B.R. GCN5L1 Functions as a Mitochondrial Acetyltransferase that Regulates Mitophagy; The University of New Mexico: Albuquerque, NM, USA, 2015. [Google Scholar]
- Grasso, D.; Garcia, M.N.; Iovanna, J.L. Autophagy in pancreatic cancer. Int. J. Cell Biol. 2012, 2012, 760498. [Google Scholar] [CrossRef] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef]
- Cayron, C.; Guillermet-Guibert, J. The type of KRAS mutation drives PI3Kα/γ signalling dependency: Implication for the choice of targeted therapy in pancreatic adenocarcinoma patients. Clin. Res. Hepatol. Gastroenterol. 2020, 45, 101473. [Google Scholar] [CrossRef] [PubMed]
- Lentsch, E.; Li, L.; Pfeffer, S.; Ekici, A.B.; Taher, L.; Pilarsky, C.; Grützmann, R. CRISPR/Cas9-mediated knock-out of krasG12D mutated pancreatic cancer cell lines. Int. J. Mol. Sci. 2019, 20, 5706. [Google Scholar] [CrossRef] [Green Version]
- Hutchings, D.; Waters, K.M.; Weiss, M.J.; Wolfgang, C.L.; Makary, M.A.; He, J.; Cameron, J.L.; Wood, L.D.; Hruban, R.H. Cancerization of the pancreatic ducts: Demonstration of a common and under-recognized process using immunolabeling of paired duct lesions and invasive pancreatic ductal adenocarcinoma for p53 and Smad4 expression. Am. J. Surg. Pathol. 2018, 42, 1556. [Google Scholar] [CrossRef]
- Sagami, R.; Yamao, K.; Nakahodo, J.; Minami, R.; Tsurusaki, M.; Murakami, K.; Amano, Y. Pre-Operative Imaging and Pathological Diagnosis of Localized High-Grade Pancreatic Intra-Epithelial Neoplasia without Invasive Carcinoma. Cancers 2021, 13, 945. [Google Scholar] [CrossRef]
- Fujikura, K.; Hutchings, D.; Braxton, A.M.; Zhu, Q.; Laheru, D.A.; Hruban, R.H.; Thompson, E.D.; Wood, L.D. Intraductal pancreatic cancer is less responsive than cancer in the stroma to neoadjuvant chemotherapy. Mod. Pathol. 2020, 33, 2026–2034. [Google Scholar] [CrossRef]
- Dardare, J.; Witz, A.; Merlin, J.-L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ pathway in patients with pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-E.; Yoo, J.E.; Kim, J.; Kim, S.; Kim, S.; Lee, H.; Cheong, H. NEDD4L downregulates autophagy and cell growth by modulating ULK1 and a glutamine transporter. Cell Death Dis. 2020, 11, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusini, M.; Nenna, A.; Chello, C.; Greco, S.M.; Gagliardi, I.; Nappi, F.; Chello, M. Role of autophagy in aneurysm and dissection of the ascending aorta. Future Cardiol. 2020, 16, 517–526. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Rahman, M.H.; Biswas, P.; Hossain, M.S.; Islam, R.; Hannan, M.A.; Uddin, M.J.; Rhim, H. Potential Therapeutic Role of Phytochemicals to Mitigate Mitochondrial Dysfunctions in Alzheimer’s Disease. Antioxidants 2020, 10, 23. [Google Scholar] [CrossRef]
- Wang, X.; Wu, R.; Liu, Y.; Zhao, Y.; Bi, Z.; Yao, Y.; Liu, Q.; Shi, H.; Wang, F.; Wang, Y. m6A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7. Autophagy 2020, 16, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Gao, S.-J. Targeting XPO1 enhances innate immune response and inhibits KSHV lytic replication during primary infection by nuclear stabilization of the p62 autophagy adaptor protein. Cell Death Dis. 2021, 12, 29. [Google Scholar] [CrossRef]
- Zhou, S.; Zhang, M.; Zhou, C.; Wang, W.; Yang, H.; Ye, W. CDKL3 Targets ATG5 to Promote Carcinogenesis of Esophageal Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 1602. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Tooze, S. The Role of Autophagy in Pancreatic Cancer—Recent Advances. Biology 2020, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, Y.H.; Wang, Y.; Kong, K.; Grzeskowiak, C.; Zagorodna, O.; Dogruluk, T.; Lu, H.; Villafane, N.; Bhavana, V.H.; Moreno, D. Differential expression of MAGEA6 toggles autophagy to promote pancreatic cancer progression. Elife 2020, 9, e48963. [Google Scholar] [CrossRef]
- New, M.; Van Acker, T.; Long, J.S.; Sakamaki, J.I.; Ryan, K.M.; Tooze, S.A. Molecular Pathways Controlling Autophagy in Pancreatic Cancer. Front. Oncol. 2017, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Papademetrio, D.L.; Garcia, M.N.; Grasso, D.; Alvarez, É. Autophagy-Mediated Exosomes as Immunomodulators of Natural Killer Cells in Pancreatic Cancer Microenvironment. Front. Oncol. 2021, 10, 3444. [Google Scholar] [CrossRef]
- Yang, M.C.; Wang, H.C.; Hou, Y.C.; Tung, H.L.; Chiu, T.J.; Shan, Y.S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 179. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Yang, J.; Ren, B.; Wang, H.; Yang, G.; Chen, Y.; You, L.; Zhao, Y. Reprogramming of Amino Acid Metabolism in Pancreatic Cancer: Recent Advances and Therapeutic Strategies. Front. Oncol. 2020, 10, 572722. [Google Scholar] [CrossRef]
- Liu, H.; Shi, Y.; Qian, F. Opportunities and delusions regarding drug delivery targeting pancreatic cancer-associated fibroblasts. Adv. Drug Deliv. Rev. 2021, 172, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Guo, H.; Cao, J. The importance of autophagy regulation in obstructive sleep apnea. Sleep Breath. 2021, 25, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011, 140, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Baba, Y.; Kitano, Y.; Miyake, K.; Zhang, X.; Yamamura, K.; Kosumi, K.; Kaida, T.; Arima, K.; Taki, K. KRAS, BRAF, and PIK3CA mutations, and patient prognosis in 126 pancreatic cancers: Pyrosequencing technology and literature review. Med. Oncol. 2016, 33, 32. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Lee, C.-S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef] [Green Version]
- Ré, A.E.L.; Fernández-Barrena, M.G.; Almada, L.L.; Mills, L.D.; Elsawa, S.F.; Lund, G.; Ropolo, A.; Molejon, M.I.; Vaccaro, M.I.; Fernandez-Zapico, M.E. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J. Biol. Chem. 2012, 287, 25325–25334. [Google Scholar]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.H.; Tian, C.; Meng, Y.; Qin, Y.W.; Du, Y.H.; Du, J.; Li, H.H. Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells. J. Cell. Physiol. 2012, 227, 127–135. [Google Scholar] [CrossRef]
- Maehara, S.I.; Tanaka, S.; Shimada, M.; Shirabe, K.; Saito, Y.; Takahashi, K.; Maehara, Y. Selenoprotein P, as a predictor for evaluating gemcitabine resistance in human pancreatic cancer cells. Int. J. Cancer 2004, 112, 184–189. [Google Scholar] [CrossRef]
- Sun, T. Long noncoding RNAs act as regulators of autophagy in cancer. Pharmacol. Res. 2018, 129, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Y.; Zhang, W.; Li, J.; Liu, W.; Lu, W. Long non-coding RNA SNHG14 exerts oncogenic functions in non-small cell lung cancer through acting as an miR-340 sponge. Biosci. Rep. 2019, 39, BSR20180941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A coordinator of membrane-associated cargo trafficking and autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Saftig, P.; Beertsen, W.; Eskelinen, E.-L. LAMP-2: A control step for phagosome and autophagosome maturation. Autophagy 2008, 4, 510–512. [Google Scholar] [CrossRef] [Green Version]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, H.; Affonso, C.; Bonomo, R.; Mañas, A.; Xiang, J. Deficiency in ubiquitin-like protein Ubl4A impairs migration of fibroblasts and macrophages. Biochem. Biophys. Res. Commun. 2017, 483, 617–623. [Google Scholar] [CrossRef]
- Chen, H.; Li, L.; Hu, J.; Zhao, Z.; Ji, L.; Cheng, C.; Zhang, G.; Li, Y.; Chen, H.; Pan, S. UBL4A inhibits autophagy-mediated proliferation and metastasis of pancreatic ductal adenocarcinoma via targeting LAMP1. J. Exp. Clin. Cancer Res. 2019, 38, 297. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Yue, B.Y. Optineurin: The autophagy connection. Exp. Eye Res. 2016, 144, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, J.; Xu, Y.; Wu, R.; Chen, X.; Song, X.; Zeh, H.; Kang, R.; Klionsky, D.J.; Wang, X. Tumor Heterogeneity in Autophagy-Dependent Ferroptosis. Autophagy 2021, 17, 3361–3374. [Google Scholar] [CrossRef]
- Xu, J.; Song, J.; Yang, X.; Guo, J.; Wang, T.; Zhuo, W. ProNGF siRNA inhibits cell proliferation and invasion of pancreatic cancer cells and promotes anoikis. Biomed. Pharmacother. 2019, 111, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Akar, U.; Mehta, K.; Lopez-Berenstein, G. PKCδ and tissue transglutaminase are novel inhibitors of autophagy in pancreatic cancer cells (Autophagy (2007) 3 (480–483)). Autophagy 2009, 5, 283. [Google Scholar] [CrossRef] [Green Version]
- Ashour, A.A.; Abdel-Aziz, A.-A.H.; Mansour, A.M.; Alpay, S.N.; Huo, L.; Ozpolat, B. Targeting elongation factor-2 kinase (eEF-2K) induces apoptosis in human pancreatic cancer cells. Apoptosis 2014, 19, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrzenjak, A.; Kremser, M.L.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef]
- Mujumdar, N.; Mackenzie, T.N.; Dudeja, V.; Chugh, R.; Antonoff, M.B.; Borja–Cacho, D.; Sangwan, V.; Dawra, R.; Vickers, S.M.; Saluja, A.K. Triptolide induces cell death in pancreatic cancer cells by apoptotic and autophagic pathways. Gastroenterology 2010, 139, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Udelnow, A.; Kreyes, A.; Ellinger, S.; Landfester, K.; Walther, P.; Klapperstueck, T.; Wohlrab, J.; Henne-Bruns, D.; Knippschild, U.; Würl, P. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PLoS ONE 2011, 6, e20143. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Bläuer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637. [Google Scholar] [CrossRef] [Green Version]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: A phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T. Protective autophagy elicited by RAF→ MEK→ ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.B.; Marchetti, K.R.; Castria, T.B.; Jardim, D.L.F.; Fernandes, G.S. Trametinib and Hydroxychloroquine (HCQ) Combination Treatment in KRAS-Mutated Advanced Pancreatic Adenocarcinoma: Detailed Description of Two Cases. J. Gastrointest. Cancer 2021, 52, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Halwani, A.A. Development of Pharmaceutical Nanomedicines: From the Bench to the Market. Pharmaceutics 2022, 14, 106. [Google Scholar] [CrossRef] [PubMed]
- Jooste, V.; Bengrine-Lefevre, L.; Manfredi, S.; Quipourt, V.; Grosclaude, P.; Facy, O.; Lepage, C.; Ghiringhelli, F.; Bouvier, A.-M. Management and Outcomes of Pancreatic Cancer in French Real-World Clinical Practice. Cancers 2022, 14, 1675. [Google Scholar] [CrossRef]
- Peddi, S.; MacKay, J.A. Berunda Polypeptides Carrying Rapalogues Inhibit Tumor mTORC1 Better than Oral Everolimus. Biomacromolecules 2020, 21, 3038–3046. [Google Scholar] [CrossRef]
- Foth, M.; Garrido-Laguna, I.; Kinsey, C.G. Therapeutic Targeting of Autophagy in Pancreatic Cancer. Surg. Oncol. Clin. 2021, 30, 709–718. [Google Scholar] [CrossRef]
- De Dosso, S.; Siebenhüner, A.R.; Winder, T.; Meisel, A.; Fritsch, R.; Astaras, C.; Szturz, P.; Borner, M. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 2021, 96, 102180. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Fanta, P.T.; Lowy, A.M. Adenocarcinoma of the pancreas. In Yamada’s Textbook of Gastroenterology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2022; pp. 1666–1688. [Google Scholar]
- Ganesh, A.N.; Heusser, C.; Garad, S.; Sánchez-Félix, M.V. Patient-centric design for peptide delivery: Trends in routes of administration and advancement in drug delivery technologies. Med. Drug Discov. 2021, 9, 100079. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477. [Google Scholar] [CrossRef]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Hannan, M.A.; Rahman, M.A.; Rahman, M.S.; Sohag, A.A.M.; Dash, R.; Hossain, K.S.; Farjana, M.; Uddin, M.J. Intermittent fasting, a possible priming tool for host defense against SARS-CoV-2 infection: Crosstalk among calorie restriction, autophagy and immune response. Immunol. Lett. 2020, 226, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ock, C.Y.; Park, J.-M.; Han, Y.-M.; Jeong, M.; Kim, M.-Y.; Lee, H.J.; Hahm, K.B. Genetic ablation or pharmacologic inhibition of autophagy mitigated NSAID-associated gastric damages. J. Mol. Med. 2017, 95, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elrahman, K.S.; Hamilton, A.; Vasefi, M.; Ferguson, S.S. Autophagy is increased following either pharmacological or genetic silencing of mGluR5 signaling in Alzheimer’s disease mouse models. Mol. Brain 2018, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Autophagy and its effects: Making sense of double-edged swords. PLoS Biol. 2014, 12, e1001967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarretta, S.; Zhai, P.; Volpe, M.; Sadoshima, J. Pharmacological modulation of autophagy during cardiac stress. J. Cardiovasc. Pharmacol. 2012, 60, 235. [Google Scholar] [CrossRef] [Green Version]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy modulation in cancer: Current knowledge on action and therapy. Oxidative Med. Cell. Longev. 2018, 2018, 8023821. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.H.; Cho, K.S.; Hwang, J.J.; Lee, S.-J.; Choi, J.A.; Koh, J.-Y. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6030–6037. [Google Scholar] [CrossRef] [Green Version]
- Plantone, D.; Koudriavtseva, T. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: A mini-review. Clin. Drug Investig. 2018, 38, 653–671. [Google Scholar] [CrossRef]
- Mo, J. Zinc Sulfide-Based Hybrid Exosome-Coated Autophagy-Mediated H2S-sensitized PDT/chemotherapeutic Synergistic Nanoplatform for Targeted Treatment of Glioblastoma Stem-Like Cells in Orthotopic Mouse Glioblastoma Model. bioRxiv 2020. [Google Scholar] [CrossRef]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45. [Google Scholar] [CrossRef] [Green Version]
- Nicastri, M.C. Design, Synthesis, and Characterization of Dimeric Lysosomal Inhibitors and Their Effect on Cancer Biology. Ph.D. Thesis, University of Pennsylvania, Philadelphia, PA, USA, 2018. [Google Scholar]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Brit. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Sohel, M.; Biswas, P.; Al Amin, M.; Hossain, M.A.; Sultana, H.; Dey, D.; Aktar, S.; Setu, A.; Khan, M.S.; Paul, P.; et al. Genistein, a Potential Phytochemical against Breast Cancer Treatment-Insight into the Molecular Mechanisms. Processes 2022, 10, 415. [Google Scholar] [CrossRef]
- Allavena, G.; Boyd, C.; Oo, K.S.; Maellaro, E.; Zhivotovsky, B.; Kaminskyy, V.O. Suppressed translation and ULK1 degradation as potential mechanisms of autophagy limitation under prolonged starvation. Autophagy 2016, 12, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Chude, C.I.; Amaravadi, R.K. Targeting autophagy in cancer: Update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Yang, F.; Wang, Q.; Shen, Q.; Huang, J.; Peng, C.; Zhang, Y.; Wan, W.; Wong, C.C.; Sun, Q. VPS34 acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy. Mol. Cell 2017, 67, 907–921.e907. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Al Abdullah, A.; Rahman, M.A.; Kabir, M.T.; Alkahtani, S.; Alanazi, I.S.; Perveen, A.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Exploring the Promise of Flavonoids to Combat Neuropathic Pain: From Molecular Mechanisms to Therapeutic Implications. Front. Neurosci. 2020, 14, 478. [Google Scholar] [CrossRef]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- Betin, V.M.; Singleton, B.K.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. Autophagy facilitates organelle clearance during differentiation of human erythroblasts: Evidence for a role for ATG4 paralogs during autophagosome maturation. Autophagy 2013, 9, 881–893. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.-Q.; Ni, T.; Hong, B.; Wang, H.-Y.; Jiang, F.-J.; Zou, S.; Chen, Y.; Zheng, X.-L.; Klionsky, D.J.; Liang, Y. Dual roles of Atg8− PE deconjugation by Atg4 in autophagy. Autophagy 2012, 8, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, B.; Benavides, A.; Shi, Y.; Yang, Y.; Frost, P.; Gera, J.; Lichtenstein, A. The PP242 mammalian target of rapamycin (mTOR) inhibitor activates extracellular signal-regulated kinase (ERK) in multiple myeloma cells via a target of rapamycin complex 1 (TORC1)/eukaryotic translation initiation factor 4E (eIF-4E)/RAF pathway and activation is a mechanism of resistance. J. Biol. Chem. 2012, 287, 21796–21805. [Google Scholar] [PubMed] [Green Version]
- Walter, K.; Rodriguez-Aznar, E.; Ferreira, M.S.V.; Frappart, P.-O.; Dittrich, T.; Tiwary, K.; Meessen, S.; Lerma, L.; Daiss, N.; Schulte, L.-A. A feedback-loop between telomerase activity and stemness factors regulates PDAC stem cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Colhado Rodrigues, B.L.; Lallo, M.A.; Perez, E.C. The controversial role of autophagy in tumor development: A systematic review. Immunol. Investig. 2020, 49, 386–396. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, L.; Zhou, H.; Wang, W.; Luo, Y.; Yang, H.; Yi, H. Inhibition of autophagy promotes cisplatin-induced apoptotic cell death through Atg5 and Beclin 1 in A549 human lung cancer cells. Mol. Med. Rep. 2018, 17, 6859–6865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Shen, X.; Tao, B.; Lin, C.; Li, K.; Luo, Z.; Cai, K. The nanoparticle-facilitated autophagy inhibition of cancer stem cells for improved chemotherapeutic effects on glioblastomas. J. Mater. Chem. B 2019, 7, 2054–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Saha, S.K.; Rahman, M.S.; Uddin, M.J.; Uddin, M.S.; Pang, M.G.; Rhim, H.; Cho, S.G. Molecular Insights Into Therapeutic Potential of Autophagy Modulation by Natural Products for Cancer Stem Cells. Front. Cell Dev. Biol. 2020, 8, 283. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.A.; Ahmed, K.R.; Rahman, M.H.; Parvez, M.A.K.; Lee, I.-S.; Kim, B. Therapeutic Aspects and Molecular Targets of Autophagy to Control Pancreatic Cancer Management. Biomedicines 2022, 10, 1459. https://doi.org/10.3390/biomedicines10061459
Rahman MA, Ahmed KR, Rahman MH, Parvez MAK, Lee I-S, Kim B. Therapeutic Aspects and Molecular Targets of Autophagy to Control Pancreatic Cancer Management. Biomedicines. 2022; 10(6):1459. https://doi.org/10.3390/biomedicines10061459
Chicago/Turabian StyleRahman, Md. Ataur, Kazi Rejvee Ahmed, MD. Hasanur Rahman, Md. Anowar Khasru Parvez, In-Seon Lee, and Bonglee Kim. 2022. "Therapeutic Aspects and Molecular Targets of Autophagy to Control Pancreatic Cancer Management" Biomedicines 10, no. 6: 1459. https://doi.org/10.3390/biomedicines10061459
APA StyleRahman, M. A., Ahmed, K. R., Rahman, M. H., Parvez, M. A. K., Lee, I. -S., & Kim, B. (2022). Therapeutic Aspects and Molecular Targets of Autophagy to Control Pancreatic Cancer Management. Biomedicines, 10(6), 1459. https://doi.org/10.3390/biomedicines10061459